Dehydrocrenatidine Inhibits Voltage-Gated Sodium Channels and Ameliorates Mechanic Allodia in a Rat Model of Neuropathic Pain


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
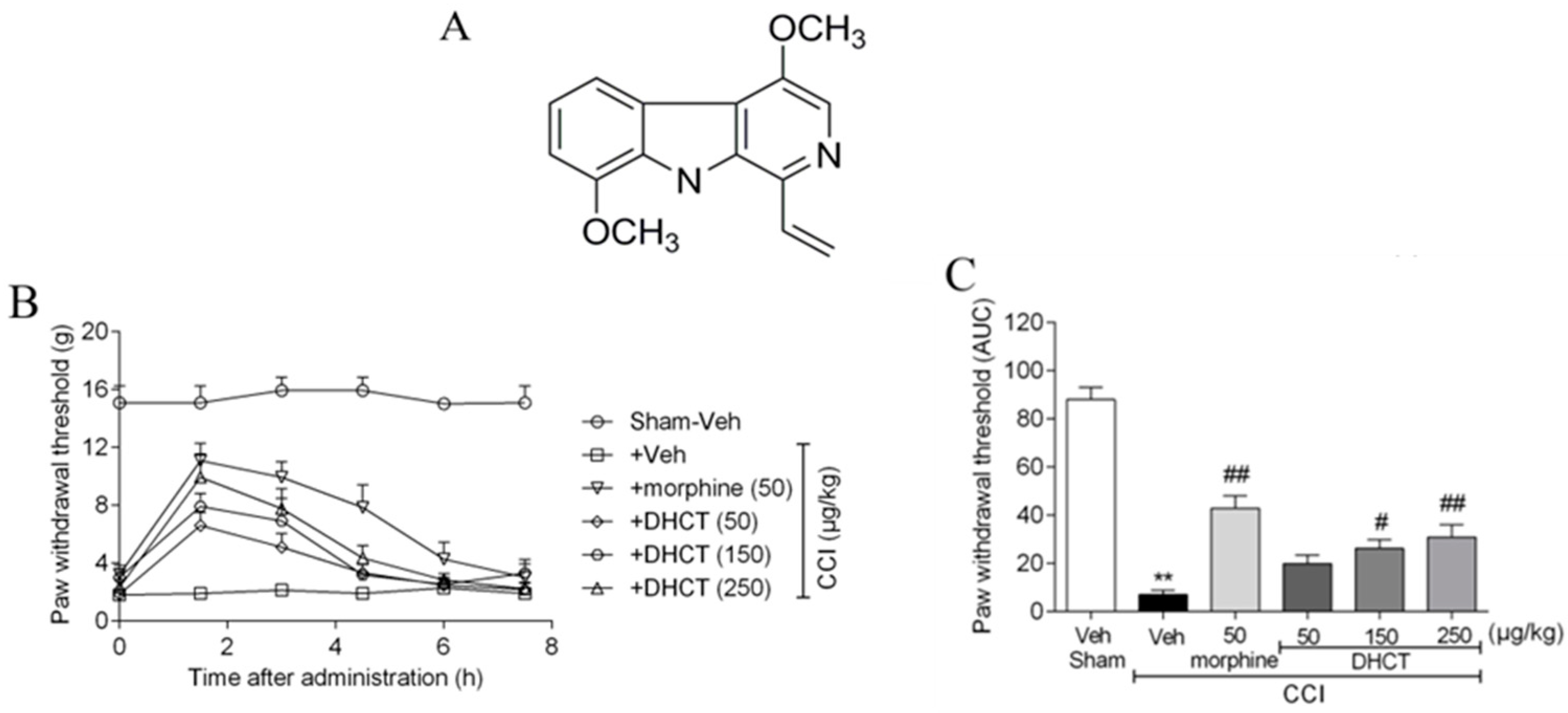
2.1. Dehydrocrenatidine Ameliorated Mechanic Allodynia in a Neuropathic Pain Model of Sciatic Nerve Partial Ligation
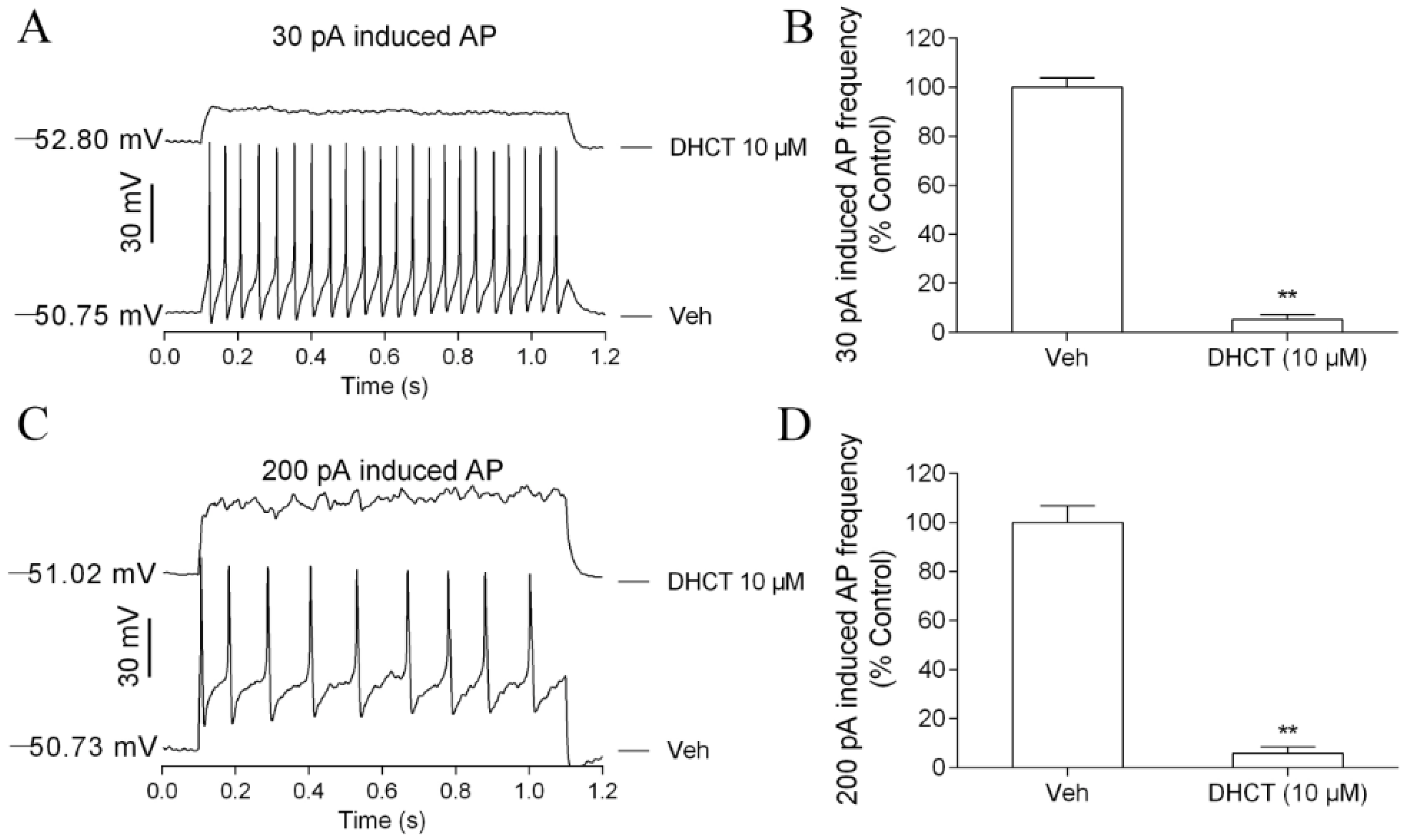
2.2. DHCT Suppressed Action Potential Generation in Acutely Dissociated Rat DRG Neurons
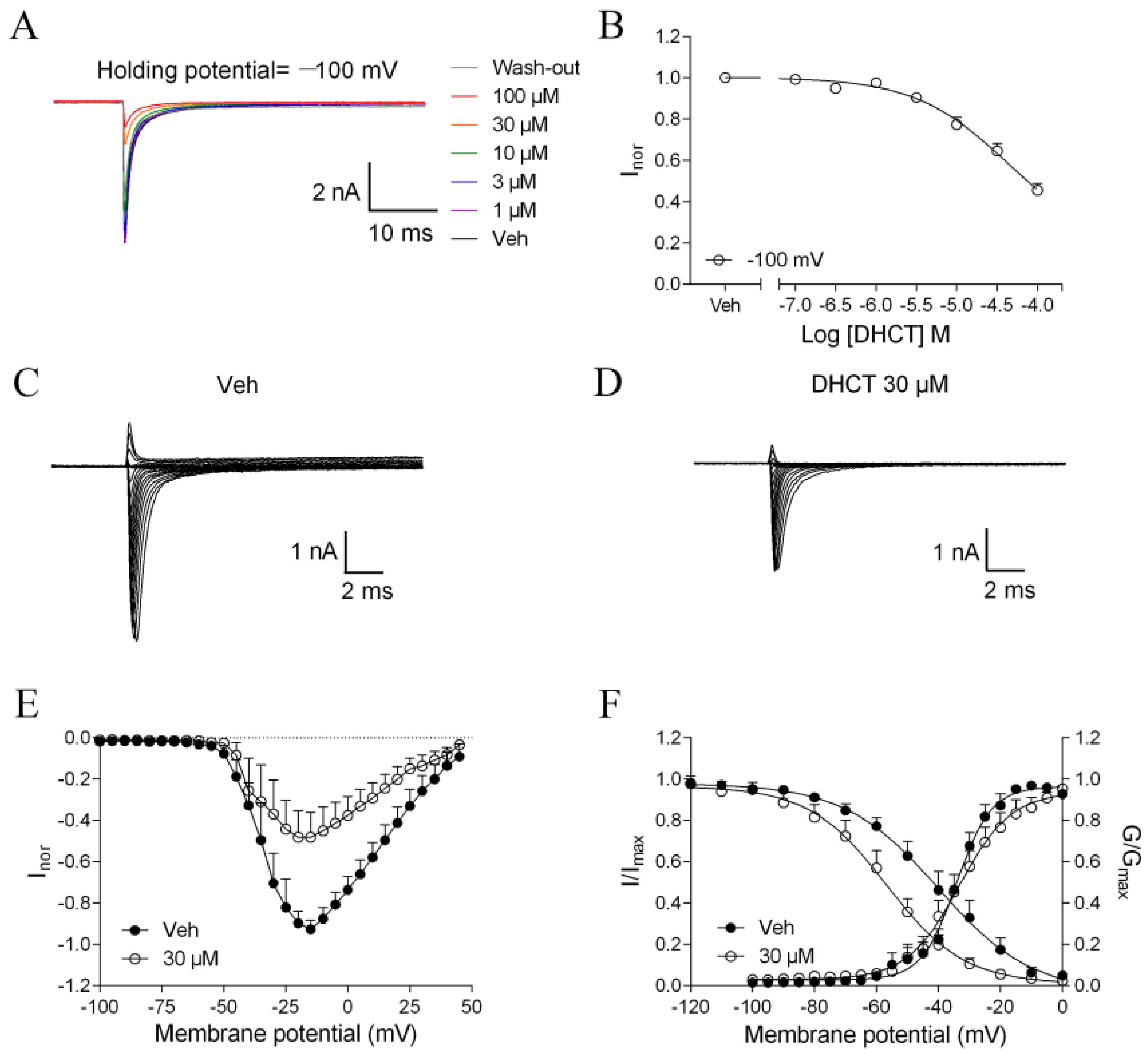
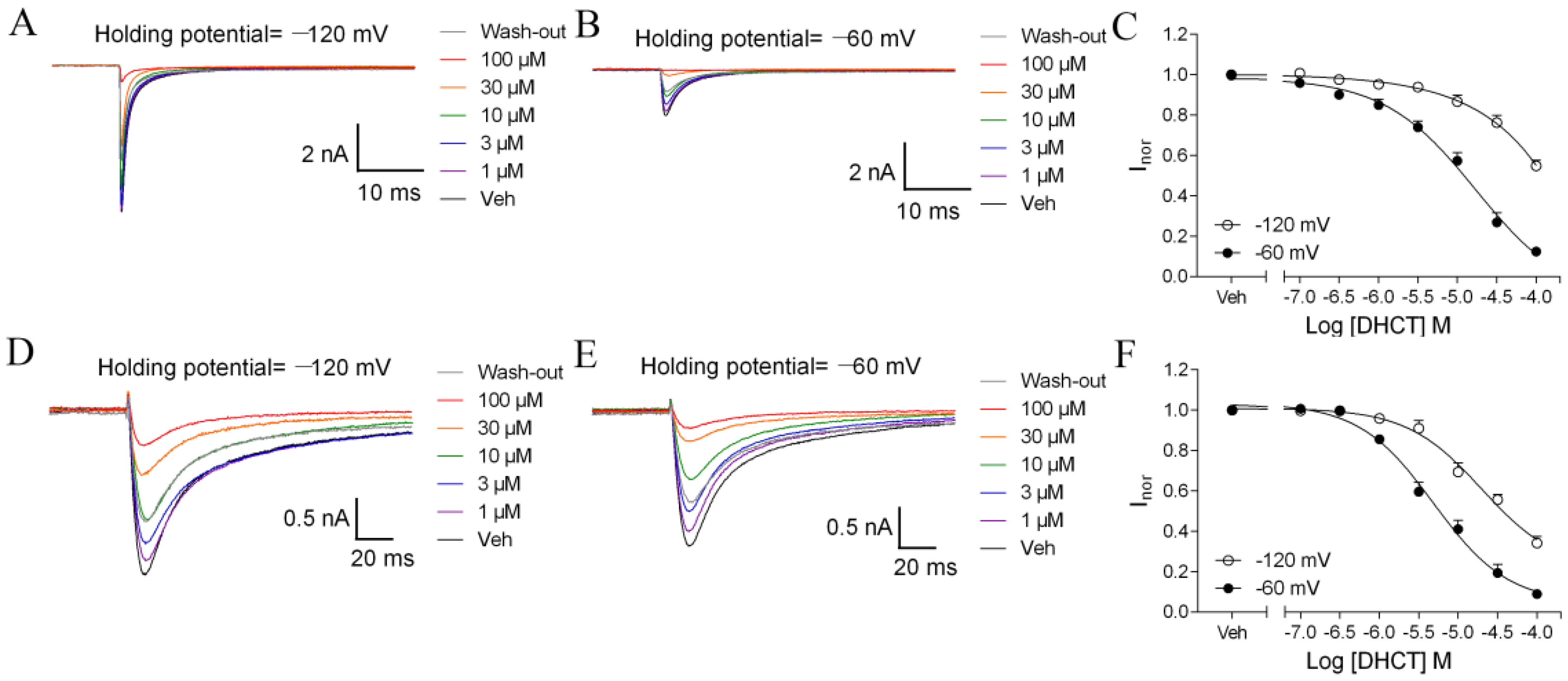
2.3. DHCT Suppressed both Tetrodotoxin-Sensitive (TTX-S) and TTX-Resistant (TTX-R) VGSC Na+ Currents in DRG Neurons
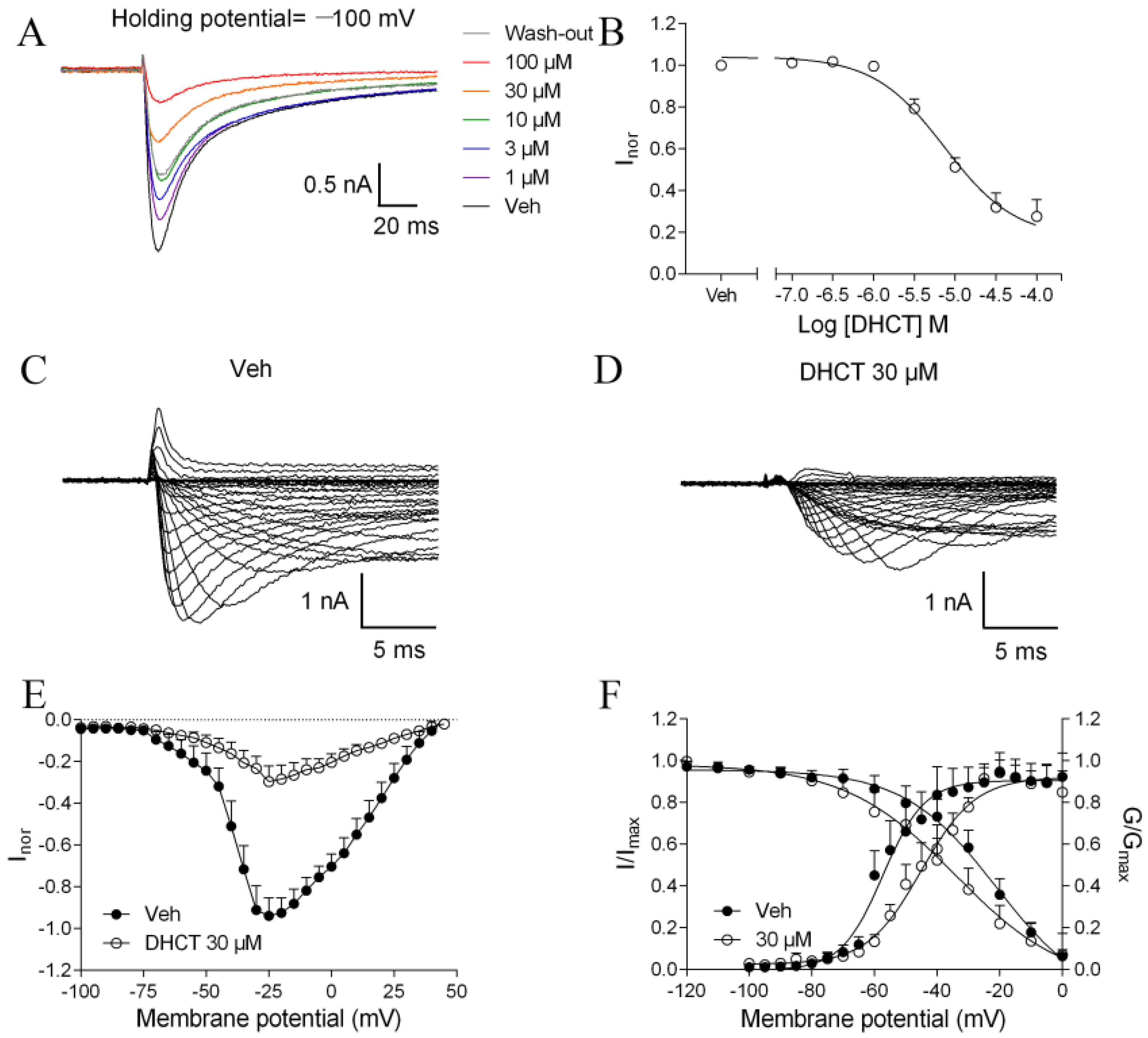
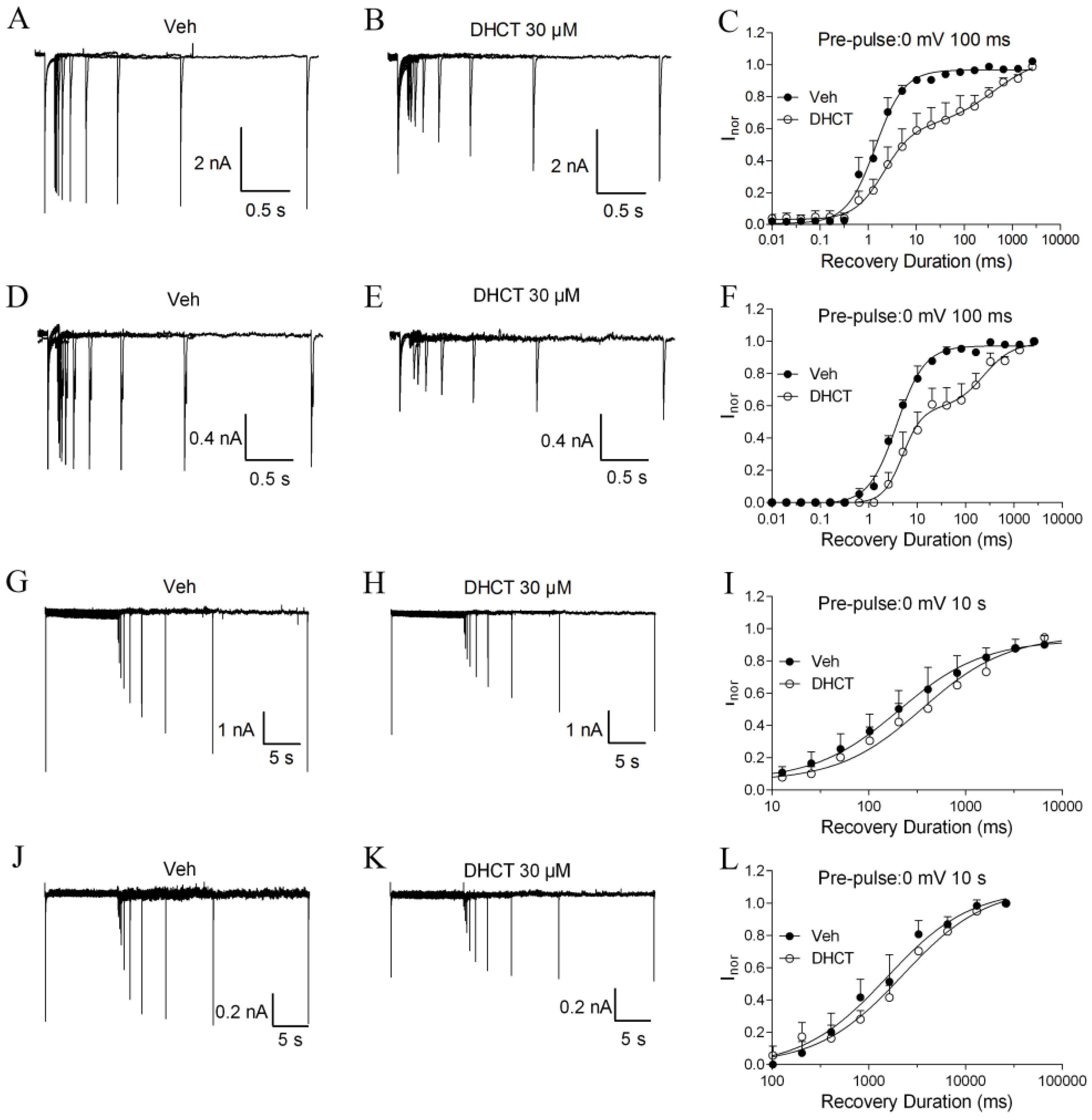
2.4. DHCT Preferred to Interact with Inactivated State of VGSCs
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Care
4.3. Acutely Dissected Rat Dorsal Root Ganglion Neurons
4.4. Patch Clamp Recording in DRG Neurons
4.5. Chronic Constrictive Injury (CCI) Neuropathic Pain Model
4.6. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mulvey, M.R.; Bennett, M.I.; Liwowsky, I.; Freynhagen, R. The role of screening tools in diagnosing neuropathic pain. Pain Manag. 2014, 4, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, I. Pain market. Dressnature Rev. Drug Discov. 2010, 9, 589–590. [Google Scholar] [CrossRef] [PubMed]
- Willis, W.D. Role of neurotransmitters in sensitization of pain responses. Ann. N. Y. Acad. Sci. 2001, 933, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Minett, M.S.; Nassar, M.A.; Clark, A.K.; Passmore, G.; Dickenson, A.H.; Wang, F.; Malcangio, M.; Wood, J.N. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun. 2012, 3, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Jay, G.W.; Barkin, R.L. Neuropathic pain: Etiology, pathophysiology, mechanisms, and evaluations. Dis. Month 2014, 60, 6–47. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.; Argoff, C.E.; Bennett, G.J.; Cummins, T.R.; Durieux, M.E.; Gerner, P.; Gold, M.S.; Porreca, F.; Strichartz, G.R. The role of sodium channels in chronic inflammatory and neuropathic pain. J. Pain 2006, 7, S1–S29. [Google Scholar] [CrossRef] [PubMed]
- Brederson, J.D.; Kym, P.R.; Szallasi, A. Targeting TRP channels for pain relief. Eur. J. Pharmacol. 2013, 716, 61–76. [Google Scholar] [CrossRef]
- Bean, B.P. The action potential in mammalian central neurons. Nat. Rev. Neurosci. 2007, 8, 451–465. [Google Scholar] [CrossRef]
- Heyer, E.J.; Macdonald, R.L. Calcium- and sodium-dependent action potentials of mouse spinal cord and dorsal root ganglion neurons in cell culture. J. Neurophysiol. 1982, 47, 641–655. [Google Scholar] [CrossRef]
- Fletcher, A. Action potential: Generation and propagation. Anaesth. Intensive Care Med. 2008, 9, 251–255. [Google Scholar] [CrossRef]
- Thakor, K.; Lin, A.; Matsuka, Y.; Meyer, M.; Ruangsri, S.; Nishimura, I.; Spigelman, I. Nav1.8, neuropathic pain. Mol. Pain 2009, 5, 14. [Google Scholar] [PubMed]
- Hong, C.J.; Hsueh, Y.P. Cask associates with glutamate receptor interacting protein and signaling molecules. Biochem. Biophys. Res. Commun. 2006, 351, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Hains, B.C.; Klein, J.P.; Saab, C.Y.; Craner, M.J.; Black, J.A.; Waxman, S.G. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 8881–8892. [Google Scholar] [CrossRef]
- Zhao, F.; Gao, Z.; Jiao, W.; Chen, L.; Chen, L.; Yao, X. In vitro anti-inflammatory effects of beta-carboline alkaloids, isolated from Picrasma quassioides, through inhibition of the inos pathway. Planta Med. 2012, 78, 1906–1911. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Chen, L.; Bi, C.; Zhang, M.; Jiao, W.; Yao, X. In vitro anti-inflammatory effect of picrasmalignan a by the inhibition of iNOS and COX-2 expression in LPS-activated macrophage RAW 264.7 cells. Mol. Med. Rep. 2013, 8, 1575–1579. [Google Scholar] [CrossRef] [PubMed]
- Jiao, W.H.; Gao, H.; Li, C.Y.; Zhou, G.X.; Kitanaka, S.; Ohmura, A.; Yao, X.S. β-carboline alkaloids from the stems of Picrasma quassioides. Magn. Reson. Chem. 2010, 48, 490–495. [Google Scholar] [PubMed]
- Xu, J.; Xiao, D.; Lin, Q.H.; He, J.F.; Liu, W.Y.; Xie, N.; Feng, F.; Qu, W. Cytotoxic tirucallane and apotirucallane triterpenoids from the stems of Picrasma quassioides. J. Natl. Prod. 2016, 79, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Hikino, H.; Ohta, T.; Takemoto, T. Stereostructure of picrasin d and e, simaroubolides of Picrasma quassioides. Chem. Pharm. Bull. 2008, 19, 212–213. [Google Scholar] [CrossRef][Green Version]
- Jiao, W.H.; Gao, H.; Li, C.Y.; Zhao, F.; Jiang, R.W.; Wang, Y.; Zhou, G.X.; Yao, X.S. Quassidines A–D, Bis-β-carboline Alkaloids from the Stems of Picrasma quassioides. J. Natl. Prod. 2010, 73, 167–171. [Google Scholar] [CrossRef]
- Yin, Y.; Heo, S.I.; Roh, K.S.; Wang, M.H. Biological activities of fractions from methanolic extract of Picrasma quassioides. J. Plant Biol. 2009, 52, 325–331. [Google Scholar] [CrossRef]
- Jiao, W.H.; Gao, H.; Zhao, F.; Lin, H.W.; Pan, Y.M.; Zhou, G.X.; Yao, X.S. Anti-inflammatory alkaloids from the stems of Picrasma quassioides bennet. Chem. Pharm. Bull. 2011, 59, 359–364. [Google Scholar] [CrossRef]
- Haghparast, A.; Farzin, D.; Ordikhani-Seyedlar, M.; Motaman, S.; Kermani, M.; Azizi, P. Effects of apomorphine and β-carbolines on firing rate of neurons in the ventral pallidum in the rats. Behav. Brain Res. 2012, 227, 109–115. [Google Scholar] [CrossRef]
- Luo, S.R.; Guo, R.; Yang, J.S. Studies on the determination of alkaloids in Prcrasma quassiodies (d.Don) benn. Acta Pharm. Sin. 1988, 23, 4. [Google Scholar]
- Zhang, J.; Zhu, N.; Du, Y.; Bai, Q.; Chen, X.; Nan, J.; Qin, X.; Zhang, X.; Hou, J.; Wang, Q. Dehydrocrenatidine is a novel janus kinase inhibitor. Mol. Pharmacol. 2015, 87, 572. [Google Scholar] [CrossRef]
- Hevers, W.; Lüddens, H. The diversity of GABAA receptors. Mol. Pharmacol. 1998, 18, 35–86. [Google Scholar] [CrossRef]
- Hollinshead, S.P.; Trudell, M.L.; Skolnick, P.; Cook, J.M. Structural requirements for agonist actions at the benzodiazepine receptor: Studies with analogs of 6-(benzyloxy)-4-(methoxymethyl)-.Beta.-carboline-3-carboxylic acid ethyl ester. J. Med. Chem. 1990, 33, 1062–1069. [Google Scholar] [CrossRef]
- Grella, B.; Teitler, M.; Smith, C.; Herrickdavis, K.; Glennon, R.A. Binding of β-carbolines at 5-HT(2) serotonin receptors. Bioorganic Med. Chem. Lett. 2003, 13, 4421–4425. [Google Scholar] [CrossRef]
- Glennon, R.A.; Grella, B.; Tyacke, R.J.; Lau, A.; Westaway, J.; Hudson, A.L. Binding of β-carbolines at imidazoline I2 receptors: A structure–affinity investigation. Bioorganic Med. Chem. Lett. 2004, 14, 999–1002. [Google Scholar] [CrossRef]
- Abdel-Fattah, A.F.; Matsumoto, K.; Gammaz, H.A.; Watanabe, H. Hypothermic effect of harmala alkaloid in rats: Involvement of serotonergic mechanism. Pharmacol. Biochem. Behav. 1995, 52, 421–426. [Google Scholar] [CrossRef]
- Ho, C.; O’Leary, M.E. Single-cell analysis of sodium channel expression in dorsal root ganglion neurons. Mol. Cell. Neurosci. 2011, 46, 159. [Google Scholar] [CrossRef]
- O’Reilly, A.O.; Eberhardt, E.; Weidner, C.; Alzheimer, C.; Wallace, B.A.; Lampert, A. Bisphenol a binds to the local anesthetic receptor site to block the human cardiac sodium channel. PLoS ONE 2012, 7, e41667. [Google Scholar] [CrossRef]
- Gaudioso, C.; Hao, J.; Martin-Eauclaire, M.F.; Gabriac, M.; Delmas, P. Menthol pain relief through cumulative inactivation of voltage-gated sodium channels. Pain 2012, 153, 473–484. [Google Scholar] [CrossRef]
- Errington, A.C.; Stöhr, T.; Heers, C.; Lees, G. The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol. Pharmacol. 2008, 73, 157–169. [Google Scholar] [CrossRef]
- Sheets, P.L.; Jarecki, B.W.; Cummins, T.R. Lidocaine reduces the transition to slow inactivation in Nav1.7 voltage-gated sodium channels. Br. J. Pharmacol. 2011, 164, 719–730. [Google Scholar] [CrossRef]
- Hildebrand, M.E.; Mezeyova, J.; Smith, P.L.; Salter, M.W.; Tringham, E.; Snutch, T.P. Identification of sodium channel isoforms that mediate action potential firing in lamina I/II spinal cord neurons. Mol. Pain 2011, 7, 1–13. [Google Scholar] [CrossRef]
- Zhao, F.; Li, X.; Jin, L.; Zhang, F.; Inoue, M.; Yu, B.; Cao, Z. Development of a rapid throughput assay for identification of hNav1.7 antagonist using unique efficacious sodium channel agonist, antillatoxin. Mar. Drugs 2016, 14, 36. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, H.; Zhang, F.; Zhang, C.L.; Zou, X.; Cao, Z. Selective voltage-gated sodium channel peptide toxins from animal venom: Pharmacological probes and analgesic drug development. ACS Chem. Neurosci. 2018, 9, 187–197. [Google Scholar] [CrossRef]
- Dibhajj, S.D.; Yang, Y.; Black, J.A.; Waxman, S.G. The Nav1.7 sodium channel: From molecule to man. Nat. Rev. Neurosci. 2012, 14, 49–62. [Google Scholar] [CrossRef]
- Decosterd, I.; Ji, R.R.; Abdi, S.; Tate, S.; Woolf, C.J. The pattern of expression of the voltage-gated sodium channels Na(v)1.8 and Na(v)1.9 does not change in uninjured primary sensory neurons in experimental neuropathic pain models. Pain 2015, 96, 269–277. [Google Scholar] [CrossRef]
- Wang, J.; Ou, S.W.; Wang, Y.J. Distribution and function of voltage-gated sodium channels in the nervous system. Channels 2017, 11, 534. [Google Scholar] [CrossRef]
- Kim, H.Y.; Park, E.J.; Joe, E.H.; Jou, I. Curcumin suppresses janus kinase-STAT inflammatory signaling through activation of Src homology 2 domain-containing tyrosine phosphatase 2 in brain microglia. J. Immunol. 2003, 171, 6072. [Google Scholar] [CrossRef]
- Natarajan, C.; Sriram, S.; Muthian, G.; Bright, J.J. Signaling through JAK2-STAT5 pathway is essential for IL-3. Glia 2004, 45, 188–196. [Google Scholar]
- Yin, L.; Dai, Q.; Jiang, P.; Lin, Z.; Dai, H.; Yao, Z.; Hua, L.; Ma, X.; Qu, L.; Jiang, J. Manganese exposure facilitates microglial JAK2-STAT3 signaling and consequent secretion of TNF-a and IL-1β to promote neuronal death. Neurotoxicology 2018, 64, 195–203. [Google Scholar] [CrossRef]
- Chi, Z.; Jie, Z.; Jing, Z.; Li, H.; Zhao, Z.; Liao, Y.; Wang, X.; Su, J.; Sang, S.; Yuan, X. Neuroprotective and anti-apoptotic effects of valproic acid on adult rat cerebral cortex through erk and akt signaling pathway at acute phase of traumatic brain injury. Brain Res. 2014, 1555, 1. [Google Scholar]
- Aedín, M.; James, B.; Marina, L. Lps-induced release of il-6 from glia modulates production of IL-1β in a JAK2-dependent manner. J. Neuroinflammation 2012, 9, 126. [Google Scholar]
- Dominguez, E.; Rivat, C.; Pommier, B.; Mauborgne, A.; Pohl, M. JAK/STAT3 pathway is activated in spinal cord microglia after peripheral nerve injury and contributes to neuropathic pain development in rat. J. Neurochem. 2008, 107, 50–60. [Google Scholar] [CrossRef]
- Cummins, T.R.; Rush, A.M.; Estacion, M.; Dibhajj, S.D.; Waxman, S.G. Voltage-clamp and current-clamp recordings from mammalian drg neurons. Nat. Protocols 2009, 4, 1103–1112. [Google Scholar] [CrossRef]
- Chen, R.; Chung, S.H. Mechanism of tetrodotoxin block and resistance in sodium channels. Biochem. Biophys. Res. Commun. 2014, 446, 370–374. [Google Scholar] [CrossRef]
- Ulbricht, W. Sodium channel inactivation: Molecular determinants and modulation. Physiol. Rev. 2005, 85, 1271. [Google Scholar] [CrossRef]
- Catterall, W.A.; Swanson, T.M. Structural basis for pharmacology of voltage-gated sodium and calcium channels. Mol. Pharmacol. 2015, 88, 141. [Google Scholar] [CrossRef]
- Wang, Y.; Mi, J.; Lu, K.; Lu, Y.; Wang, K. Comparison of gating properties and use-dependent block of Nav1.5 and Nav1.7 channels by anti-arrhythmics mexiletine and lidocaine. PLoS ONE 2015, 10, e0128653. [Google Scholar] [CrossRef]
- Chevrier, P.; Vijayaragavan, K.; Chahine, M. Differential modulation of Nav1.7 and Nav1.8 peripheral nerve sodium channels by the local anesthetic lidocaine. Br. J. Pharmacol. 2004, 142, 576–584. [Google Scholar] [CrossRef]
- Capes, D.L.; Arcisio-Miranda, M.; Jarecki, B.W.; French, R.J.; Chanda, B. Gating transitions in the selectivity filter region of a sodium channel are coupled to the domain IV voltage sensor. Proc. Natl. Acad. Sci. USA 2012, 109, 2648–2653. [Google Scholar] [CrossRef]
- Yang, Y.; Luo, S.; Shen, X.; Li, Y. [chemical investigation of the alkaloids of Ku-Mu [picrasma quassioides (d. Don) benn.] (author’s transl)]. Yao Xue Xue Bao 1979, 14, 167–177. [Google Scholar]
- Dibhajj, S.D.; Choi, J.S.; Macala, L.J.; Tyrrell, L.; Black, J.A.; Cummins, T.R.; Waxman, S.G. Transfection of rat or mouse neurons by biolistics or electroporation. Nat. Protocols 2009, 4, 1118. [Google Scholar] [CrossRef]
- Chen, Y.; Stevens, B.; Chang, J.; Milbrandt, J.; Barres, B.A.; Hell, J.W. NS21: Re-defined and modified supplement B27 for neuronal cultures. J. Neurosci. Methods 2008, 171, 239–247. [Google Scholar] [CrossRef]
- He, Y.; Zou, X.; Li, X.; Chen, J.; Liang, J.; Fan, Z.; Yu, B.; Cao, Z. Activation of sodium channels by α-scorpion toxin, BmK NT1, produced neurotoxicity in cerebellar granule cells: An association with intracellular Ca2+ overloading. Arch. Toxikol. 2016, 91, 935–948. [Google Scholar] [CrossRef]
- Roy, M.L.; Narahashi, T. Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J. Neurosci. 1992, 12, 2104–2111. [Google Scholar] [CrossRef]
- Austin, P.J.; Wu, A.; Moalem-Taylor, G. Chronic constriction of the sciatic nerve and pain hypersensitivity testing in rats. J. Vis. Exp. Jove 2012, 61, e3393. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, F.; Tang, Q.; Xu, J.; Wang, S.; Li, S.; Zou, X.; Cao, Z. Dehydrocrenatidine Inhibits Voltage-Gated Sodium Channels and Ameliorates Mechanic Allodia in a Rat Model of Neuropathic Pain. Toxins 2019, 11, 229. https://doi.org/10.3390/toxins11040229
Zhao F, Tang Q, Xu J, Wang S, Li S, Zou X, Cao Z. Dehydrocrenatidine Inhibits Voltage-Gated Sodium Channels and Ameliorates Mechanic Allodia in a Rat Model of Neuropathic Pain. Toxins. 2019; 11(4):229. https://doi.org/10.3390/toxins11040229
Chicago/Turabian StyleZhao, Fang, Qinglian Tang, Jian Xu, Shuangyan Wang, Shaoheng Li, Xiaohan Zou, and Zhengyu Cao. 2019. "Dehydrocrenatidine Inhibits Voltage-Gated Sodium Channels and Ameliorates Mechanic Allodia in a Rat Model of Neuropathic Pain" Toxins 11, no. 4: 229. https://doi.org/10.3390/toxins11040229
APA StyleZhao, F., Tang, Q., Xu, J., Wang, S., Li, S., Zou, X., & Cao, Z. (2019). Dehydrocrenatidine Inhibits Voltage-Gated Sodium Channels and Ameliorates Mechanic Allodia in a Rat Model of Neuropathic Pain. Toxins, 11(4), 229. https://doi.org/10.3390/toxins11040229