The Key Role of Phosphate on Vascular Calcification

 ,
, 

{kind=link}
{kind=link}
Abstract
1. Introduction
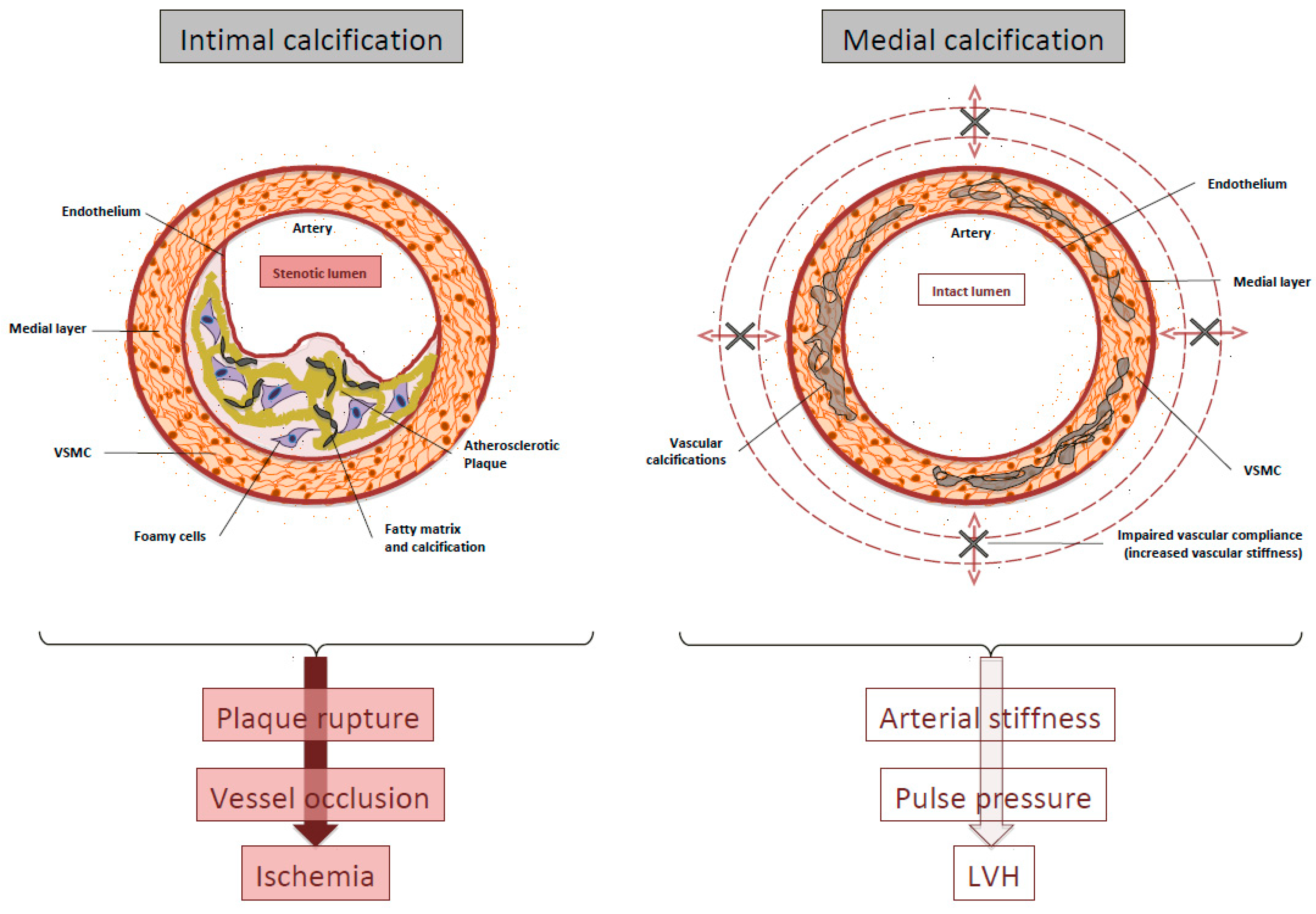
2. Definitions and Clinical Significance of VC
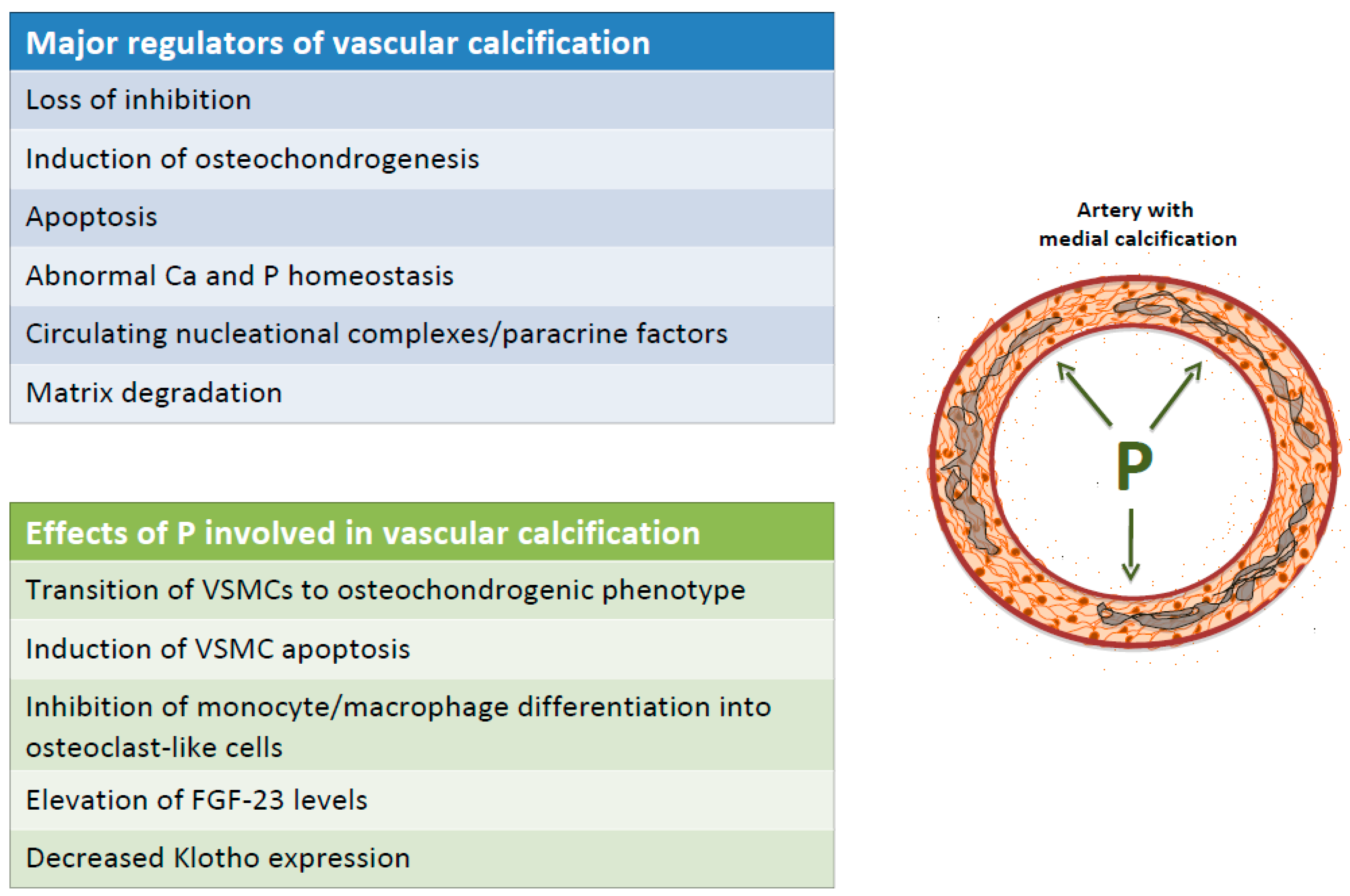
3. VC is An Actively Regulated Process
4. VSMCs: A Model System to Investigate Mechanisms of VC In Vitro
5. The Role of Phosphorus in the Development and Progression of VC
6. Phosphate-Induced Apoptosis of VSMCs
7. Phosphate and Autophagy in VSMCs
8. Iron, Phosphate, and VC
9. Phosphate and VC in the Healthy Population
10. Phosphate and VC in CKD
11. Phosphate and VC in ESRD
Author Contributions
Funding
Conflicts of Interest
References
- Okuno, S.; Ishimura, E.; Kitatani, K.; Fujino, Y.; Kohno, K.; Maeno, Y.; Maekawa, K.; Yamakawa, T.; Imanishi, Y.; Inaba, M.; et al. Presence of abdominal aortic calcification is significantly associated with all-cause and cardiovascular mortality in maintenance hemodialysis patients. Am. J. Kidney Dis. 2007, 49, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Oldendorf, M.; Moshage, W.; Heidler, R.; Zeitler, E.; Luft, F.C. Electron beam computed tomography in the evaluation of cardiac calcification in chronic dialysis patients. Am. J. Kidney Dis. 1996, 27, 394. [Google Scholar] [CrossRef]
- Goodman, W.G.; Goldin, J.; Kuizon, B.D.; Yoon, C.; Gales, B.; Sider, D.; Wang, Y.; Chung, J.; Emerick, A.; Greaser, L.; et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N. Engl. J. Med. 2000, 342, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- London, G.M.; Guérin, A.P.; Marchais, S.J.; Métivier, F.; Pannier, B.; Adda, H. Arterial media calcification in end-stage renal disease: Impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transplant. 2003, 18, 1731–1740. [Google Scholar] [CrossRef]
- Vervloet, M.; Cozzolino, M. Vascular calcification in chronic kidney disease: Different bricks in the wall? Kidney Int. 2017, 91, 808–817. [Google Scholar] [CrossRef]
- Jablonski, K.L.; Chonchol, M. Vascular calcification in end-stage renal disease. Hemodial. Int. 2013, 17 (Suppl. 1), S17. [Google Scholar] [CrossRef]
- Shantouf, R.; Kovesdy, C.P.; Kim, Y.; Ahmadi, N.; Luna, A.; Luna, C.; Rambod, M.; Nissenson, A.R.; Budoff, M.J.; Kalantar-Zadeh, K. Association of serum alkaline phosphatase with coronary artery calcification in maintenance hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1106–1114. [Google Scholar] [CrossRef]
- Moe, S.M.; O’Neill, K.D.; Duan, D.; Ahmed, S.; Chen, N.X.; Leapman, S.B.; Fineberg, N.; Kopecky, K. Medial artery calcification in ESRD patients is associated with deposition of bone matrix proteins. Kidney Int. 2002, 61, 638–647. [Google Scholar] [CrossRef]
- Cozzolino, M.; Gallieni, M.; Brancaccio, D. Vascular calcification in uremic conditions: New insights into pathogenesis. Semin. Nephrol. 2006, 26, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Cianciolo, G.; Galassi, A.; Capelli, I.; Schillaci, R.; La Manna, G.; Cozzolino, M. Klotho-FGF23, Cardiovascular Disease, and Vascular Calcification: Black or White? Curr. Vasc. Pharmacol. 2018, 16, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shi, M.; Zhang, J.; Quiñones, H.; Griffith, C.; Kuro-o, M.; Moe, O.W. Klotho deficiency causes vascular calcification in chronic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Houben, E.; Neradova, A.; Schurgers, L.J.; Vervloet, M. The influence of phosphate, calcium and magnesium on matrix Gla-protein and vascular calcification: A systematic review. G. Ital. Nefrol. 2016, 33, 1724–5590. [Google Scholar]
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: A potential mechanism for accelerated vascular calcification in ESRD. J. Am. Soc. Nephrol. 2004, 15, 2857–2867. [Google Scholar] [CrossRef] [PubMed]
- Bernelot Moens, S.J.; Verweij, S.L.; van der Valk, F.M.; van Capelleveen, J.C.; Kroon, J.; Versloot, M.; Verberne, H.J.; Marquering, H.A.; Duivenvoorden, R.; Vogt, L.; et al. Arterial and Cellular Inflammation in Patients with CKD. J. Am. Soc. Nephrol. 2017, 28, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Huveneers, S.; Daemen, M.J.; Hordijk, P.L. Between Rho(k) and a hard place: The relation between vessel wall stiffness, endothelial contractility, and cardiovascular disease. Circ. Res. 2015, 116, 895–908. [Google Scholar] [CrossRef]
- Cozzolino, M.; Galassi, A.; Apetrii, M.; Covic, A. What would we like to know, and what do we not know about fibroblast growth factor 23? J. Nephrol. 2011, 24, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Y.; Woo, J.; Wang, M. Association of inflammation and malnutrition with cardiac valve calcification in continuous ambulatory peritoneal dialysis patients. J. Am. Soc. Nephrol. 2001, 12, 1927–1936. [Google Scholar] [PubMed]
- Fukuda, D.; Aikawa, M. Intimal smooth muscle cells: The context-dependent origin. Circulation 2010, 122, 2005–2008. [Google Scholar] [CrossRef]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Gomez, D.; Bell, R.D.; Campbell, J.H.; Clowes, A.W.; Gabbiani, G.; Giachelli, C.M.; Parmacek, M.S.; Raines, E.W.; Rusch, N.J.; et al. Smooth muscle cell plasticity: Fact or fiction? Circ. Res. 2013, 112, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Joner, M.; Sakakura, K. Recent highlights of ATVB: Calcification. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1329–1332. [Google Scholar] [CrossRef]
- Ix, J.H.; De Boer, I.H.; Peralta, C.A.; Adeney, K.L.; Duprez, D.A.; Jenny, N.S.; Siscovick, D.S.; Kestenbaum, B.R. Serum phosphorus concentrations and arterial stiffness among individuals with normal kidney function to moderate kidney disease in MESA. Clin. J. Am. Soc. Nephrol. 2009, 4, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Laddu, D.R.; Rana, J.S.; Murillo, R.; Sorel, M.E.; Quesenberry, C.P., Jr.; Allen, N.B.; Gabriel, K.P.; Carnethon, M.R.; Liu, K.; Reis, J.P.; et al. 25-Year Physical Activity Trajectories and Development of Subclinical Coronary Artery Disease as Measured by Coronary Artery Calcium: The Coronary Artery Risk Development in Young Adults (CARDIA) Study. Mayo Clin. Proc. 2017, 92, 1660–1670. [Google Scholar] [CrossRef] [PubMed]
- Linefsky, J.P.; O’Brien, K.D.; Katz, R.; de Boer, I.H.; Barasch, E.; Jenny, N.S.; Siscovick, D.S.; Kestenbaum, B. Association of serum phosphate levels with aortic valve sclerosis and annular calcification: The cardiovascular health study. J. Am. Coll. Cardiol. 2011, 58, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Adeney, K.L.; Siscovick, D.S.; Ix, J.H.; Seliger, S.L.; Shlipak, M.G.; Jenny, N.S.; Kestenbaum, B.R. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J. Am. Soc. Nephrol. 2009, 20, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Shang, D.; Xie, Q.; Ge, X.; Yan, H.; Tian, J.; Kuang, D.; Hao, C.M.; Zhu, T. Hyperphosphatemia as an independent risk factor for coronary artery calcification progression in peritoneal dialysis patients. BMC Nephrol. 2015, 16, 107. [Google Scholar] [CrossRef]
- Zoccali, C.; London, G. Con: Vascular calcification is a surrogate marker, but not the cause of ongoing vascular disease, and it is not a treatment target in chronic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Bornfeldt, K.E. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ. Res. 2016, 118, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Price, P.A.; Lim, J.E. The inhibition of calcium phosphate precipitation by fetuin is accompanied by the formation of a fetuin-mineral complex. J. Biol. Chem. 2003, 278, 22144–22152. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, P.; Elli, F.; Cappelletti, L.; Tosi, D.; Savi, F.; Bulfamante, G.; Cozzolino, M. Osteonectin (SPARC) Expression in Vascular Calcification: In Vitro and Ex Vivo Studies. Calcif. Tissue Int. 2016, 99, 472–480. [Google Scholar] [CrossRef]
- Fadini, G.P. Task for Today: Complete the Puzzle of Circulating Stem Cells and the Atherosclerotic Burden. Circ. Res. 2016, 119, 502–504. [Google Scholar] [CrossRef] [PubMed]
- Cianciolo, G.; Capelli, I.; Cappuccilli, M.; Scrivo, A.; Donadei, C.; Marchetti, A.; Rucci, P.; La Manna, G. Is chronic kidney disease-mineral and bone disorder associated with the presence of endothelial progenitor cells with a calcifying phenotype? Clin. Kidney J. 2017, 10, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Albiero, M.; Menegazzo, L.; Boscaro, E.; Agostini, C.; de Kreutzenberg, S.V.; Rattazzi, M.; Avogaro, A. Procalcific phenotypic drift of circulating progenitor cells in type 2 diabetes with coronary artery disease. Exp. Diabetes Res. 2012, 2012, 921685. [Google Scholar] [CrossRef]
- Gössl, M.; Mödder, U.I.; Atkinson, E.J.; Lerman, A.; Khosla, S. Osteocalcin expression by circulating endothelial progenitor cells in patients with coronary atherosclerosis. J. Am. Coll. Cardiol. 2008, 52, 1314–1325. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.X.; O’Neill, K.D.; Moe, S.M. Matrix vesicles induce calcification of recipient vascular smooth muscle cells through multiple signaling pathways. Kidney Int. 2018, 93, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Raggi, P.; Bellasi, A.; Kooienga, L.; Spiegel, D.M. Mortality effect of coronary calcification and phosphate binder choice in incident hemodialysis patients. Kidney Int. 2007, 71, 438–441. [Google Scholar] [CrossRef]
- Chen, N.X.; O’Neill, K.D.; Duan, D.; Moe, S.M. Phosphorus and uremic serum up-regulate osteopontin expression in vascular smooth muscle cells. Kidney Int. 2002, 62, 1724–1731. [Google Scholar] [CrossRef] [PubMed]
- Patidar, A.; Singh, D.K.; Winocour, P.; Farrington, K.; Baydoun, A.R. Human uraemic serum displays calcific potential in vitro that increases with advancing chronic kidney disease. Clin. Sci. 2013, 125, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Zickler, D.; Willy, K.; Girndt, M.; Fiedler, R.; Martus, P.; Storr, M.; Schindler, R. High cut-off dialysis in chronic haemodialysis patients reduces serum procalcific activity. Nephrol. Dial. Transplant. 2016, 31, 1706–1712. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, P.; Galassi, A.; Alfieri, C.; Messa, P.; Cozzolino, M. Uremic Patients with Increased Vascular Calcification Score Have Serum with High Calcific Potential: Role of Vascular Smooth Muscle Cell Osteoblastic Differentiation and Apoptosis. Blood Purif. 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ayoubi, S.; Sheikh, S.P.; Eskildsen, T.V. Human induced pluripotent stem cell-derived vascular smooth muscle cells: Differentiation and therapeutic potential. Cardiovasc. Res. 2017, 113, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Cho, J.G.; Lee, A.; Chang, W.; Lee, M.-S.; Kim, J. Endothelial to Mesenchymal Transition Represents a Key Link in the Interaction between Inflammation and Endothelial Dysfunction. Front. Immunol. 2018, 9, 294. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Tang, R.N.; Liu, H.; Pan, M.M.; Lv, L.L.; Zhang, J.D.; Crowley, S.D.; Liu, B.C. Cinacalcet ameliorates cardiac fibrosis in uremic hearts through suppression of endothelial-to-mesenchymal transition. Int. J. Cardiol. 2014, 171, e65–e69. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; O’Neill, K.D.; Hood, A.F.; Evan, A.P.; Moe, S.M. Calciphylaxis is associated with hyperphosphatemia and increased osteopontin expression by vascular smooth muscle cells. Am. J. Kidney Dis. 2001, 37, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, P.; Elli, F.; Cappelletti, L.; Tosi, D.; Braidotti, P.; Bulfamante, G.; Cozzolino, M. A new in vitro model to delay high phosphate-induced vascular calcification progression. Mol. Cell. Biochem. 2015, 410, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Hénaut, L.; Mary, A.; Chillon, J.M.; Kamel, S.; Massy, Z.A. The Impact of Uremic Toxins on Vascular Smooth Muscle Cell Function. Toxins 2018, 10, 218. [Google Scholar] [CrossRef] [PubMed]
- Coen, G.; Manni, M.; Agnoli, A.; Balducci, A.; Dessi, M.; De Angelis, S.; Jankovic, L.; Mantella, D.; Morosetti, M.; Naticchia, A.; et al. Cardiac calcifications: Fetuin-A and other risk factors in hemodialysis patients. ASAIO J. 2006, 52, 150–156. [Google Scholar] [CrossRef]
- Ciceri, P.; Volpi, E.; Brenna, I.; Elli, F.; Borghi, E.; Brancaccio, D.; Cozzolino, M. The combination of lanthanum chloride and the calcimimetic calindol delays the progression of vascular smooth muscle cells calcification. Biochem. Biophys. Res. Commun. 2012, 418, 770–773. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, P.; Elli, F.; Brenna, I.; Volpi, E.; Romagnoli, S.; Tosi, D.; Braidotti, P.; Brancaccio, D.; Cozzolino, M. Lanthanum prevents high phosphate-induced vascular calcification by preserving vascular smooth muscle lineage markers. Calcif. Tissue Int. 2013, 92, 521–530. [Google Scholar] [CrossRef]
- Leu, H.J.; Brunner, U. [Calcified and ossified phlebosclerosis]. Vasa 1992, 21, 11–14. [Google Scholar] [PubMed]
- O’Neill, W.C.; Sigrist, M.K.; McIntyre, C.W. Plasma pyrophosphate and vascular calcification in chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 187–191. [Google Scholar] [CrossRef]
- O’Neill, W.C.; Lomashvili, K.A.; Malluche, H.H.; Faugere, M.C.; Riser, B.L. Treatment with pyrophosphate inhibits uremic vascular calcification. Kidney Int. 2011, 79, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Farzaneh-Far, A.; Shanahan, C.M.; Weissberg, P.L. The role of apoptosis in the initiation of vascular calcification. Z. Kardiol. 2001, 90 (Suppl. 3), 43–46. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.C.; McNair, R.; Figg, N.; Skepper, J.N.; Schurgers, L.; Gupta, A.; Hiorns, M.; Donald, A.E.; Deanfield, J.; Rees, L.; et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation 2008, 118, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Wunsch, R.; Turzer, M.; Bahner, M.; Raggi, P.; Querfeld, U.; Mehls, O.; Schaefer, F. Advanced coronary and carotid arteriopathy in young adults with childhood-onset chronic renal failure. Circulation 2002, 106, 100–105. [Google Scholar] [CrossRef]
- Meema, H.E.; Oreopoulos, D.G.; deVeber, G.A. Arterial calcifications in severe chronic renal disease and their relationship to dialysis treatment, renal transplant, and parathyroidectomy. Radiology 1976, 121, 315–321. [Google Scholar] [CrossRef]
- Dai, X.Y.; Zhao, M.M.; Cai, Y.; Guan, Q.C.; Zhao, Y.; Guan, Y.; Kong, W.; Zhu, W.G.; Xu, M.J.; Wang, X. Phosphate-induced autophagy counteracts vascular calcification by reducing matrix vesicle release. Kidney Int. 2013, 83, 1042–1051. [Google Scholar] [CrossRef]
- Negri, A.L.; Ureña Torres, P.A. Iron-based phosphate binders: Do they offer advantages over currently available phosphate binders? Clin. Kidney J. 2015, 8, 161–167. [Google Scholar] [CrossRef] [PubMed][Green Version]
- KDOQI; National Kidney Foundation. KDOQI clinical practice guidelines and clinical practice recommendations for anemia in chronic kidney disease. Am. J. Kidney Dis. 2006, 47 (5 suppl 3), S11–S145. [Google Scholar]
- Zarjou, A.; Jeney, V.; Arosio, P.; Poli, M.; Antal-Szalmás, P.; Agarwal, A.; Balla, G.; Balla, J. Ferritin prevents calcification and osteoblastic differentiation of vascular smooth muscle cells. J. Am. Soc. Nephrol. 2009, 20, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, P.; Elli, F.; Braidotti, P.; Falleni, M.; Tosi, D.; Bulfamante, G.; Block, G.A.; Cozzolino, M. Iron citrate reduces high phosphate-induced vascular calcification by inhibiting apoptosis. Atherosclerosis 2016, 254, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Becs, G.; Zarjou, A.; Agarwal, A.; Kovács, K.É.; Becs, Á.; Nyitrai, M.; Balogh, E.; Bányai, E.; Eaton, J.W.; Arosio, P.; et al. Pharmacological induction of ferritin prevents osteoblastic transformation of smooth muscle cells. J. Cell. Mol. Med. 2016, 20, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Short, R.A. Longitudinal relationships among coronary artery calcification, serum phosphorus, and kidney function. Clin. J. Am. Soc. Nephrol. 2009, 4, 1968–1973. [Google Scholar] [CrossRef] [PubMed]
- Bundy, J.D.; Chen, J.; Yang, W. Risk factors for progression of coronary artery calcification in patients with chronic kidney disease: The CRIC study. Atherosclerosis 2018, 271, 53–60. [Google Scholar] [CrossRef]
- Kestenbaum, B.R.; Adeney, K.L.; de Boer, I.H.; Ix, J.H.; Shlipak, M.G.; Siscovick, D.S. Incidence and progression of coronary calcification in chronic kidney disease: The Multi-Ethnic Study of Atherosclerosis. Kidney Int. 2009, 76, 991–998. [Google Scholar] [CrossRef]
- Pasch, A.; Farese, S.; Gräber, S.; Wald, J.; Richtering, W.; Floege, J.; Jahnen-Dechent, W. Nanoparticle-based test measures overall propensity for calcification in serum. J. Am. Soc. Nephrol. 2012, 23, 1744–1752. [Google Scholar] [CrossRef]
- Smith, E.R.; Ford, M.L.; Tomlinson, L.A. Serum calcification propensity predicts all-cause mortality in predialysis CKD. J. Am. Soc. Nephrol. 2014, 25, 339–348. [Google Scholar] [CrossRef]
- Shantouf, R.S.; Budoff, M.J.; Ahmadi, N.; Ghaffari, A.; Flores, F.; Gopal, A.; Noori, N.; Jing, J.; Kovesdy, C.P.; Kalantar-Zadeh, K. Total and individual coronary artery calcium scores as independent predictors of mortality in hemodialysis patients. Am. J. Nephrol. 2010, 31, 419–425. [Google Scholar] [CrossRef]
- Ohtake, T.; Ishioka, K.; Honda, K.; Oka, M.; Maesato, K.; Mano, T.; Ikee, R.; Moriya, H.; Hidaka, S.; Kobayashi, S. Impact of coronary artery calcification in hemodialysis patients: Risk factors and associations with prognosis. Hemodial. Int. 2010, 14, 218–225. [Google Scholar] [CrossRef]
- Noordzij, M.; Cranenburg, E.M.; Engelsman, L.F.; Hermans, M.M.; Boeschoten, E.W.; Brandenburg, V.M.; Bos, W.J.; Kooman, J.P.; Dekker, F.W.; Ketteler, M.; et al. Progression of aortic calcification is associated with disorders of mineral metabolism and mortality in chronic dialysis patients. Nephrol. Dial. Transplant. 2011, 26, 1662–1669. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Song, S.W.; Kim, T.Y.; Kim, Y.O. Risk factors for progression of aortic arch calcification in patients on maintenance hemodialysis and peritoneal dialysis. Hemodial. Int. 2011, 15, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Nasrallah, M.M.; El-Shehaby, A.R.; Salem, M.M.; Osman, N.A.; El Sheikh, E.; Sharaf El Din, U.A. Fibroblast growth factor-23 (FGF-23) is independently correlated to aortic calcification in haemodialysis patients. Nephrol. Dial. Transplant. 2010, 25, 2679–2685. [Google Scholar] [CrossRef] [PubMed]
- Ozkok, A.; Kekik, C.; Karahan, G.E.; Sakaci, T.; Ozel, A.; Unsal, A.; Yildiz, A. FGF-23 associated with the progression of coronary artery calcification in hemodialysis patients. BMC Nephrol. 2013, 14, 241. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cozzolino, M.; Ciceri, P.; Galassi, A.; Mangano, M.; Carugo, S.; Capelli, I.; Cianciolo, G. The Key Role of Phosphate on Vascular Calcification. Toxins 2019, 11, 213. https://doi.org/10.3390/toxins11040213
Cozzolino M, Ciceri P, Galassi A, Mangano M, Carugo S, Capelli I, Cianciolo G. The Key Role of Phosphate on Vascular Calcification. Toxins. 2019; 11(4):213. https://doi.org/10.3390/toxins11040213
Chicago/Turabian StyleCozzolino, Mario, Paola Ciceri, Andrea Galassi, Michela Mangano, Stefano Carugo, Irene Capelli, and Giuseppe Cianciolo. 2019. "The Key Role of Phosphate on Vascular Calcification" Toxins 11, no. 4: 213. https://doi.org/10.3390/toxins11040213
APA StyleCozzolino, M., Ciceri, P., Galassi, A., Mangano, M., Carugo, S., Capelli, I., & Cianciolo, G. (2019). The Key Role of Phosphate on Vascular Calcification. Toxins, 11(4), 213. https://doi.org/10.3390/toxins11040213