NMR-Based Metabolic Profiles of Intact Zebrafish Embryos Exposed to Aflatoxin B1 Recapitulates Hepatotoxicity and Supports Possible Neurotoxicity



Abstract
:1. Introduction
2. Results
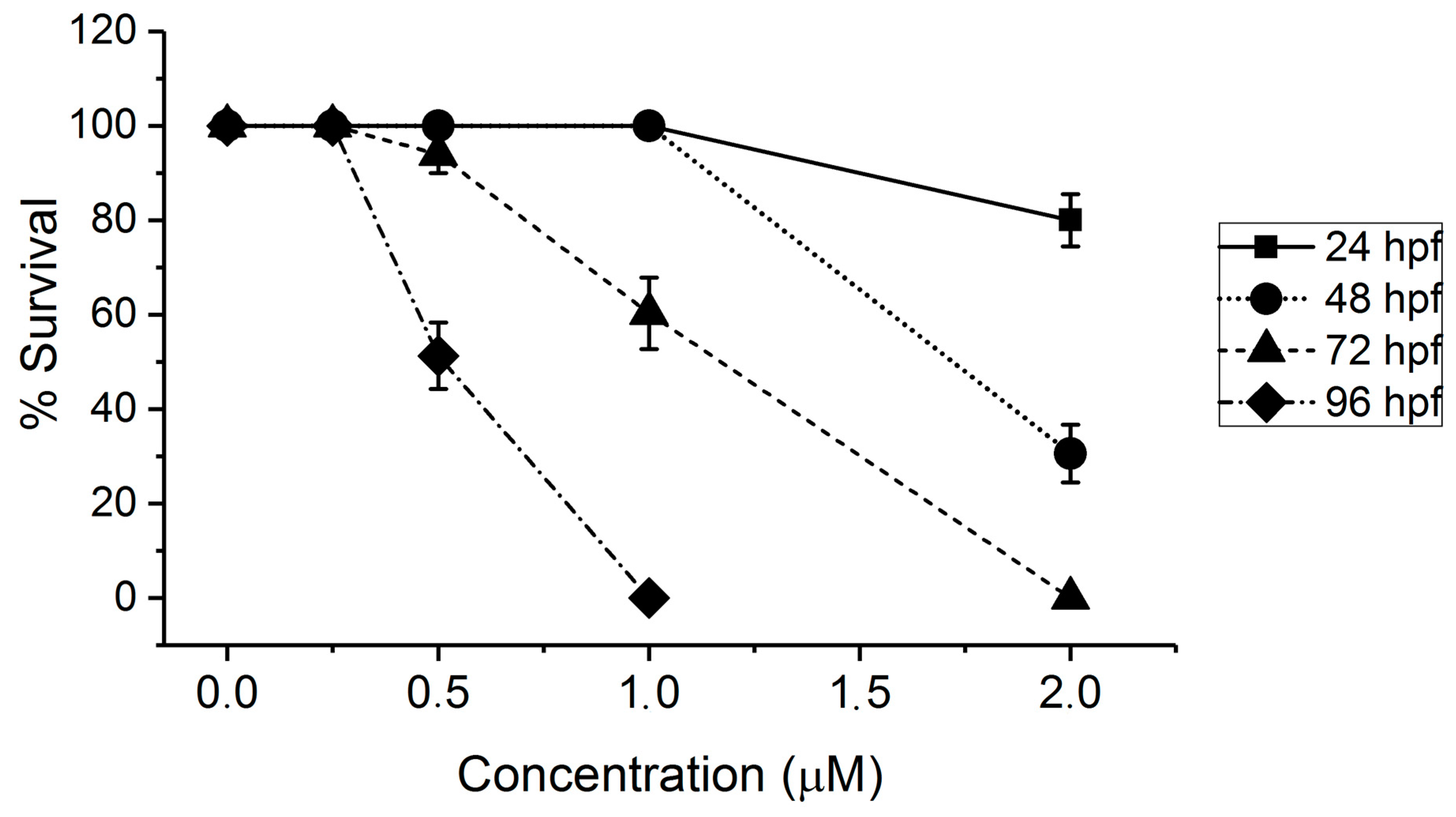
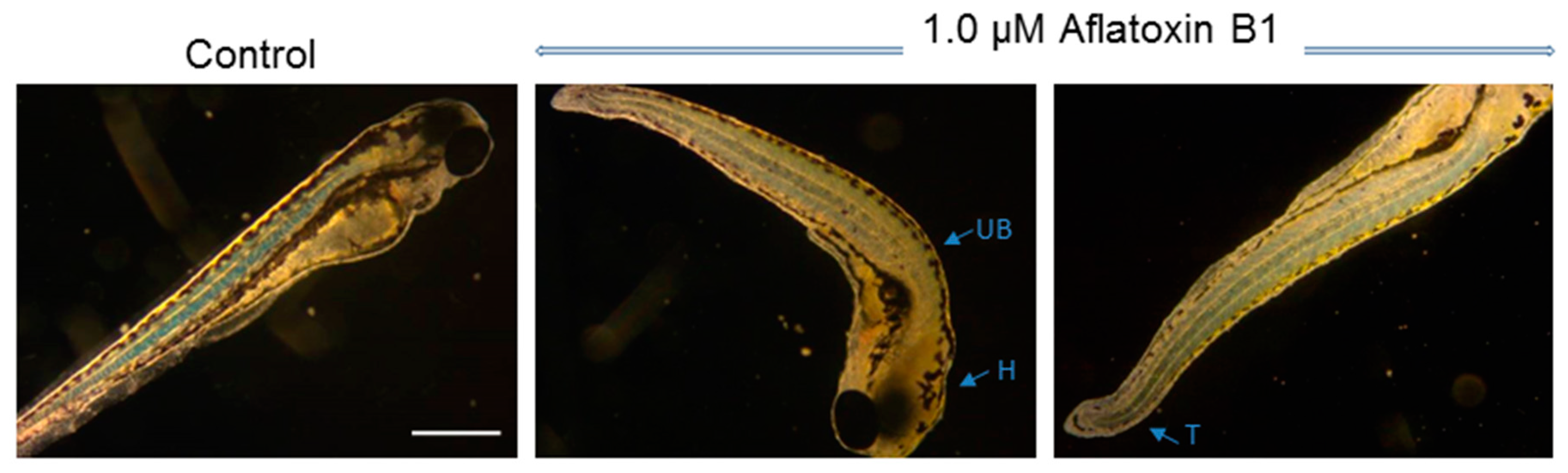
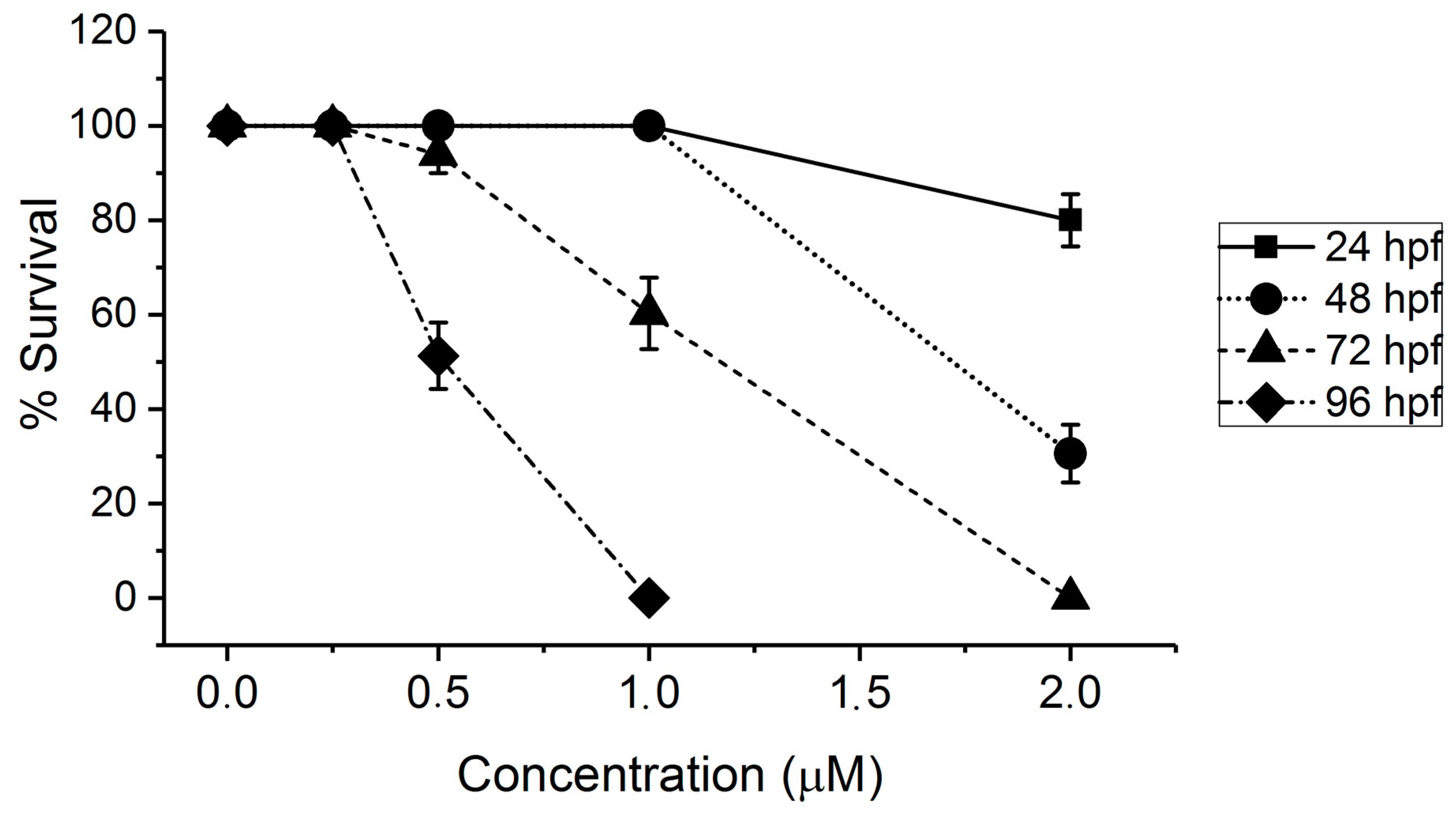
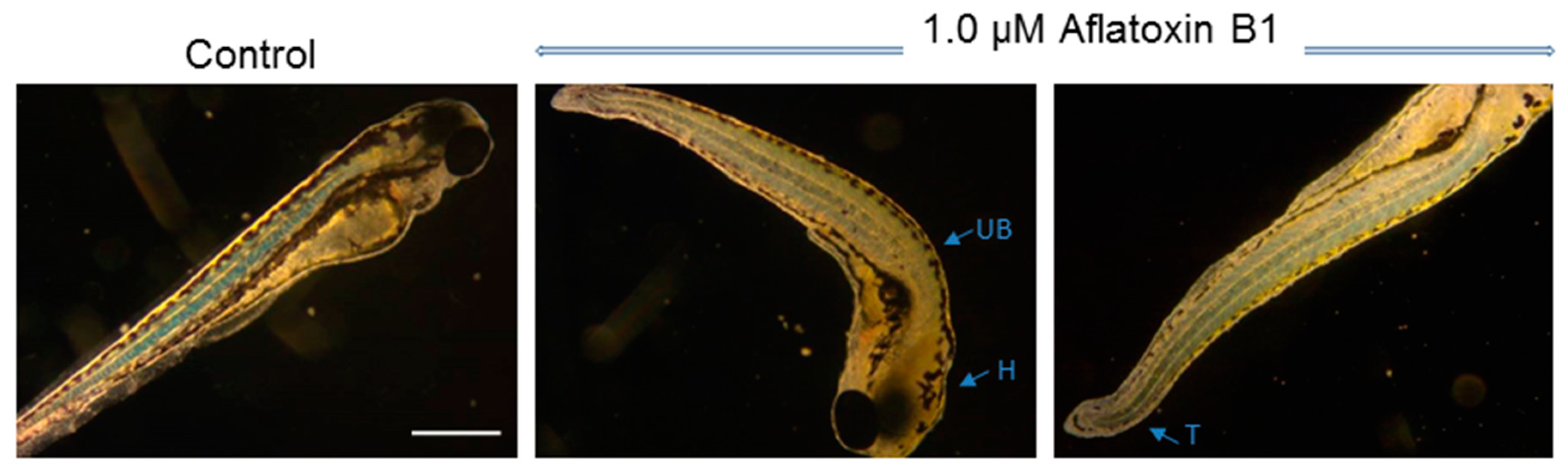
2.1. Toxicity of AFB1 in the Zebrafish Embryo Model
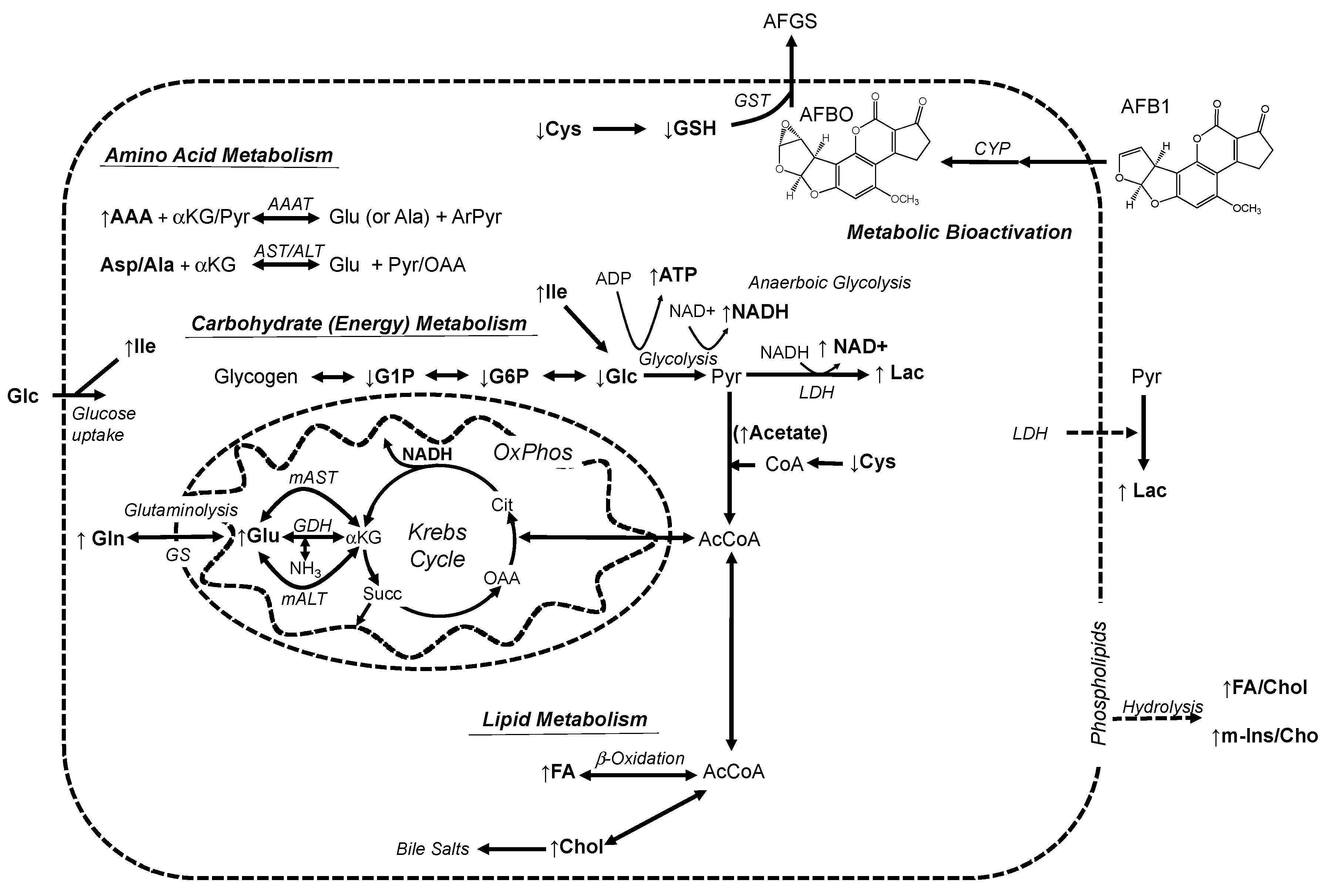
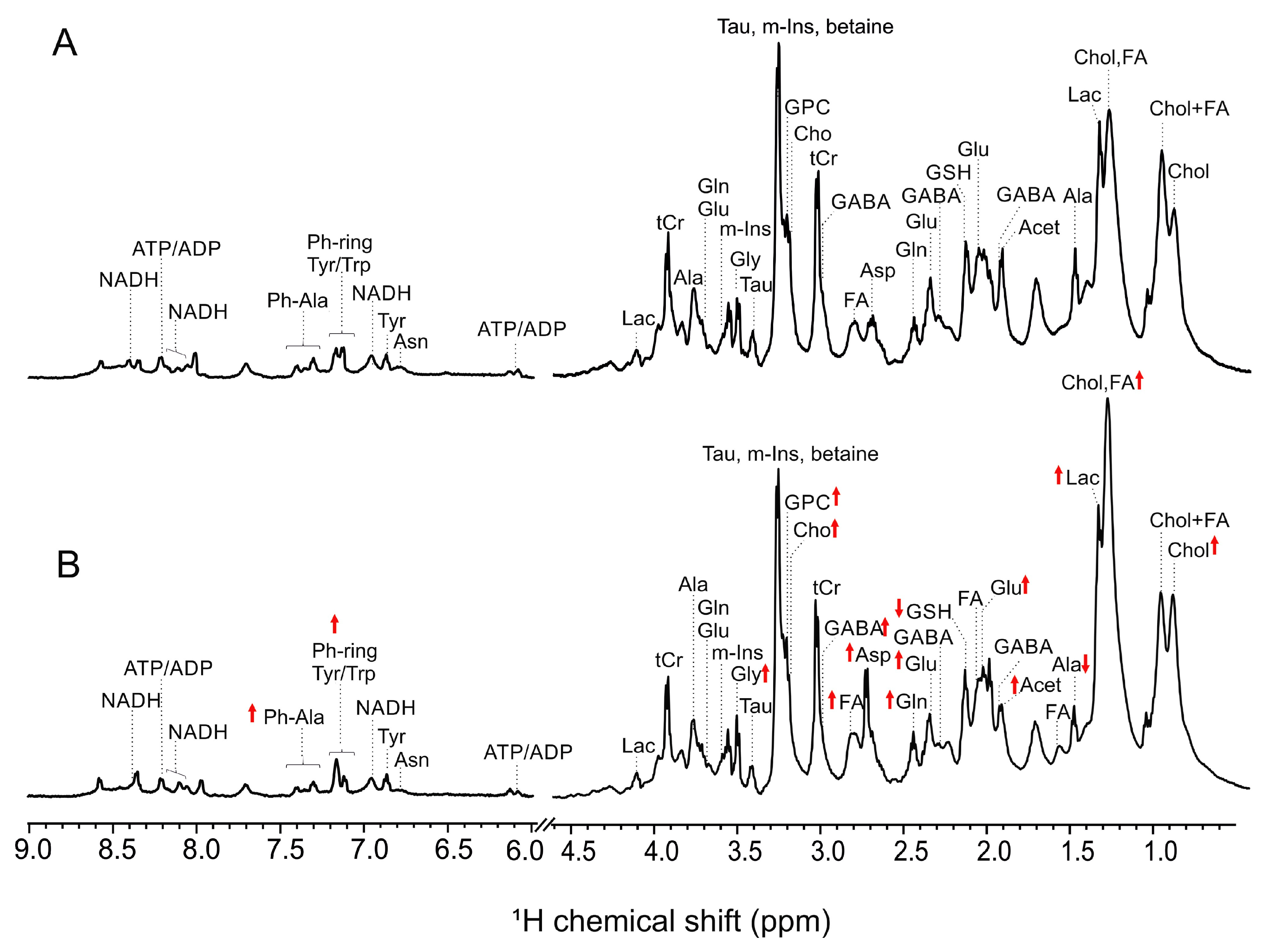
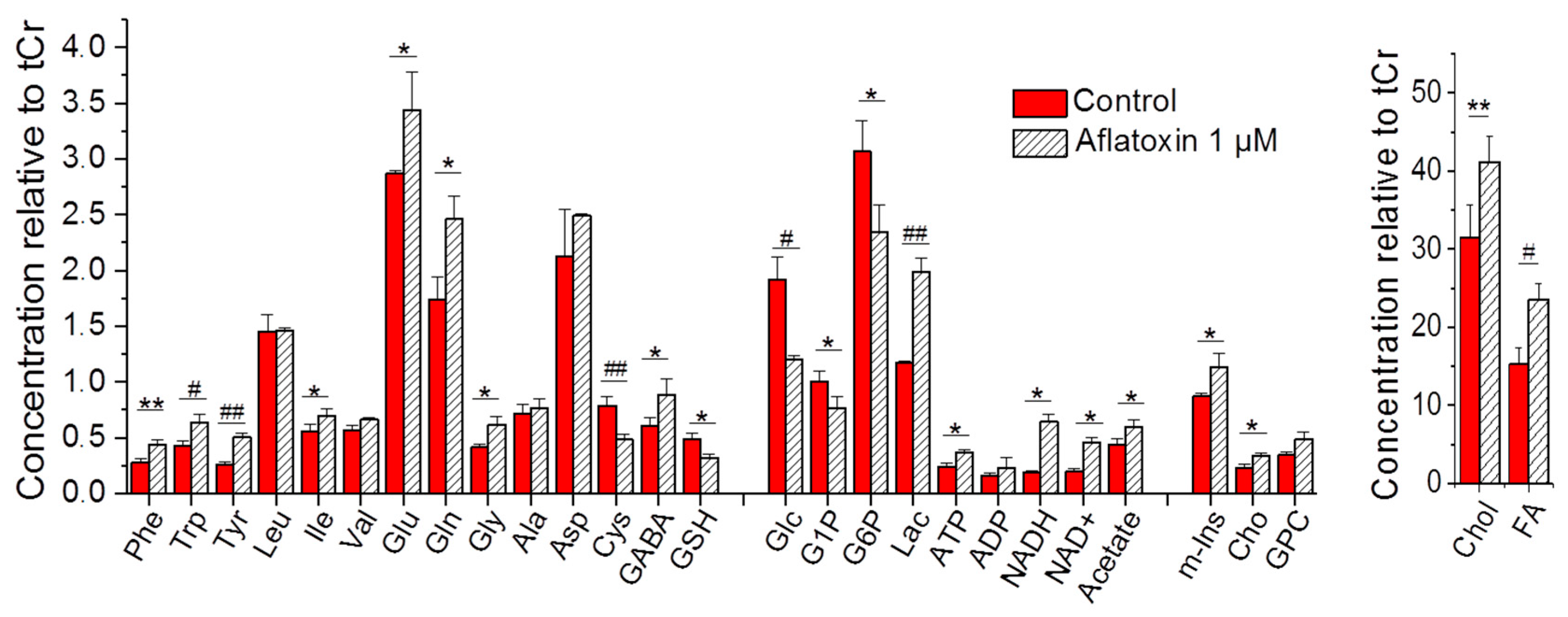
2.2. NMR-Based Metabolic Profiles of Zebrafish Exposed to AFB1
3. Discussion
3.1. Toxicity of AFB1 in the Zebrafish Embryo Model
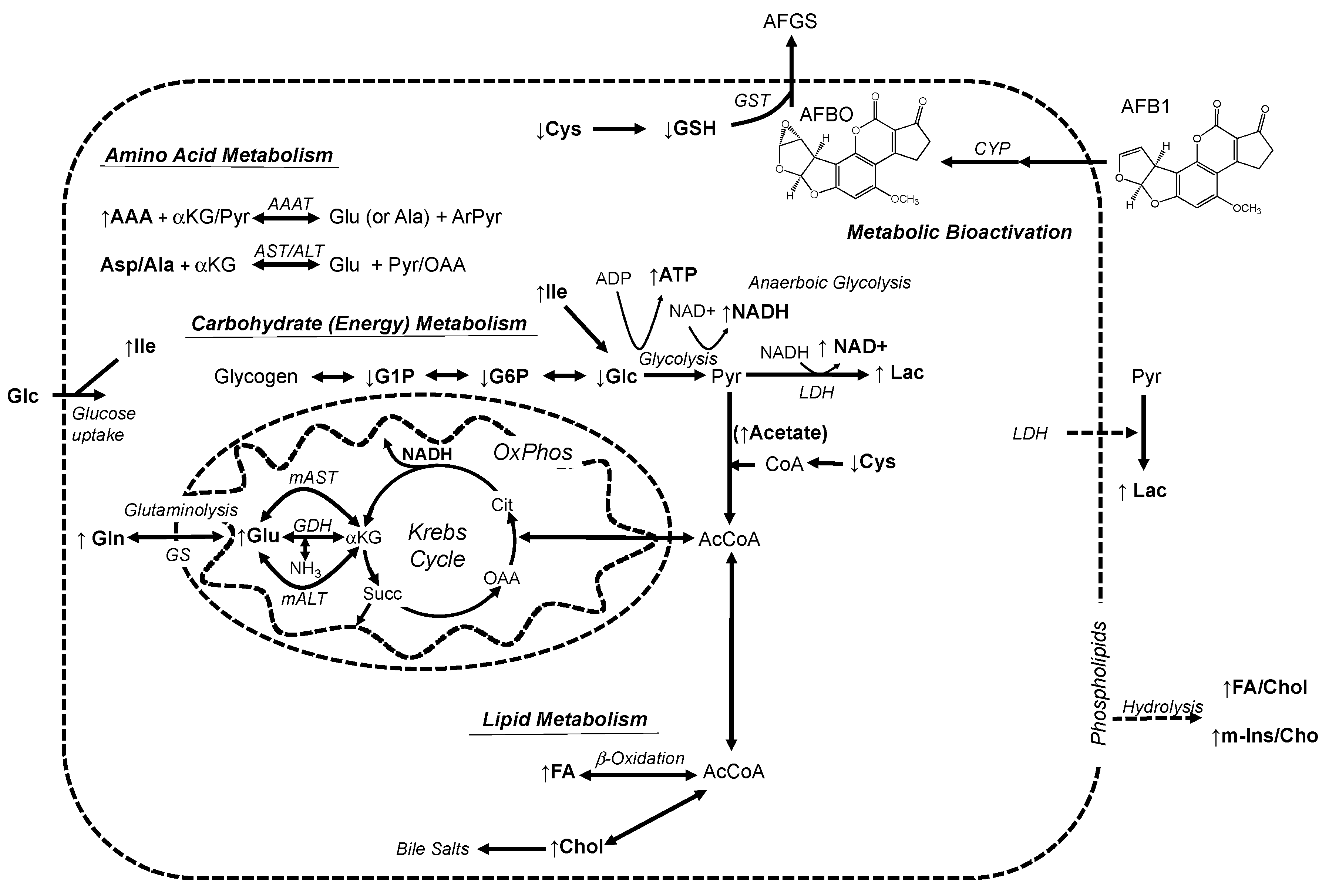
3.2. Alteration of Metabolic Profiles of Zebrafish Embryos by AFB1: Development of a Toxicological Model
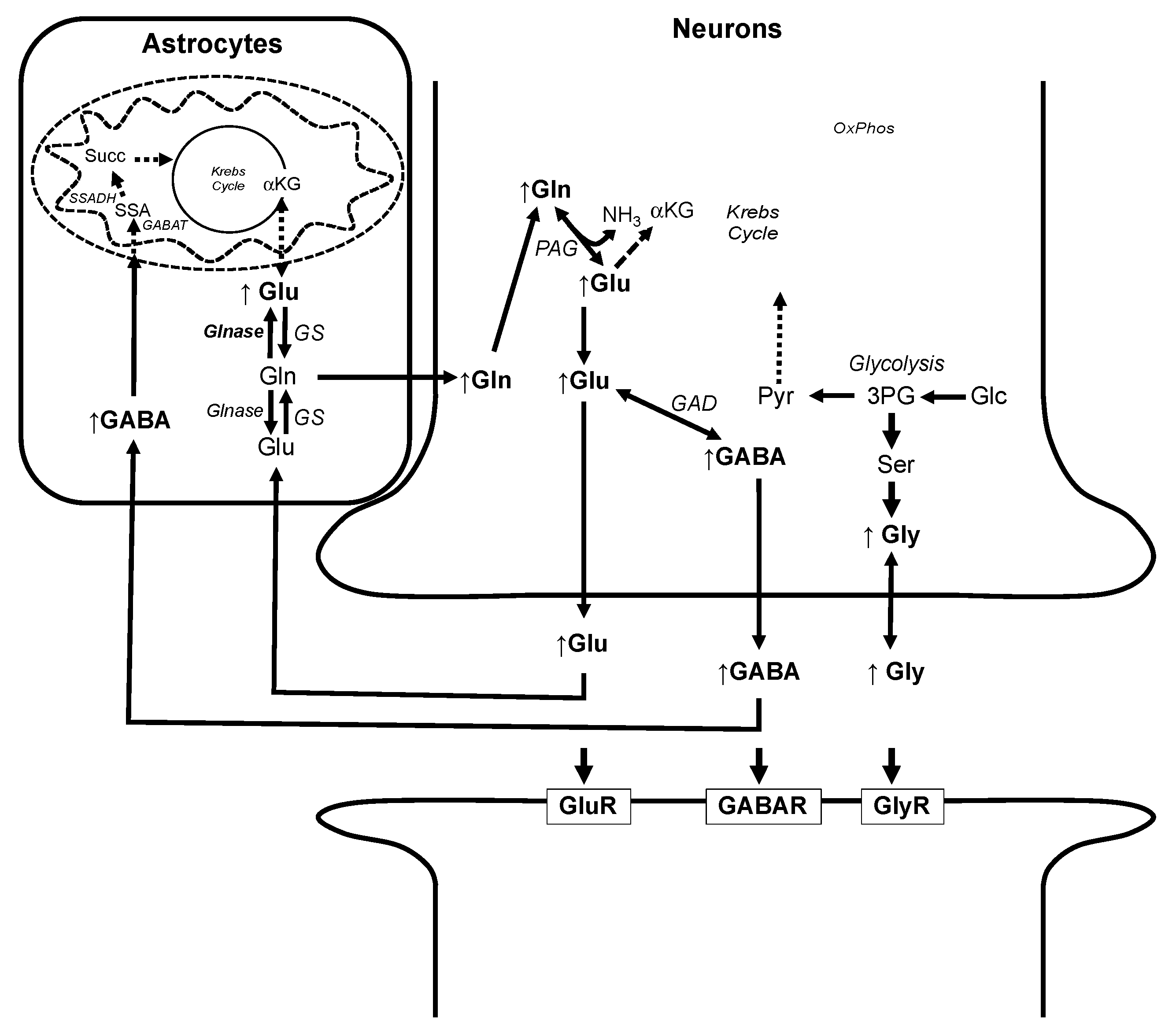
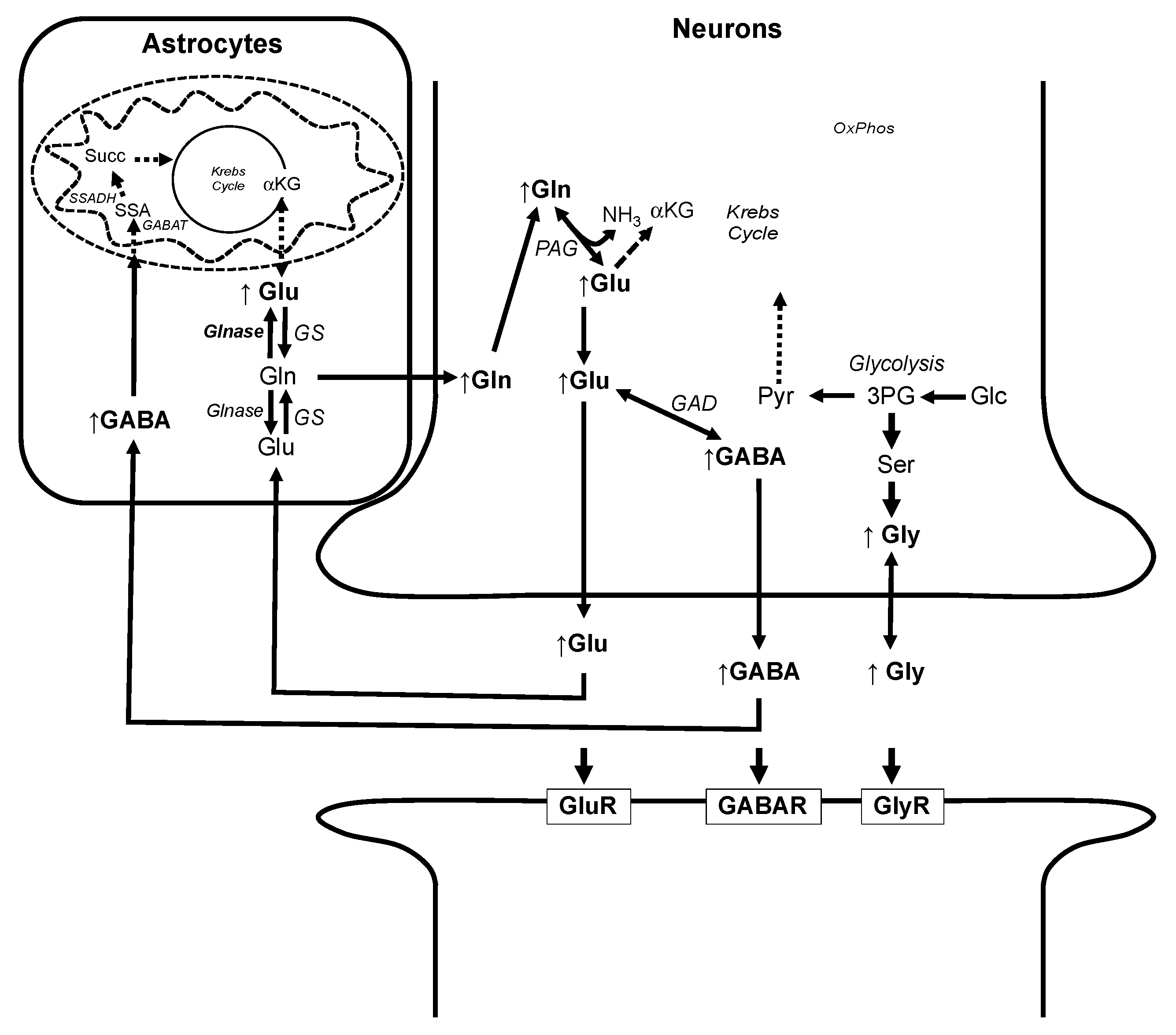
3.3. Alteration of Metabolic Profiles in Relation to Neurotoxicity of AFB1
4. Conclusions
5. Materials and Methods
5.1. Chemicals
5.2. Zebrafish Embryos
5.3. Zebrafish Embryo Toxicity Assays
5.4. HRMAS NMR
5.5. 1H NMR Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rushing, B.R.; Selim, M.I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food Chem. Toxicol. 2019, 124, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.L. Discovery of aflatoxins and significant historical features. Toxins Rev. 2008, 27, 171–201. [Google Scholar] [CrossRef]
- Monson, M.S.; Coulombe, R.A.; Reed, K.M. Aflatoxicosis: Lessons from toxicity and responses to aflatoxin B1 in poultry. Agriculture 2015, 5, 742–777. [Google Scholar] [CrossRef]
- Food and Agriculture Organization of the United Nations (FAO). Worldwide Regulations for Mycotoxins in Food and Feed in 2003. 2004. Available online: http://www.fao.org/3/y5499e/y5499e00.htm (accessed on 27 March 2019).
- Williams, J.H.; Phillips, T.D.; Jolly, P.E.; Stiles, J.K.; Jolly, C.M.; Aggarwal, D. Human aflatoxicosis in developing countries: A review of the toxicology, exposure, potential health consequences, and interventions. Am. J. Clin. Nutr. 2004, 80, 1106–1122. [Google Scholar] [CrossRef]
- Probst, C.; Njapau, H.; Cotty, P.J. Outbreak of an acute aflatoxicosis in Kenya in 2004: Identification of a causal agent. Appl. Environ. Microbiol. 2007, 73, 2762–2764. [Google Scholar] [CrossRef]
- Kamala, A.; Shirima, C.; Jani, B.; Bakari, M.; Sillo, H.; Rusibamayila, N.; De Saeger, S.; Kimanya, M.; Gong, Y.Y.; Simba, A. Outbreak of an acute aflatoxicosis in Tanzania during 2016. World Mycotoxin J. 2018, 11, 311–320. [Google Scholar] [CrossRef]
- Magnussen, A.; Parsi, M.A. Aflatoxins, hepatocellular carcinoma and public health. World J. Gastroenterol. 2013, 19, 1508–1512. [Google Scholar] [CrossRef] [PubMed]
- Ostry, V.; Malir, F.; Toman, J.; Grosse, Y. Mycotoxins as human carcinogens – the IARCH monographs classification. Mycotoxin Res. 2017, 33, 65–73. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, F. Global burden of aflatoxin-induced hepatocellular carcinoma: A risk assessment. Environ. Health Perspect. 2010, 118, 818–824. [Google Scholar]
- Weigt, S.; Huebler, N.; Strecker, R.; Braunbeck, T.; Broschard, T.H. Zebrafish (Danio rerio) embryos as model for testing proteratogens. Toxicology 2011, 281, 25–36. [Google Scholar] [CrossRef]
- Dhanapal, J.; Ravindraan, M.B.; Baskar, S.K. Toxic effects of aflatoxin B1 on embryonic development of zebrafish (Danio rerio): Potential activity of piceatannol encapsulated chitosan/poly (lactic acid) nanoparticles. Anticancer Agents Med. Chem. 2015, 15, 248–257. [Google Scholar] [CrossRef]
- Wu, T.S.; Cheng, Y.C.; Chen, P.J.; Huang, Y.T.; Yu, F.Y.; Liu, B.H. Exposure to aflatoxin B1 interferes with locomotion and neural development in zebrafish embryos and larvae. Chemosphere 2019, 217, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Anater, A.; Manyes, L.; Meca, G.; Ferrer, E.; Luciano, F.B.; Pimpão, C.T. Mycotoxins and their consequences in aquaculture: A review. Aquaculture 2016, 451, 1–10. [Google Scholar] [CrossRef]
- Kabli, S.; Spaink, H.P.; De Groot, H.J.M.; Alia, A. In vivo metabolite profile of adult zebrafish brain obtained by high-resolution localized magnetic resonance spectroscopy. J. Magnet. Reson. Imaging 2009, 29, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.; Champagne, D.L.; Alia, A.; Richardson, M.K. Large-scale analysis of acute ethanol exposure in zebrafish development: A critical time window. PLoS ONE 2011, 6, e20037. [Google Scholar] [CrossRef] [PubMed]
- van Amerongen, Y.F.; Roy, U.; Spaink, H.P.; de Groot, H.J.; Huster, D.; Schiller, J.; Alia, A. Zebrafish brain lipid characterization and quantification by 1H nuclear magnetic resonance spectroscopy and MALDI-TOF mass spectrometry. Zebrafish 2014, 11, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.P.; Roy, U.; Jaja-Chimedza, A.; Sanchez, K.; Matysik, J.; Alia, A. High-resolution magic angle spinning nuclear magnetic resonance of intact zebrafish embryos detects metabolic changes following exposure to teratogenic polymethoxyalkenes from algae. Zebrafish 2016, 13, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Roy, U.; Conklin, L.; Schiller, J.; Matysik, J.; Berry, J.P.; Alia, A. Metabolic profiling of zebrafish (Danio rerio) embryos by NMR spectroscopy reveals multifaceted toxicity of β-methylamino-L-alanine (BMAA). Sci. Rep. 2017, 7, 17305. [Google Scholar] [CrossRef]
- Zhang, L.; Ye, Y.; Tian, Y.; Wang, Y.; Tang, H. Systems responses of rats to aflatoxin B1 exposure revealed with metabonomics changes in multiple biological matrices. J. Proteome Res. 2011, 10, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Hu, B.; Shao, L.; Tian, Y.; Jin, T.; Jin, Y.; Ji, S.; Fan, X. Integrated analysis of transcriptomics and metabonomics profiles in aflatoxin B1-induced hepatotoxicity in rat. Food Chem. Toxicol. 2013, 55, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Huang, S.; Fan, C.; Zheng, N.; Zhang, N.; Li, S.; Wang, J. Metabolomic analysis of alterations in lipid oxidation, carbohydrate and amino acid metabolism in dairy goats caused by exposure to aflatoxin B1. J. Dairy Res. 2017, 84, 401–406. [Google Scholar] [CrossRef]
- Ogunade, I.; Jiang, Y.; Adeyami, J.; Oliveira, A.; Vyas, D.; Adesogan, A. Biomarker of aflatoxin ingestion: 1H NMR-based plasma metabolomics of dairy cows fed aflatoxin B1 with or without sequestering agents. Toxins 2018, 10, 545. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Peng, J. Liver development in zebrafish (Danio rerio). J. Genet. Genom. 2009, 36, 325–334. [Google Scholar] [CrossRef]
- Andraesen, E.A.; Spitsbergen, J.M.; Tanguay, R.L.; Stegeman, J.J.; Heideman, W.; Peterson, R.E. Tissue-specific expression of AHR2, ARNT2 and CYP1A in zebrafish embryos and larvae: Effects of developmental stage and 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure. Toxicol. Sci. 2002, 68, 403–419. [Google Scholar] [CrossRef]
- Tseng, H.P.; Hseu, T.H.; Buhler, D.R.; Wang, W.D.; Hu, C.H. Constitutive and xenobiotics-induced expression of a novel CYP3A gene from zebrafish larva. Toxicol. Appl. Pharmacol. 2005, 205, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Ziglari, T.; Allameh, A. The significance of glutathione conjugation in aflatoxin metabolism. In Aflatoxins—Recent Advances and Future Prospects; Razzaghi-Abyaneh, M., Ed.; Intech: London, UK, 2013; pp. 267–286. [Google Scholar]
- Klein, J. Membrane breakdown in acute and chronic neurodegeneration: Focus on choline-containing phospholipids. J. Neural Transm. 2000, 107, 1027–1063. [Google Scholar] [CrossRef]
- Leeman, W.R.; van de Gevel, I.A.; Rutten, A.A. Cytototoxicity of retinoic acid, menadione and aflatoxin B1 in rat liver slices using Netwell inserts as a new culture system. Toxicol. In Vitro 1995, 9, 291–298. [Google Scholar] [CrossRef]
- Dejong, C.H.; van de Poll, M.C.; Soeters, P.B.; Jalan, R.; Olde Damink, S.W. Aromatic amino acid metabolism during liver failure. J. Nutr. 2007, 137, 1579S–1585S. [Google Scholar] [CrossRef]
- Ishikawa, T. Branched-chain amino acids to tyrosine ratio value as a potential prognostic factor for hepatocellular carcinoma. World J. Gastroenterol. 2012, 18, 2005–2008. [Google Scholar] [CrossRef]
- Tajiri, K.; Shimizu, Y. Branched-chain amino acids in liver diseases. World J. Gastroenterol. 2013, 19, 7620–7629. [Google Scholar] [CrossRef]
- Monirujjaman, M.; Ferdouse, A. Metabolic and physiological roles of branched-chain amino acids. Adv. Mol. Biol. 2014, 364976. [Google Scholar] [CrossRef]
- Doi, M.; Yamaoka, I.; Fukunaga, T.; Nakayam, M. Isoleucine, a potent plasma glucose-lowering amino acid, stimulates glucose uptake in C2C12. Biochem. Biophys. Res. Commun. 2003, 312, 1111–1117. [Google Scholar] [CrossRef]
- Doi, M.; Yamaoka, I.; Nakayama, M.; Mochizuki, S.; Sugahara, K.; Yoshizawa, F. Isoleucine, a blood glucose-lowering amino acid, increases glucose uptake in rat skeletal muscle in the absence of increases in AMP-activated protein kinase activity. J. Nutr. 2005, 135, 2103–2108. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, S.; Takehana, K.; Fujitani, S.; Sonaka, I. Branched-chain amino acids improve glucose metabolism in rats with liver cirrhosis. Am. J. Physiol. Gastroenterol. Liver Physiol. 2005, 288, G1291–G1300. [Google Scholar] [CrossRef] [PubMed]
- Doi, M.; Yamaoka, I.; Nakayama, M.; Sugahara, K.; Yoshizawa, F. Hypoglycemic effect of isoleucine involves increased muscle glucose uptake and whole body glucose oxidation and decreased hepatic gluconeogenesis. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1683–E1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zeng, X.; Ren, M.; Xianbing, M.; Qiao, S. Novel metabolic and physiological functions of branched amino acids: A review. J. Anim. Sci. Biotechnol. 2017, 8, 10. [Google Scholar] [CrossRef]
- Wilson, R.P.; Halver, J.E. Protein and amino acid requirements of fishes. Ann. Rev. Nutr. 1986, 6, 225–244. [Google Scholar] [CrossRef]
- Krisher, R.L.; Prather, R.S. A role for the Warburg Effect in preimplantation embryo development: Metabolic modification to support rapid cell proliferation. Mol. Reprod. Dev. 2012, 79, 311–320. [Google Scholar] [CrossRef]
- Smith, D.G.; Sturmey, R.G. Parallels between embryo and cancer cell metabolism. Biochem. Soc. Trans. 2013, 41, 664–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Yang, W.; Xiao, B.; Zhang, H.; Lei, X.; Ou, H.; Renan, Q.; Jin, R. Study on early biomarkers of zebrafish liver injury induced by acetaminophen. Toxin Rev. 2015, 34, 28–36. [Google Scholar] [CrossRef]
- McGill, M.R. The past and present of serum aminotransferases and the future of liver injury biomarkers. EXCLI J. 2016, 15, 817–828. [Google Scholar]
- Liu, Y.; Wang, W. Aflatoxin B1 impairs mitochondrial functions, activates ROS generation, induces apoptosis and involves Nrf2 signal pathway in primary broiler hepatocytes. Animal Sci. J. 2016, 87, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Aspects Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLife 2016, 5, e13374. [Google Scholar] [CrossRef] [PubMed]
- Rotimi, O.A.; Rotimai, S.O.; Duru, C.U.; Ogheneworo, J.E.; Abiodun, A.O.; Oyeniyi, B.O.; Faduyile, F.A. Acute aflatoxin B1-induced hepatotoxicity alters gene expression and disrupts lipid and lipoprotein metabolism in rats. Toxicol. Rep. 2017, 4, 408–414. [Google Scholar] [CrossRef]
- Avila, A.; Nguyen, L.; Rigo, J.-M. Glycine receptors and brain development. Front. Cell Neurosci. 2013, 7, 184. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.J.; Mirnics, K. Neurodevelopment, GABA system dysfunction, and schizophrenia. Neuropsychopharmacology 2015, 40, 190–206. [Google Scholar] [CrossRef]
- Moretto, E.; Murru, L.; Martano, G.; Sassone, J.; Passafaro, M. Glutamatergic synapses in neurodevelopmental disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 84, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Ikegwuonu, F.I. The neurotoxicity of aflatoxin B1 in the rat. Toxicology 1983, 28, 247–259. [Google Scholar] [CrossRef]
- The Zebrafish Information Network. Available online: http://www.zfin.org (accessed on 6 May 2019).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite 1 | % Change 2 | p-Value | FC 3 (T/C) | p-Value | ||
|---|---|---|---|---|---|---|
| Amino Acids | AAA:BCAA | +1.5 | <0.01 | |||
| Phe | +58.7 | <0.01 | Tyr:BCAA | +1.8 | <0.01 | |
| Trp | +48.3 | <0.001 | ||||
| Tyr | +97.8 | <0.0001 | ||||
| Leu | +0.4 | n.s. | ||||
| Ile | +26.8 | <0.05 | ||||
| Val | +16.4 | n.s. | ||||
| Glu | +20.0 | <0.05 | ||||
| Gln | +41.9 | <0.05 | ||||
| Gly | +49.3 | <0.05 | ||||
| Ala | +8.3 | n.s. | ||||
| Asp | +17.3 | n.s. | ||||
| Cys | −38.9 | <0.0001 | ||||
| GABA 4 | +45.8 | <0.05 | ||||
| Carbohydrates and Energy Metabolism | ||||||
| Glc | −37.2 | <0.001 | ||||
| G1P | −23.6 | <0.01 | ||||
| G6P | −23.6 | <0.01 | ||||
| Lac | +70.4 | <0.0001 | ||||
| Acetate | +36.3 | <0.05 | ||||
| ATP | +52.4 | <0.05 | ||||
| ADP | +49.4 | n.s. | ||||
| NADH | +234.1 | <0.05 | ||||
| NAD+ | +132.5 | <0.05 | ||||
| Lipids (and Polar Head Groups) | ||||||
| Chol | +30.7 | <0.01 | ||||
| FA | +54.5 | <0.001 | ||||
| m-Ins | +29.1 | <0.05 | ||||
| Cho | +48.6 | <0.05 | ||||
| GPC | +42.2 | n.s. | ||||
| Glutathione | −34.8 | <0.05 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuberi, Z.; Eeza, M.N.H.; Matysik, J.; Berry, J.P.; Alia, A. NMR-Based Metabolic Profiles of Intact Zebrafish Embryos Exposed to Aflatoxin B1 Recapitulates Hepatotoxicity and Supports Possible Neurotoxicity. Toxins 2019, 11, 258. https://doi.org/10.3390/toxins11050258
Zuberi Z, Eeza MNH, Matysik J, Berry JP, Alia A. NMR-Based Metabolic Profiles of Intact Zebrafish Embryos Exposed to Aflatoxin B1 Recapitulates Hepatotoxicity and Supports Possible Neurotoxicity. Toxins. 2019; 11(5):258. https://doi.org/10.3390/toxins11050258
Chicago/Turabian StyleZuberi, Zain, Muhamed N. H. Eeza, Joerg Matysik, John P. Berry, and A. Alia. 2019. "NMR-Based Metabolic Profiles of Intact Zebrafish Embryos Exposed to Aflatoxin B1 Recapitulates Hepatotoxicity and Supports Possible Neurotoxicity" Toxins 11, no. 5: 258. https://doi.org/10.3390/toxins11050258
APA StyleZuberi, Z., Eeza, M. N. H., Matysik, J., Berry, J. P., & Alia, A. (2019). NMR-Based Metabolic Profiles of Intact Zebrafish Embryos Exposed to Aflatoxin B1 Recapitulates Hepatotoxicity and Supports Possible Neurotoxicity. Toxins, 11(5), 258. https://doi.org/10.3390/toxins11050258