1. Introduction
The ability of a variety of aggregated amyloid-like proteins to interact with lipid bilayers in organelles and cell membranes can occur under certain pathological conditions and in neurodegenerative diseases, which can lead to brain degeneration. For instance, in Parkinson’s disease (PD), neuronal dysfunction and degeneration in affected areas of the brain involves the aberrant folding and aggregation of the α-synuclein (αS) protein [
1,
2]. Although the precise physiological function of αS protein remains to be elucidated, several studies suggest that αS is involved in facilitating the assembly of large complexes at the presynaptic plasma membrane required for neurotransmission [
3,
4]. On the other hand, the aberrant folding of αS protein exposes hydrophobic domains on its surface, which promote self-assembly of αS into the stable toxic oligomeric structures that contain antiparallel β-sheets [
5], one of the cardinal features in the neuropathology of PD [
6]. The pathogenesis of PD largely relates to the ability of toxic oligomeric αS to disrupt the functions of cellular membranes [
7], which include a generalized increase in membrane conductance, the formation of membrane channels and pores, and a disruption of the membrane structural integrity [
8], including that of mitochondrial membranes (reviewed in [
9]).
The molecular mechanisms by which αS binds to mitochondrial membranes have also been investigated. For instance, in dopaminergic cell lines and primary neurons that overexpress αS intracellularly, mitochondrial pathology (mitochondrial swelling, fission and loss of mitochondrial membrane potential, and release of cytochrome c), elevated levels of reactive oxygen species (ROS), an increase of mitochondrial Ca
2+, subsequent bioenergetic failure, and a decrease in cell viability were observed [
10,
11,
12]. Similar effects were also observed when neuronal cell lines were treated with oligomeric αS [
10,
12,
13]. These effects are linked to the ability of αS oligomers to bind with cardiolipin (CL), an abundant phospholipid of the inner mitochondrial membrane (IMM), which results in the perturbation of the IMM structure [
9]. Another consequence of the binding of oligomeric αS to CL is the loss of the function of the mitochondrial adenonsine diphosphate/ adenosine triphosphate (ADP/ATP) carrier, a membrane-bound protein, which is normally stabilized by CL molecules [
14,
15]. In addition, the association of oligomeric αS with CL leads to the formation of amyloid or lipidic pores within mitochondrial membranes [
16]. Apart from oligomeric αS, other aggregated forms of amyloidogenic proteins, including amyloid beta (Aβ) and tau, can compromise the integrity of membranes that have a high amount of CL [
17].
As amyloid-like proteins, cytotoxins and cardiotoxins derived from cobra venoms—which can form oligomeric proteins in a similar manner as amyloidogenic proteins [
18]—can also interact with mitochondria by physically associating with CL in IMMs [
19]. A previous study raised the possibility that amyloidogenic proteins and natural cytotoxins may share a common pore-forming mechanism involving the piercing of mitochondria via CL binding [
9]. Cobra venom cardiotoxins (CVCs) have been used as a molecular tool to probe the structure and functions of biological membranes [
20,
21] by studying the structure of model membranes [
22,
23,
24]. Importantly, CVCs have also been investigated as potential therapeutic agents to treat various diseases, including cancer [
20,
21,
25,
26,
27]. Recently, CVCs have been described as potential therapeutic agents to restore mitochondrial bioenergetics in age-associated neurodegenerative diseases [
19,
20].
CVCs, also known as cardiotoxins or cytotoxins, are basic amphipathic proteins that contain a rigid spatial structure, including anti-parallel β-sheets (stabilized by four conserved disulfide bonds) and three elongated finger-like loops, each with apolar tips that are flanked by small stretches of highly conserved positively charged Lys and Arg residues (reviewed in [
20]). In addition, cytotoxins have been classified as either S-type cytotoxins (contain Ser in residue position 28 of loop II) or as P-type cytotoxins (contain Pro in residue position 30 of loop II) [
24]. As a rule, P-type cytotoxins have a higher net positive charge than S-type cytotoxins. P-type cytotoxins, having apolar Pro, but not polar Ser residues, have higher hydrophobicity in loop II compared to S-type cytotoxins, and this is believed to account for the higher cytotoxicity of P-type cytotoxins [
24].
Although not widely known for their neurotoxicity, the pathophysiological effects of cytotoxins in neurons include their ability to promote neurotoxicity by depolarizing excitable membranes of neurons, deregulate the activity of cell membrane-bound enzymes and receptors, and thereby disrupt neurotransmission and promote axonal degeneration. Beyond the central nervous system, cytotoxins can inhibit platelet aggregation, and promote cardiac arrest (reviewed in [
20]). These pathological effects are linked to the ability of cytotoxins to electrostatically interact with anionic lipids on the cell membrane to form stable oligomeric complexes that can act as pore-forming structures or to induce the formation of non-bilayer structures (reviewed in [
20]).
The molecular mechanisms by which CVCs promote cellular pathology remain to be elucidated. Recent evidence has shown that cytotoxins can target the membranes of intracellular organelles, such as lysosomes [
26,
28] and mitochondria [
27,
29]. For instance,
Naja naja atra cardiotoxin 3 (CTX3) has been shown to target mitochondria to induce oxidative stress, leading to a collapse of the mitochondrial transmembrane potential, release of cytochrome C, and eventual activation of apoptosis [
27].
We have shown that cytotoxins have the ability to remodel the lipid membranes of mitochondria to modulate mitochondrial bioenergetics. By employing a battery of biophysical, biochemical, and computer modeling assays, our recently published studies showed that two cytotoxins from
Naja naja oxiana cobra venom bind specifically to CL in model membranes and induce the formation of non-bilayer structures in membranes in intact mitochondria (reviewed in [
20]). Additionally, we show that the same two cytotoxins (CTI and CTII) induce the formation of a transient non-bilayer phase in mitochondrial membranes at very low concentrations, a phenomenon that leads to increased ATP synthase activity [
30,
31]. This observation suggests that the formation of transient non-bilayer structures is a physiological event that occurs to support the proper structure and function of mitochondria [
30,
31]. Interestingly, higher concentrations of cytotoxins induced a significant amount of a non-bilayer phase in IMMs, which surpassed that of the lamellar phase and completely abolished ATP synthase activity, which was due to the ability of the cytotoxin to disrupt the IMM [
30,
31]. However, the molecular mechanism by which S-type cardiotoxins can bind to mitochondrial membranes to elicit mitochondrial dysfunction in cells has not been elucidated.
The molecular surface features of amyloidegenic proteins and cobra cardiotoxins are particularly similar—both proteins have a positively charged N-terminal region, contain a central region with predominantly hydrophobic residues having a high propensity for adopting the β-sheet secondary structure, and harbor acidic residue(s) in the C-terminal domain—all of which likely underlie their shared membrane-active properties, such as the formation of transmembrane pores, disruption of membrane packing, and targeting of mitochondrial CL [
8,
9,
12,
30,
31]. Given these shared membrane-active properties, we hypothesized that cardiotoxin VII4 (CTX3) from
Naja mossambica mossambica, similarly to amyloidegenic proteins, promotes neurotoxicity by translocating mitochondria to disrupt mitochondrial function via a mechanism that involves binding to CL. In this study, we show that cardiotoxin VII4, an S-type cardiotoxin that possesses an unusually high positive charge, translocates to mitochondria in mouse primary cortical neurons and in SH-SY5Y neuroblastoma cells to promote aberrant mitochondrial fragmentation, a reduction in oxidative phosphorylation, and overt neurodegeneration. By utilizing EPR and
1H-NMR spectroscopy and phosphorescence quenching of erythrosine in model membranes, we further show that cardiotoxin VII4 binds to anionic CL, but not to zwitterionic phosphatidylcholine (PC), to induce the formation of non-bilayer structures in CL enriched membranes that model the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM). Finally, molecular dynamics and in silico docking studies revealed the molecular mechanism by which cardiotoxin from
Naja mossambica mossambica interacts with CL to intercalate into mitochondrial membranes.
3. Discussion
Membrane-active proteins from cobra venom, also known as cardiotoxins or cytotoxins, have been previously employed as valuable molecular tools to study the structure and functions of biological membranes [
19,
20,
21,
22,
23,
24,
30,
31,
35]. Indeed, there is a wealth of experimental data that shows that cardiotoxins are membrane-remodeling proteins as they can bind to anionic phospholipids of cell membranes to alter the natural packing of lipids, leading to the formation of transmembrane pores and/or non-bilayer structures [
21], and eventual cell death [
20]. A study showed that cobra venom cardiotoxin targets mitochondria to induce mitochondrial dysfunction and ensuing apoptosis [
36]. This observation poses the question of whether the cytotoxic action of cobra venom cardiotoxins is produced via its interaction with the cell membrane or via its interaction with the membrane of subcellular organelles—namely mitochondria. In our recent studies, we have shown that cytotoxins from
Naja oxiana cobra venom specifically target CL in isolated mitochondria and in model membranes enriched with CL to induce the formation of non-bilayer structures within mitochondrial membranes, presumably in the IMM [
19,
30,
36]. Intriguingly, at very low concentrations, cobra cytotoxins increased the activity of mitochondrial ATP-synthase whereas higher concentrations of cytotoxins completely abolished ATP-synthase activity, presumably by permeabilizing and consequently disintegrating mitochondrial membranes via the formation of toroidal pores [
30,
35].
In this study, we have shown that cardiotoxin VII4 from
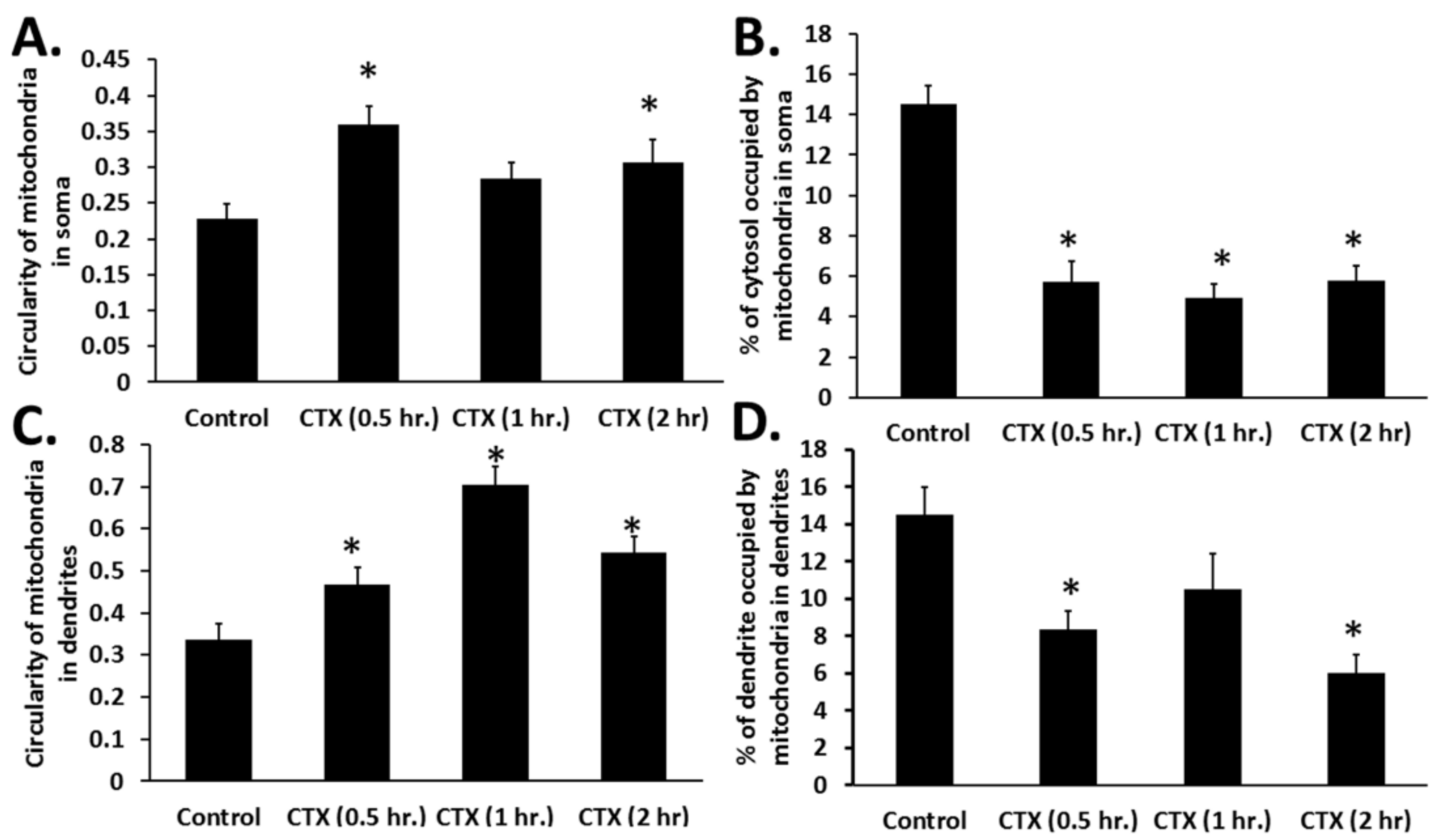
Naja mossambica mossambica cobra venom translocates to mitochondria (colocalization of Rhodamine-conjugated cardiotoxin with respiring mitochondria) in mouse cortical neurons and SH-SY5Y neuroblastoma cells to promote aberrant mitochondrial fragmentation/swelling (
Figure 5), reduce mitochondrial respiration (
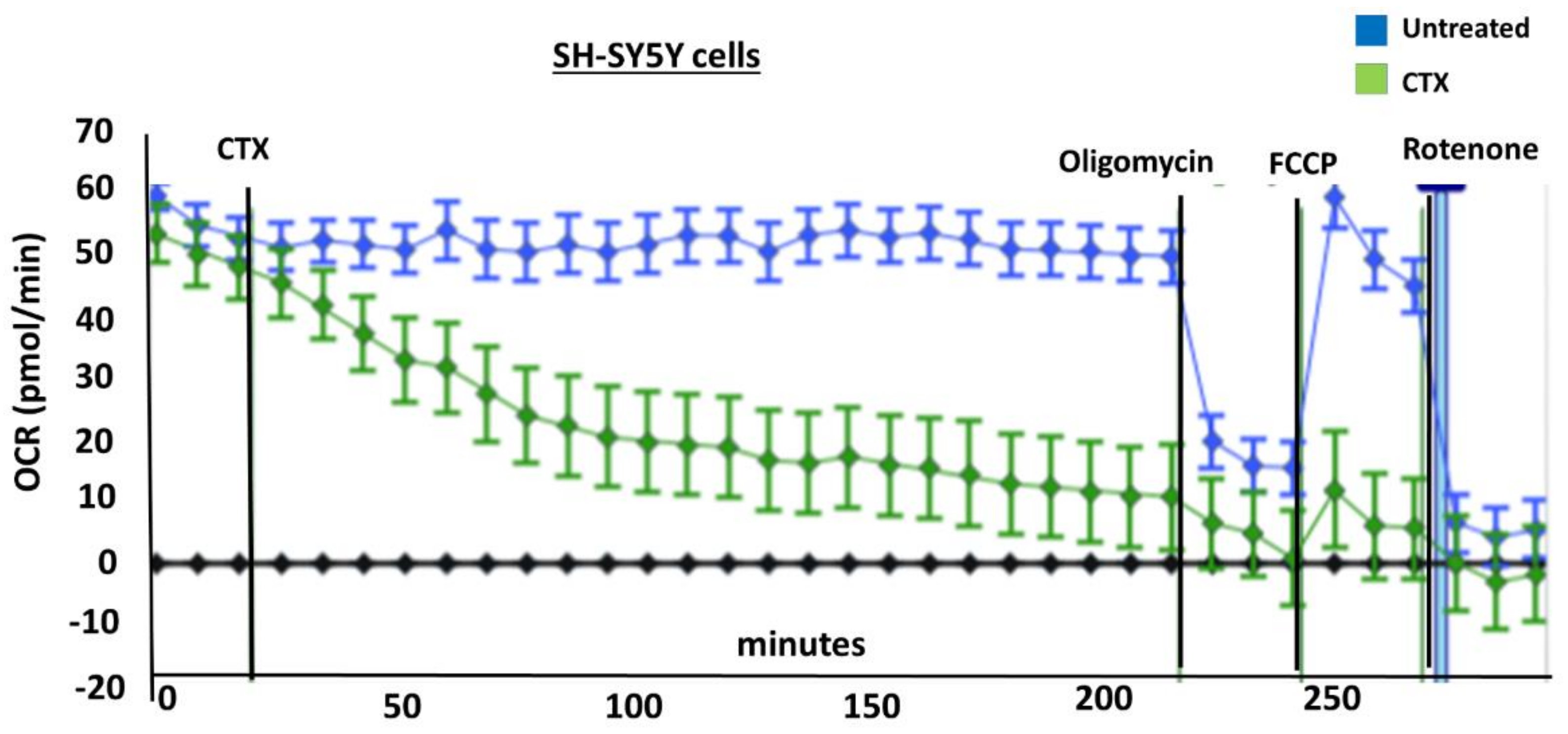
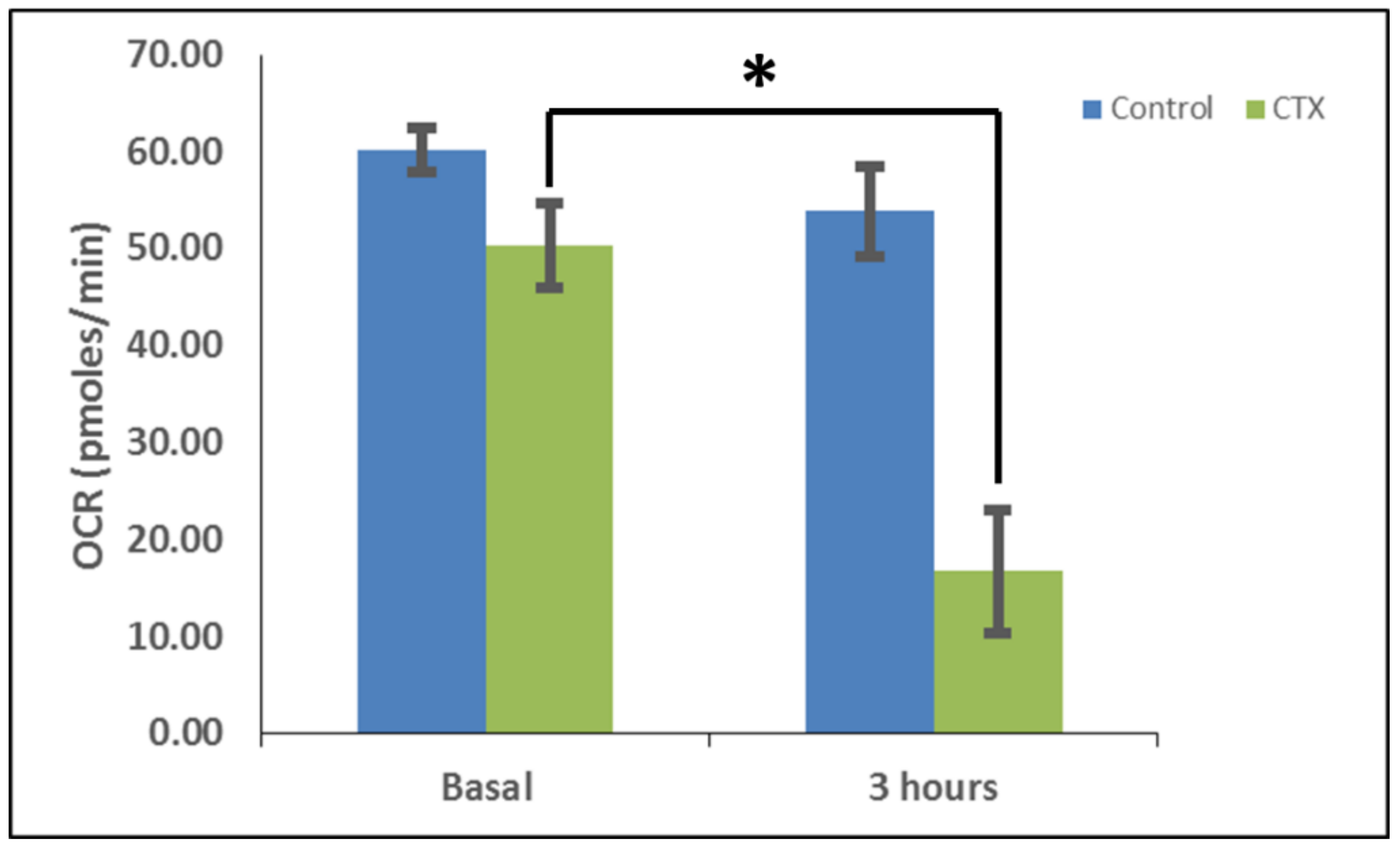
Figure 6 and
Figure 7) and ATP synthesis (
Figure 2), and elicit neurodegeneration (
Figure 1,
Figure 2,
Figure 3 and
Figure 4). In neurons, the rapid translocation of cardiotoxin VII4 to mitochondria (<2 h.) coincides with a concomitant robust decrease in oxidative phosphorylation and of mitochondrial-derived ATP levels at 4 h post-treatment, early events that are upstream of neurodegeneration. We further showed that the ability of cardiotoxin VII4 to colocalize with mitochondria (
Figure 3 and
Figure 4) in live neurons/neuronal cells is associated with its ability to interact with CL in model membranes (
Figure 8,
Figure 9,
Figure 10,
Figure 11 and
Figure 12,
Table 1 and
Table 2). Overall, our imaging and biophysical data support the notion that cobra cardiotoxin VII4 exhibit mito-toxic activity at high concentrations, presumably by interacting with CL to form non-bilayer structures (e.g., toroidal pores) to disrupt the mitochondrial membrane and structure.
Like CTI and CTII from
Naja naja oxiana cobra venom, we found that cardiotoxin VII4 preferentially binds to anionic phospholipids (CL), but not PC. Our data support the concept that highly basic three-fingered S and P-type cardiotoxins target the mitochondrion. Indeed, our erythrosine phosphorescence quenching data showed that cardiotoxin VII4 does not bind to pure PC membranes, but binds to PC membranes containing molar concentrations of CL as low as 3% to 5% (
Table 1), a molar concentration of CL as found in the OMM [
9]. The method of EPR of oriented multibilayer films, which we employed to investigate the effects of cardiotoxin VII4 on the phospholipid packing in pure PC membranes containing CL, showed that cardiotoxin VII4 does not affect phospholipid packing in pure PC membranes, presumably because cardiotoxin VII4 does bind to lipid membranes that consist of PC. However, we observed that cardiotoxin VII4 binds to PC membranes containing CL to decrease phospholipid mobility and induce the formation of non-bilayer structures (
Table 2 and
Figure 8). Most notably, the ability of cardiotoxin VII4 to induce non-bilayer structures was more pronounced in PC membranes containing 20 molar percent of CL than in the PC membranes containing 5 molar percent of CL (
Figure 8 and
Table 2).
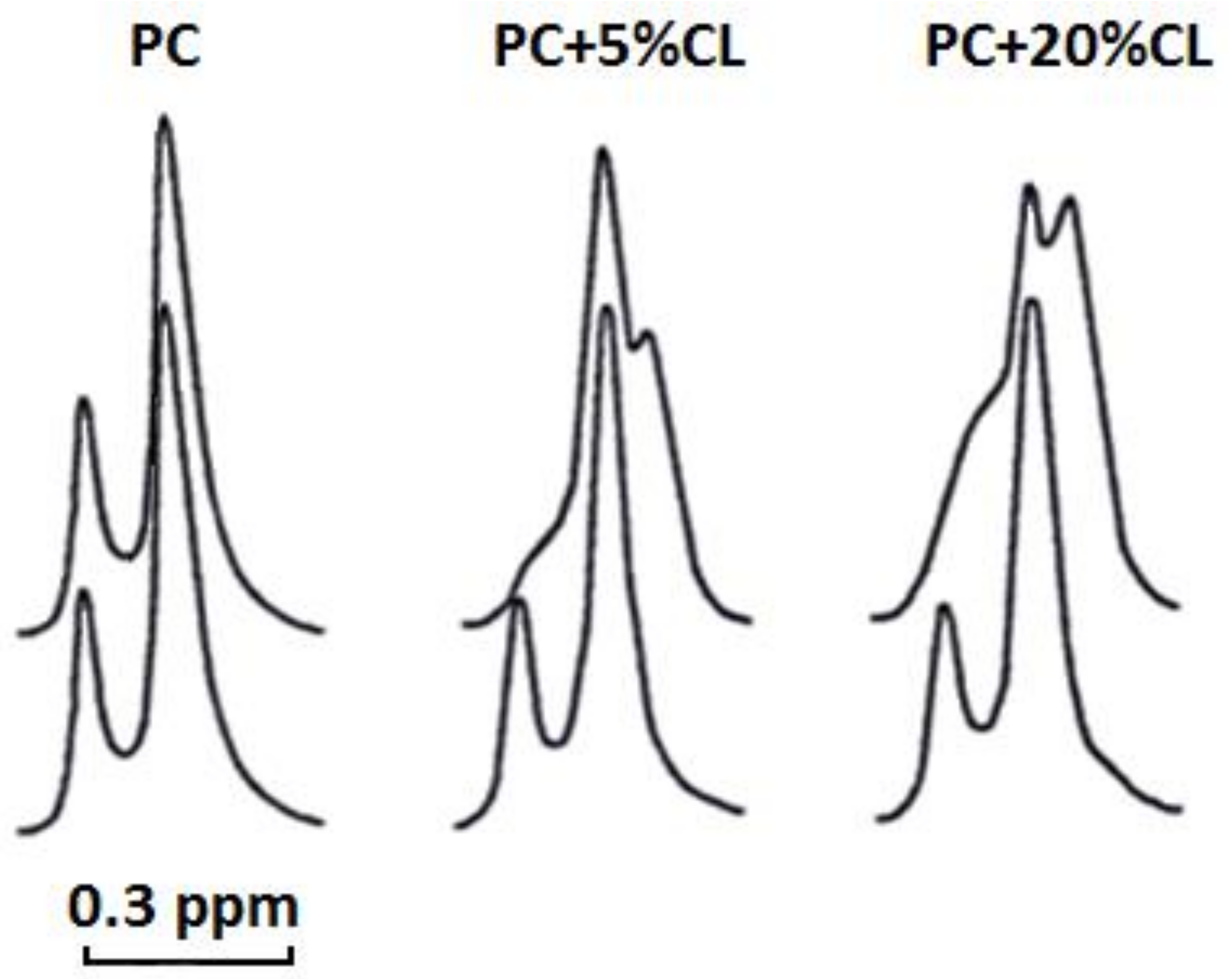
Our
1H-NMR spectroscopy data showed that cardiotoxin VII4 does not interact with pure PC liposomes, but interacts with CL to enhance the permeability of PC liposomes containing CL. Cardiotoxin VII4 also potentiates the formation of non-bilayer structures in CL containing liposomes as evident by the appearance of the high-field resonance peak for the outer N
+(CH
3)
3 signal, which is more pronounced in liposomes containing 20 mol% CL relative to liposomes containing 5 mol% CL (
Figure 9). This interesting observation suggests that PC and CL molecules contribute to the formation non-bilayer structures induced by the interaction of cardiotoxin VII4 with PC membranes containing CL.
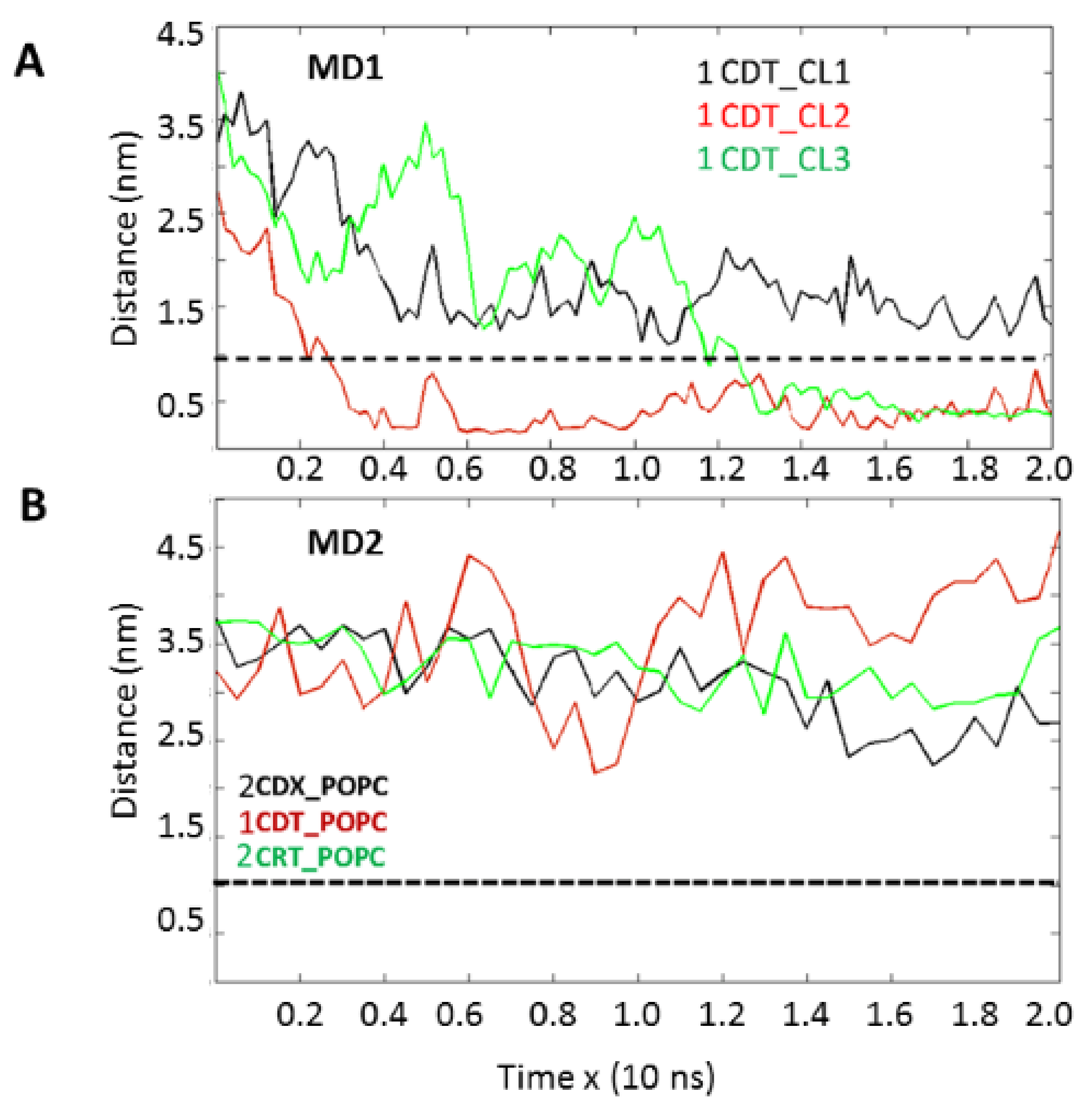
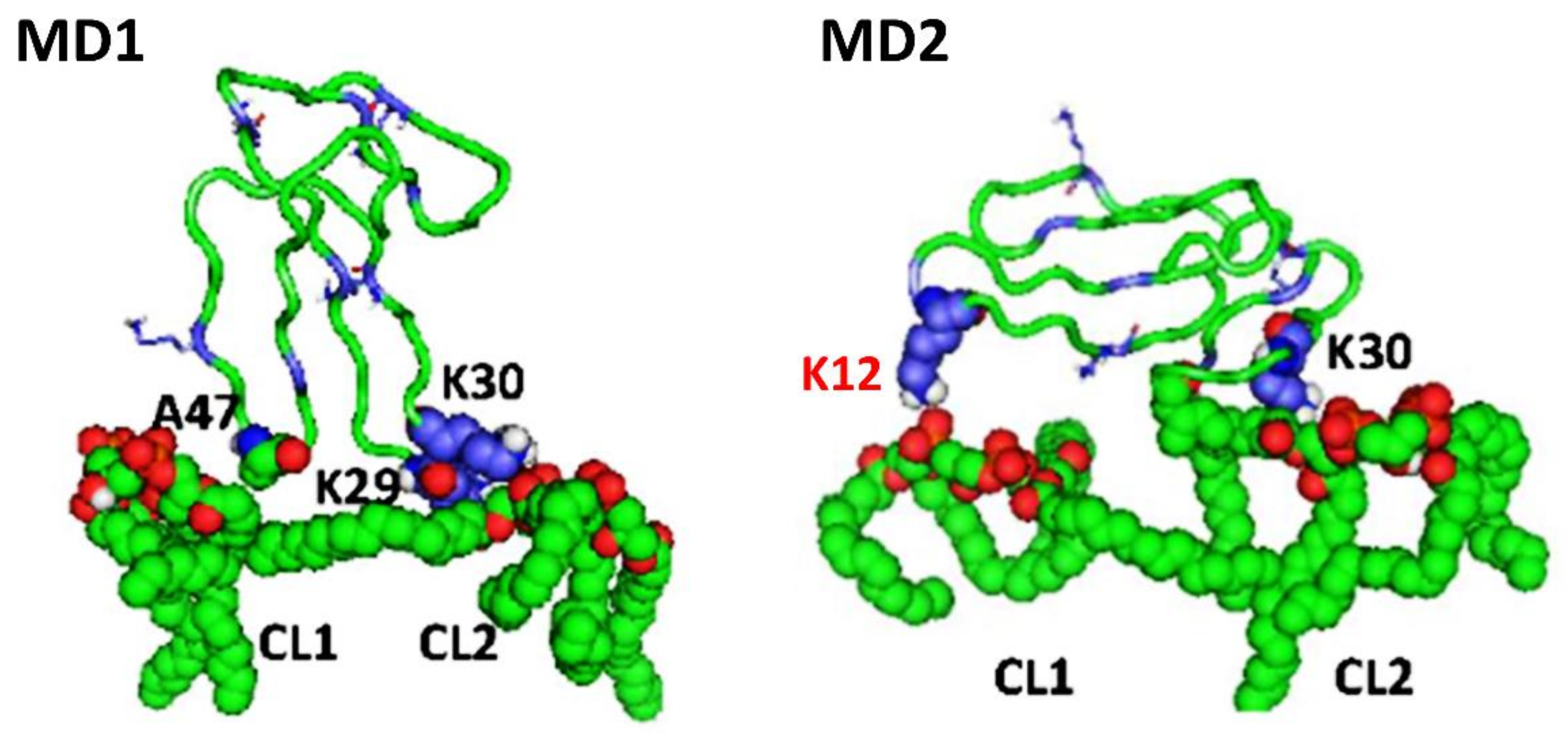
Importantly, our molecular dynamics data agree with our biophysical data in that cardiotoxin VII4 binds more avidly to CL relative to PC in lipid membranes of a similar phospholipid as that found in the OMM (PC + CL). Indeed, our molecular dynamics and biophysical data showed that cardiotoxin VII4 was unable to interact with PC lipid bilayers that lacked CL. In addition, our molecular dynamics data showed that cardiotoxin VII4 binds to CL molecules via two modes of interactions (
Figure 12). The first mode of interaction (
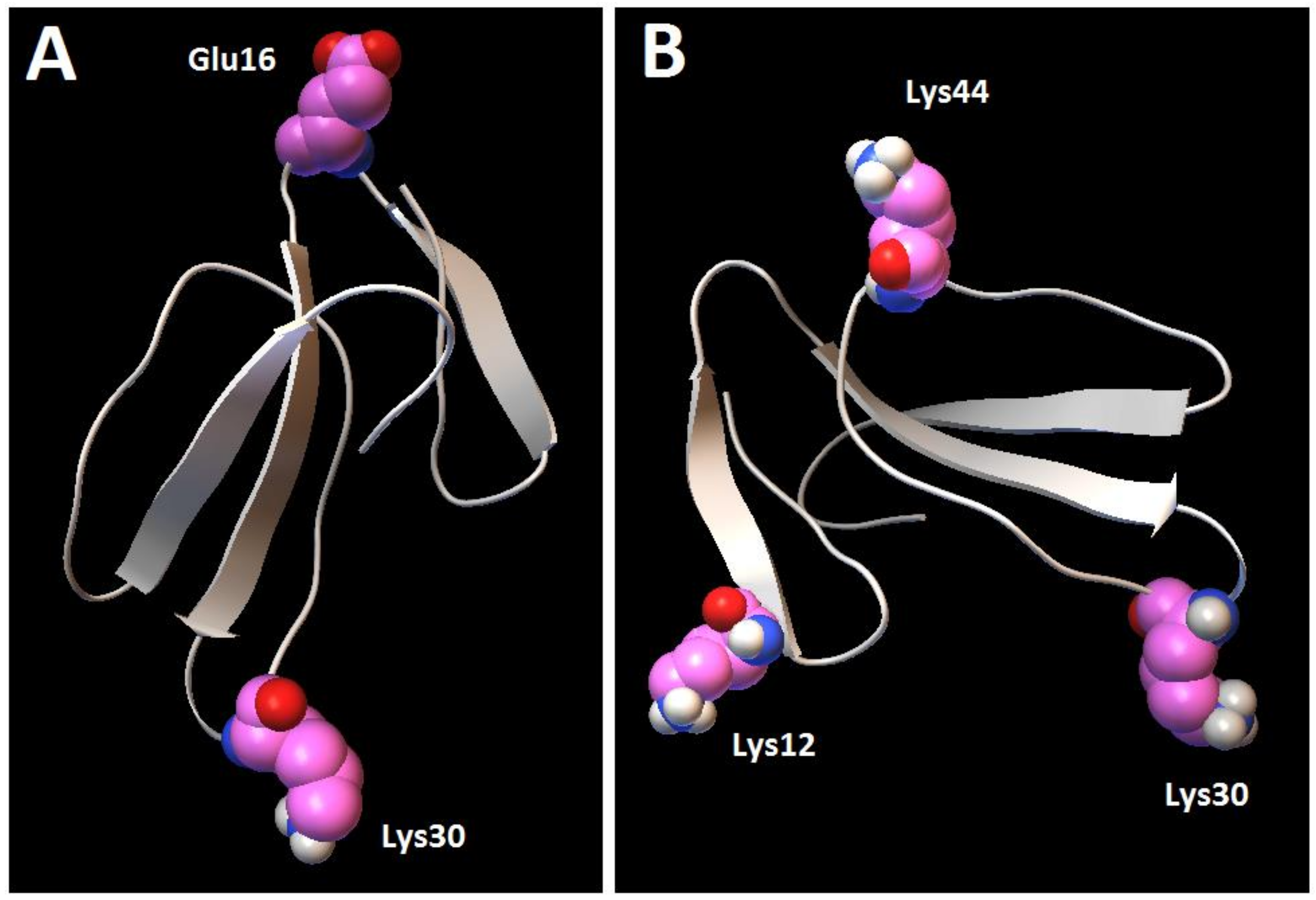
Figure 12, MD1) involves the binding of cardiotoxin VII4 to two CL molecules through electrostatic (via Lys 30 of the loop 2) and hydrogen bonds (via Ala47 of the loop 3), which ensures an orientation in which the long axis of the cardiotoxin is parallel to the membrane surface normal. Hence, should cardiotoxin VII4 embed into the lipid bilayer in this orientation, it will expose the acidic amino acid residue, Glu16, located on the opposite side of cardiotoxin VII4 (
Figure 14A), presumably to interact with the basic choline groups of PC from neighboring regions of the lipid bilayer. Therefore, this mode of cardiotoxin–membrane interaction should facilitate intermembrane contacts, and may lead to the formation of inverted micelles, which contain a cardiotoxin at the core of a micelle, in a similar mechanism as described in previous studies [
19,
20,
30]. When cardiotoxin is dissolved in solution, Glu16 cannot interact with choline groups of PC on the surface of the lipid bilayer due to the high positive force field on the surface of cardiotoxin. However, following the initial electrostatic interactions with CL on the OMM, it is likely that the solvent-exposed Glu 16 may facilitate the penetration of cardiotoxin into the lipid bilayer by interacting with the choline groups of PC from neighboring regions of the OMM.
In the second mode of interaction (
Figure 12, MD2), cardiotoxin VII4 binds to two CL molecules at the surface of the lipid bilayer via electrostatic forces that involve the interaction of two basic residues (Lys 30 and Lys12) in cardiotoxin with the phosphate groups of CL. Hence, in this mode, the long axis of cardiotoxin VII4 is oriented perpendicular to the membrane surface normal, thus exposing Lys44 to the cytosolic side of the OMM, which allows Lys44 to interact with the phosphate groups of CL of a neighboring lipid region (
Figure 14B) in a similar manner as described in a previous study [
19]. Interestingly, both basic amino acid residues located within loop 2 of cardiotoxin VII4 are not conserved among cobra venom cardiotoxins/cytotoxins (
Figure 13), suggesting a unique mechanism by which the basic molecular surface of cardiotoxin VII4 can bind to the OMM. Interestingly, it is worth noting that cardiotoxin VII4 is the only known S-type cardiotoxin/cytotoxin that contains basic residues within loop 2. In contrast, other P-type cytotoxins (e.g., 1CD9 and 2CRT) contain positively charged amino acid residues (His31 or Lys31) within loop 2, which are distinct from VII4 (
Figure 13). It is believed that basic residues and surrounding apolar amino acid residues with loop 2 facilitate the insertion of cytotoxins into lipid bilayers, whereas acidic and polar residues in loop 2 of most S-type cytotoxins lack lipid bilayer-penetrating activity [
20]. However, it is conceivable that Lys30, which considerably extends from the molecular surface of cardiotoxin VII4 (
Figure 14A), targets the phosphate groups of CL to ensure the insertion of cardiotoxin in an orientation that involves positioning the long axis of cardiotoxin VII4 parallel to the membrane surface normal. Hence, it is not likely that Lys29 directly interacts with a phosphate group of CL (
Figure 12), but may facilitate the initial electrostatic interactions of cardiotoxin with the phosphate groups’ CL of the lipid bilayer prior to insertion into the OMM.
In another attempt to characterize the CL and PC biding sites on the molecular surface of cardiotoxin VII4, we performed molecular docking studies. Hence, by cosidering the ability of cardiotoxin to interact with the phospholipid head groups, our docking data provides insight on how cardiotoxin interacts with the phosphate groups of CL prior to embedding into the hydrophobic regions of the simulated IMM. In brief, our molecular docking data show that four basic residues (Lys12, Lys18, Lys35, and His36) formed ionic bonds with the phosphate groups of CL (
Table S1) whereas three basic residues (Lys12, Lys18, and His36) formed ionic bonds with a phosphate group of PC (
Table S3). These results differ from our molecular dynamics data, which show that VII4 does not bind to PC, but preferentially binds to CL via long-range ionic interactions between the phosphate groups of CL and the −N
+H
3 group of Lys12 and Lys30. This apparent discrepancy between our molecular docking and molecular dynamics data is likely due to the fact that the alkyl chains of lipids, whose molecular motion is restricted within hydrophobic regions of the lipid bilayers, does not allow the docking of a lipid polar head with the cardiotoxin, and likely resembles interactions between the protein and a membrane surface. Therefore, our molecular docking data likely informs on the interaction of cardiotoxins with the alkyl chains of PC and CL once it has successfully penetrated the hydrophobic core of the lipid bilayer (OMM or IMM). Nevertheless, the most stable docked structures of cardiotoxin VII4 with the CL polar head involved ionic interactions between Lys 12 and the phosphate group (
Table S1), which is consistent with our MD simulations data (
Figure 12, MD2). On the other hand, Lys30 of cardiotoxin VII4 was not identified as an interactive amino acid residue in the docked structures of cardiotoxin with the polar heads of CL or PC, presumably because the −N
+H
3 group of Lys30 is far from the surface of cardiotoxin. It is conceivable that the small positively charged surface area contributed by the –N
+H
3 group of Lys30 may interact with the choline groups of PC to enable the cardiotoxin to penetrate the membrane surface.
It should be noted that the binding enthalpy energy released for all putative docked cardiotoxin VII4-CL polar head complexes was observed to be relatively higher than the enthalpy energy released when cardiotoxin VII4 docked to the polar heads of PC (
Tables S1 and S3). These data support the notion that cardiotoxin VII4 preferentially binds to CL on the surface of lipid bilayers of the OMM. Additionally, the binding enthalpy energy released for all putative docked structures for cardiotoxin VII4 complexed to the complete structure of CL was observed to be lower than that for the docked structures of cardiotoxin VII4 complexed to the complete PC molecule (
Tables S2 and S4). These data suggest that PC molecules bind more strongly to cardiotoxin VII4 compared to CL when cardiotoxin VII4 embeds into the membrane hydrophobic area of lipid membranes. In addition, our in silico data suggest that hydrophobic interactions generated by the two long alkyl chains of PC play a major role in enabling the cardiotoxin to penetrate the lipid bilayer. In addition, the four alkyl chains from CL may facilitate the formation of non-bilayer structures known to permeabilize CL-containing lipid bilayers.
An inherent limitation of our docking simulations is the lack of flexibility of the protein, which is held fixed in most docking algorithms. While MD simulations can in principle be used to evaluate molecular flexibility, for large proteins, the conformational changes may occur over several dozens of nanoseconds. A way to overcome this limitation is to generate multiple structures (conformations) of the protein using various softwares (e.g., Confab, OMEGA, and PC Spartan Pro). While it would be preferable to probe the conformational profile of ligands in the enzyme active site to significantly improve the accuracy of docking studies [
37,
38], we employed the highest level of flexibility allowed by AutoDock by using the exhaustiveness level 16, and performed three docking simulations for each ligand for statistical purposes. Hence, running docking studies using the aforementioned paramaters can add more rigor and accuracy to our docking studies and can partially account for the flexibility and conformational changes that lipids can undergo (~dozens of nanoseconds).
Overall, based on our compiled and in silico data, we suggest the following molecular mechanism by which cardiotoxin VII4 targets mitochondria to disrupt mitochondrial function. First, our collective data suggest that cardiotoxin VII4 penetrates the plasma membranes via a similar mechanism previously described for cytotoxins derived from
Naja oxiana cobra venom [
19]. In brief, cardiotoxin VII4 binds to anionic phospholipids on the plasma membrane of a neuron to dehydrate the lipid surface, alter the asymmetry of the lipid bilayer to form immobile non-bilayer structures, and subsequently inserts into hydrophobic area of the membrane with the long axis oriented parallel to the membrane surface normal. However, unlike cytotoxins (CTI and CTII), an acidic residue, Gly16, located on the opposite side of cardiotoxin VII4 may attract the basic choline groups of PC from a neighboring membrane (e.g., a membrane of the synaptic juncture), presumably to facilitate the formation of non-bilayer structures, including inverted micelles that may contain cardiotoxin VII4 within the center of the inverted micelle. The inverted micelle then releases cardiotoxin VII4 to the cytosolic compartment of neurons to target the OMM, and subsequently interact with OMM-IMM contact sites enriched in CL. Therefore, the biochemical environment of the OMM, which is enriched with inverted CL, may facilitate the translocation of cardiotoxin VII4 into a junction between OMM and IMM, where it generates inverted micelles in the intermembrane contact sites via interactions of the conserved basic residues, Lys12, Lys30, and Lys44 (
Figure 14), with CL molecules of the OMM and IMM. An inverted micelle containing cardiotoxin VII4 within a contact site between the OMM and IMM may thereby release the cardiotoxin into the intermembrane space or into the mitochondrial matrix, where cardiotoxin may bind to the mitochondrial cristae to disrupt the IMM, leading to mitochondrial fission/swelling of the mitochondrial complexes, leading to a reduction in mitochondrial oxidative phosphorylation and ATP production.
It is worth noting that the membrane-active properties of cardiotoxin VII4, as supported by our collective biophysical data, are highly similar to the membrane-active properties of cytotoxin CTII from
Naja oxiana cobra venom [
19]. Therefore, it is conceivable that cardiotoxin VII4, similarly to cytotoxin CTII, may affect mitochondrial structure and function in a concentration-dependent manner. At low concentrations, cytotoxin CTII can increase ATP-synthase activity to increase the steady-state level of ATP [
30,
31], whereas high concentrations of cytotoxin are detrimental to the mitochondrial structure by the formation of oligomers. Future directions warrant investigating the effects of very low concentrations of cardiotoxin VII4 on ATP-synthase activity and mitochondrial structure in intact mitochondria as previously described [
30]. Should cardiotoxin VII4 prove to increase mitochondrial ATP-synthase activity at very low concentrations while maintaining an intact mitochondrial structure, cardiotoxin VII4 may represent a promising pharmacological avenue for improving the mitochondrial bioenergetics in neurons, which can be employed for the treatment of patients with the age-depended neurodegeneration conditions.
5. Materials and Methods
5.1. Molecular Reagents
Egg yolk L-α-phosphatidylcholine (PC), cardiolipin (CL) from Escherichia coli, the EPR spin-labeled probes 5-doxylstearic acid (5-DSA), and potassium ferricyanide were all purchased from Sigma Chemical Co. (St. Louis, MO, USA). All phospholipids were further purified on silica columns. Erythrosine (Sigma-Aldrich, St. Louis, UK) was used as a phosphorescent probe while ferrocene (Sigma Chemical Co., St. Louis, MO, USA) was used as a quencher of erythrosine phosphorescence.
Pure cardiotoxin from Naja naja mossambica was purchased from Sigma-Aldrich (St.Louis, MO, USA) (C9759). The NHS-Rhodamine conjugate alone and a NHS-Rhodamine Antibody Labeling kit (53031) were purchased from Thermo Scientific (Waltham, MA, USA). A CytoTox-ONETM Homogenous Membrane Integrity Assay (G7890) was purchased from Promega (Madison, WI, USA). All other reagents were purchased from Life Technologies (Grand Island, NY, USA), including MitoTracker Green, Dulbecco’s Modified Eagle’s Medium (DMEM), Neurobasal Medium, B-27 Supplement, Glutamax, poly-L-lysine, fetal bovine serum, and sodium pyruvate. D-glucose, glutamic acid, and D-galactose were obtained from Thermo Scientific.
5.2. Handling of Timed-Pregnant Mice and Ethics Statement
All experiments involving mice were done in accordance to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines and in adherence to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health). All animal procedures used to minimize distress were specifically approved for research by the Institutional Animal Care and Use Committee of the University of Nevada, Reno (Protocol # 00572, approved on January 18, 2019 for three years, Animal Welfare Assurance Number: Issued by OLAW: A3500-01). Specific pathogen free (SPF) 14 day timed pregnant female C57/BL6 mice were ordered from Charles River (Wilmington, MA) and housed for two to three days upon arrival at the specific-pathogen free-approved Center for Molecular Medicine Animal Facility (UNR).
5.3. Cell Culture
Primary cortical neurons were prepared by extracting the cortices of C57BL/6J fetal mice as previously described [
32,
39]. In brief, primary cortical neurons were plated on poly-L-lysine pre-coated plates at a cell density of 200,000 cells per well in 24 well plates, in 96 well plates for the analyses of cell survival, and in 4 well chambered coverglasses (Nunc (Thermo Scientific)) for performing confocal microscopy-mediated analysis of the colocalization of NHS-Rhodamine labeled-cardiotoxin with mitochondria labeled with MitoTracker Green. The primary cortical neurons were initially plated in Neurobasal plating media, (94.8% neurobasal medium, 2% B-27 supplement, 0.2% glutamic acid, 2% fetal bovine serum, and 2 mM Glutamax). Three days later, two thirds of the media was removed and replaced with neurobasal maintenance media (97% neurobasal medium, 2% B-27 supplement, and 2.0 mM Glutamax). SH-SY5Y neuroblastoma cells (ATCC, Manassas, VA, USA) from pre-existing passages were purchased from the American Tissue Culture Collection and maintained up to 27 passages. SH-SY5Y neuroblastoma were maintained in complete DMEM medium (DMEM 90%, 10% fetal bovine serum, 2 mM Glutamax and supplemented with 5 mM HEPES) per the cell culture instructions published by American Tissue Culture Collection (ATCC).
5.4. LDH Assay
To analyze for the effects of snake venom cardiotoxin VII4 on cell viability, the CytoTox-ONETM Homogenous Membrane Integrity Assay kit (G7890) (Madison, WI, USA) was utilized per the manufacturer’s instructions. After primary cortical neurons or neuronal cells (SH-SY5Y cells) reached up to 85% confluency on black-sided Corning 96-well plates (Fisher Scientific, Waltham, MA, USA), the cells were exposed to increasing concentrations of VII4 (0.5 mM–2.0 μM, and 2.5 μM) in primary neurons or SH-SY5Y neuroblastoma cells (1.0 mM–10.0 μM ) for 24 h prior to measuring the release of lactate dehydrogenase (LD) as measured by fluorescence spectroscopy using an excitation wavelength of 560 nm and emission wavelength of 590 nm on a Molecular Devices SpectraMax M3 plate reader.
5.5. ATP Assays
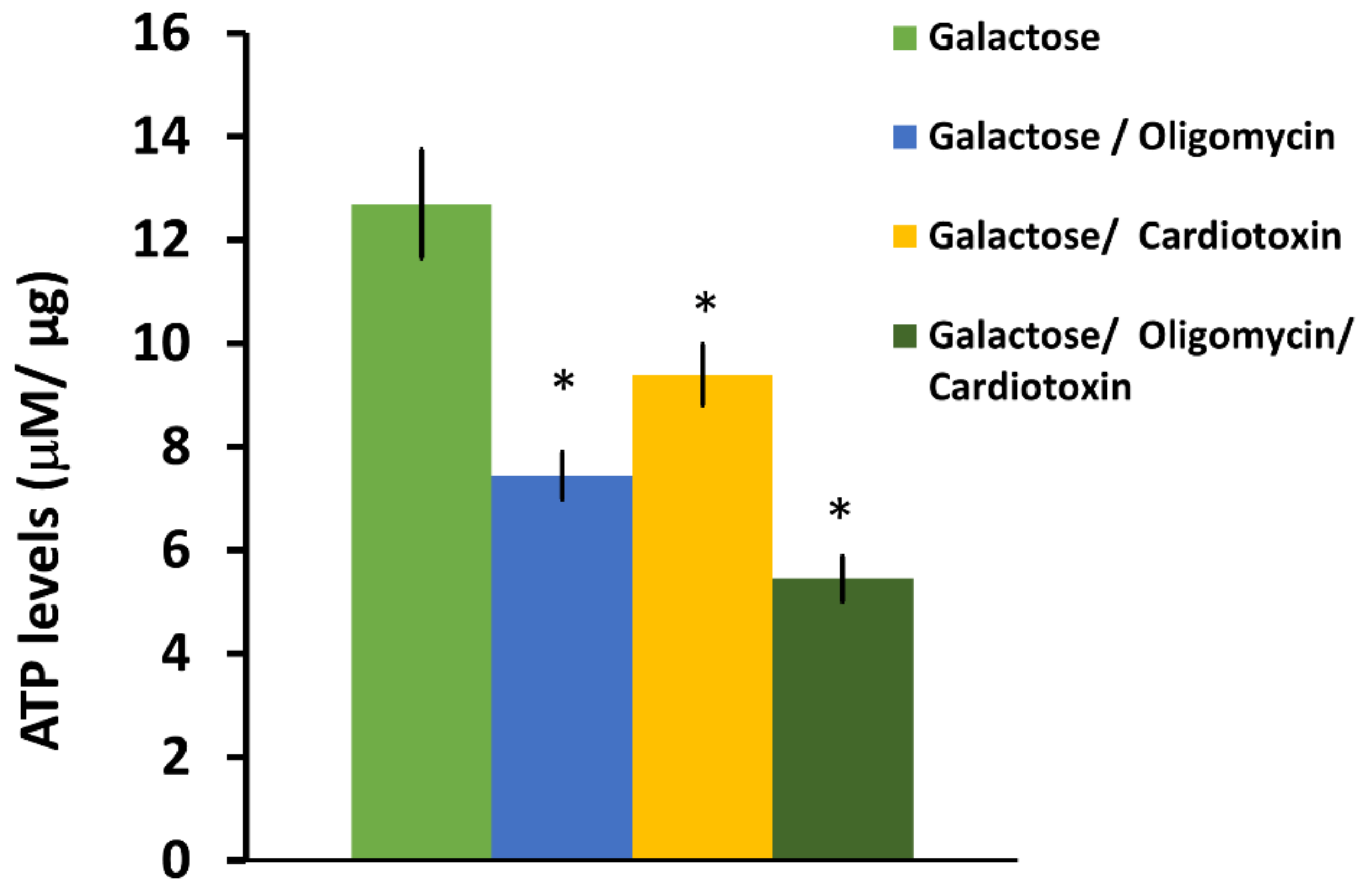
SH-SY5Y neuroblastoma cells were plated at a density of 100,000 cells per well on a black-sided, clear bottom 96-well plate. Each condition was set up in triplicate wells. After 24 h of culture, the initial complete DMEM media was removed and the cells were gently washed with DMEM containing 10 mM galactose, 2 mM sodium pyruvate, 2 mM glutamine, and 10% FBS (DMEM-Gal). The cells were then left in DMEM-Gal media for another 24 h to simulate enhanced mitochondrial respiration and thereby increase ATP derived from mitochondria [
40]. For some wells, cardiotoxin VII4 was added to designated wells with cells for 4 h before the assay analysis. Four hours following exposure with DMEM-Gal media and VII4, another set of designated wells was replaced with DMEM-Gal media containing 1 μM oligomycin as the final treatment. Oligomycin is an ATP-synthase inhibitor, and is a molecular tool used to inhibit ATP synthase-dependent ATP production and thereby calculate the amount of ATP derived from mitochondria. For each ATP assay, the final experimental conditions were: 1. DMEM-Gal only; 2. DMEM-Gal + VII4; 3. DMEM-Gal + Oligomycin; and 4. DMEM-Gal + Oligomycin + VII4. ATP standards were prepared in 1:10 serial dilutions in DMEM-Gal media to generate a standard curve (10 μM, 1 μM, 0.1 μM, 0.01 μM, and 0.001 mM). 100 μL of Cell Titer-Glo
® Reagent (Promega, Madison, WI, USA) was added to all well conditions, the ATP standards, and the plate blank wells. The plate was then wrapped in aluminum foil and incubated at room temperature (22 °C) for 2 min on an orbital shaker. Lastly, the plate was read by employing a Molecular Devices SpectraMax M3 (Molecular Devices, San Jose, CA, USA) plate reader on the luminometry setting for one reading and normalized to the protein concentration as previously described [
40,
41].
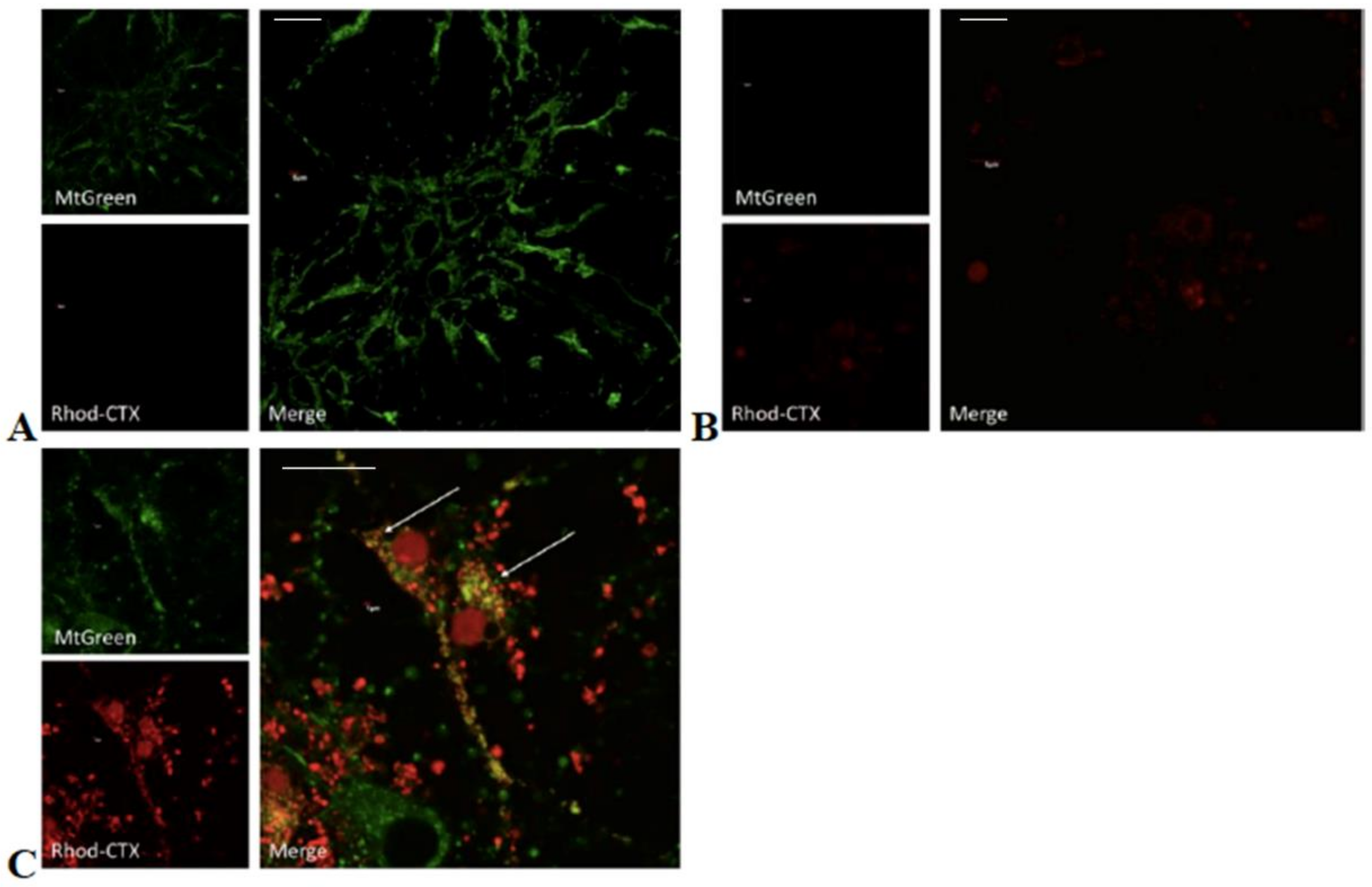
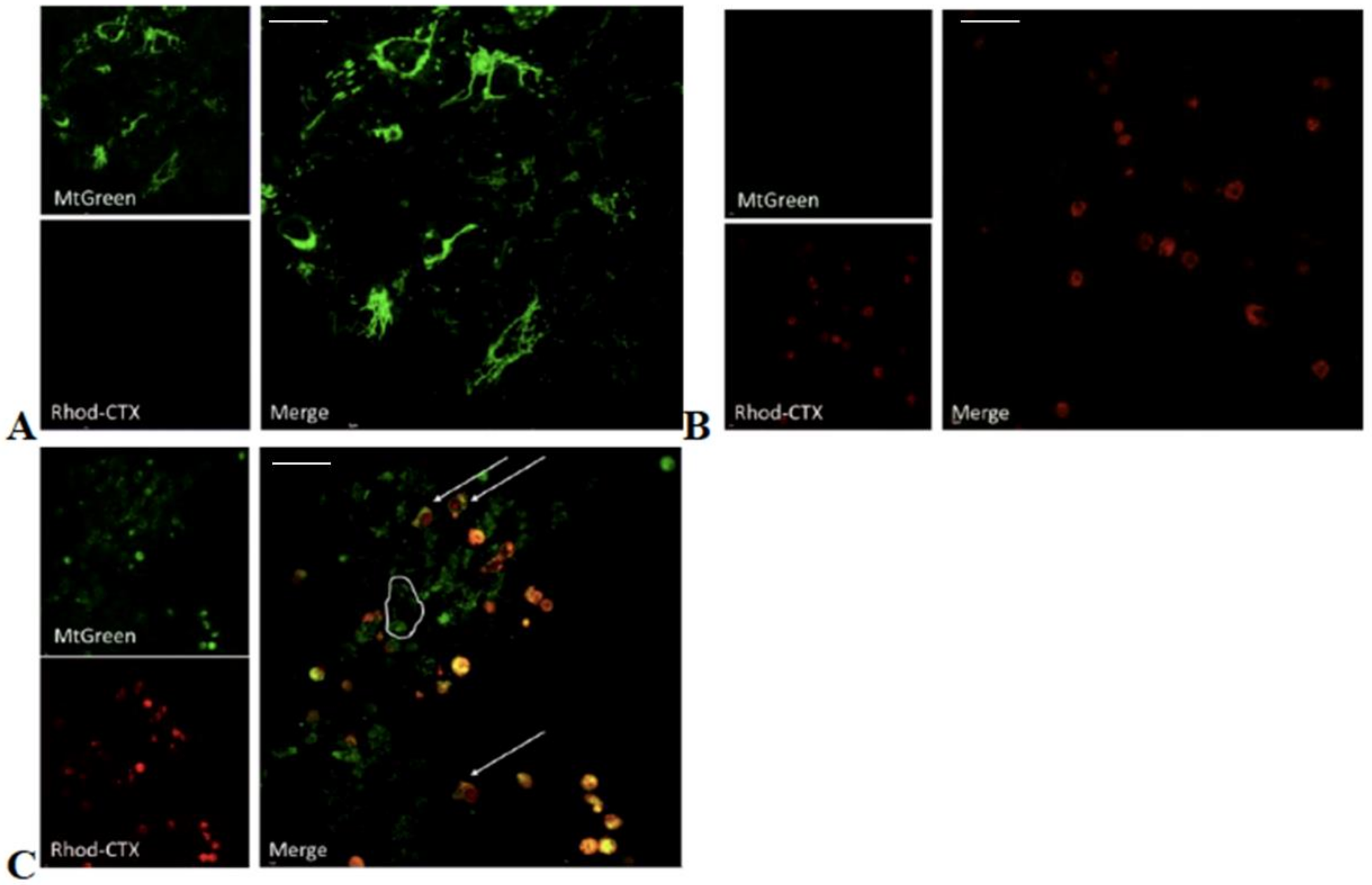
5.6. Chemical Labeling of Cardiotoxin with Rhodamine and Confocal Microscopy
In order to follow the ability of VII4 to translocate to mitochondria by employing confocal microscopy, rhodamine was conjugated to the cardiotoxin by following the manufacturer’s protocol for the NHS-Rhodamine Antibody Labeling Kit (Pierce, Rockford, IL, USA). In brief, 1 mg of solid VII4 was used for each conjugation to make a final concentration of a 295.7 μM stock solution. The chemical conjugation of NHS-Rhodamine with cardiotoxin VII4 was successfully corroborated by using the following formula:
To follow the uptake of VII4 and colocalization with mitochondria in cells by confocal microscopy, the mitochondria in primary cortical neurons and SH-SY5Y neuroblastoma were stained with a 150 nM solution of MitoTracker Green FM (Thermo Fisher Scientific, Waltham, MA, USA) in Neurobasal media or with complete DMEM media, respectively. Up to 10–15 immunofluorescent TIFF images per condition were captured by employing an Olympus Fluoview 1000 confocal microscope (Olympus) at a 60 × magnification, 4.0 digital zoom factor, sequential capture setting, and by applying the Kollman’s filter. The percentage of mitochondria (green channel) that colocalize (yellow) with Rhodamine labeled-VII4 (red channel) was analyzed by using NIH Image J version 1.44 and by using similar fluorescent imaging-based quantifications previously described for neurons [
32].
5.7. Bioenergetic Analysis: Mitochondrial Oxygen Consumption Assays
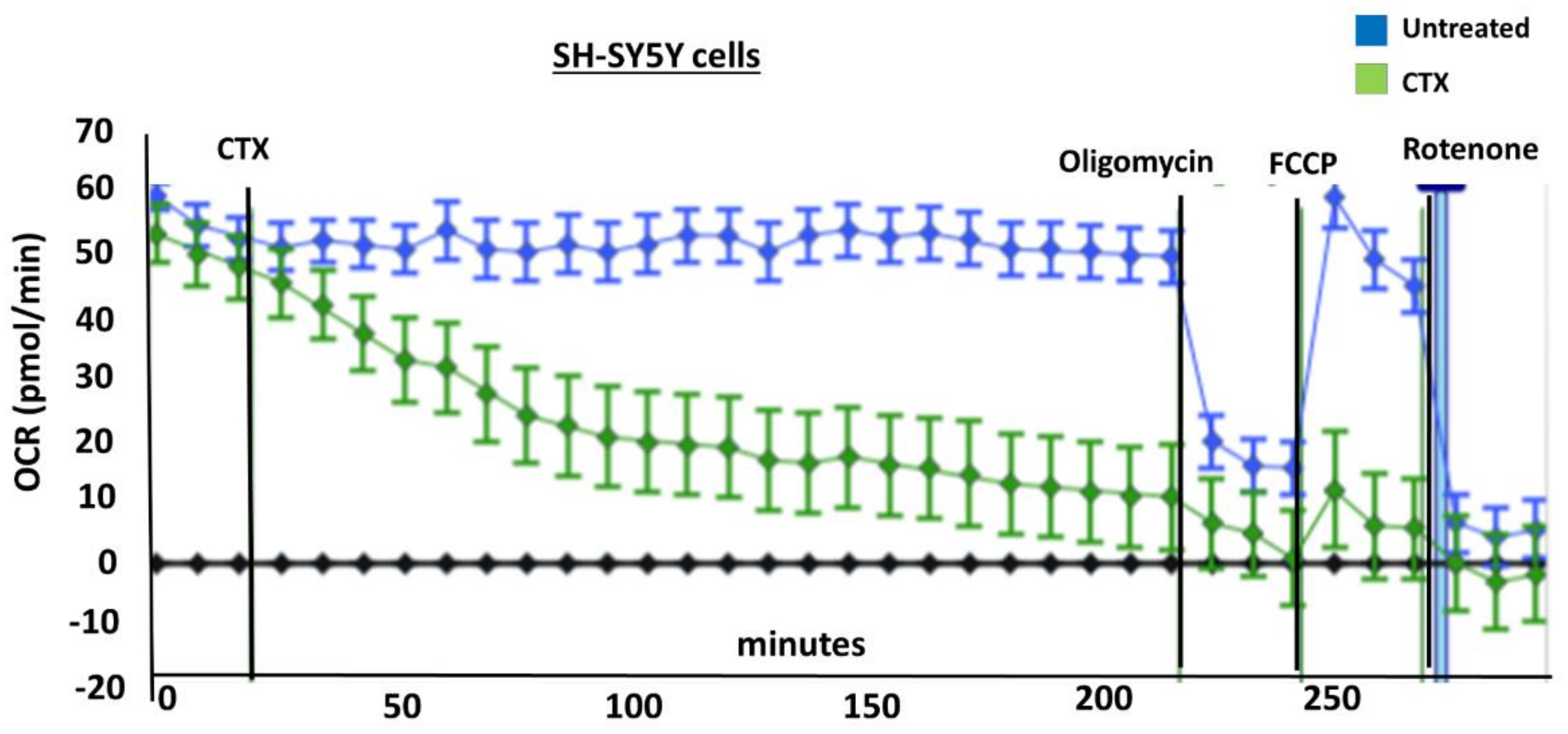
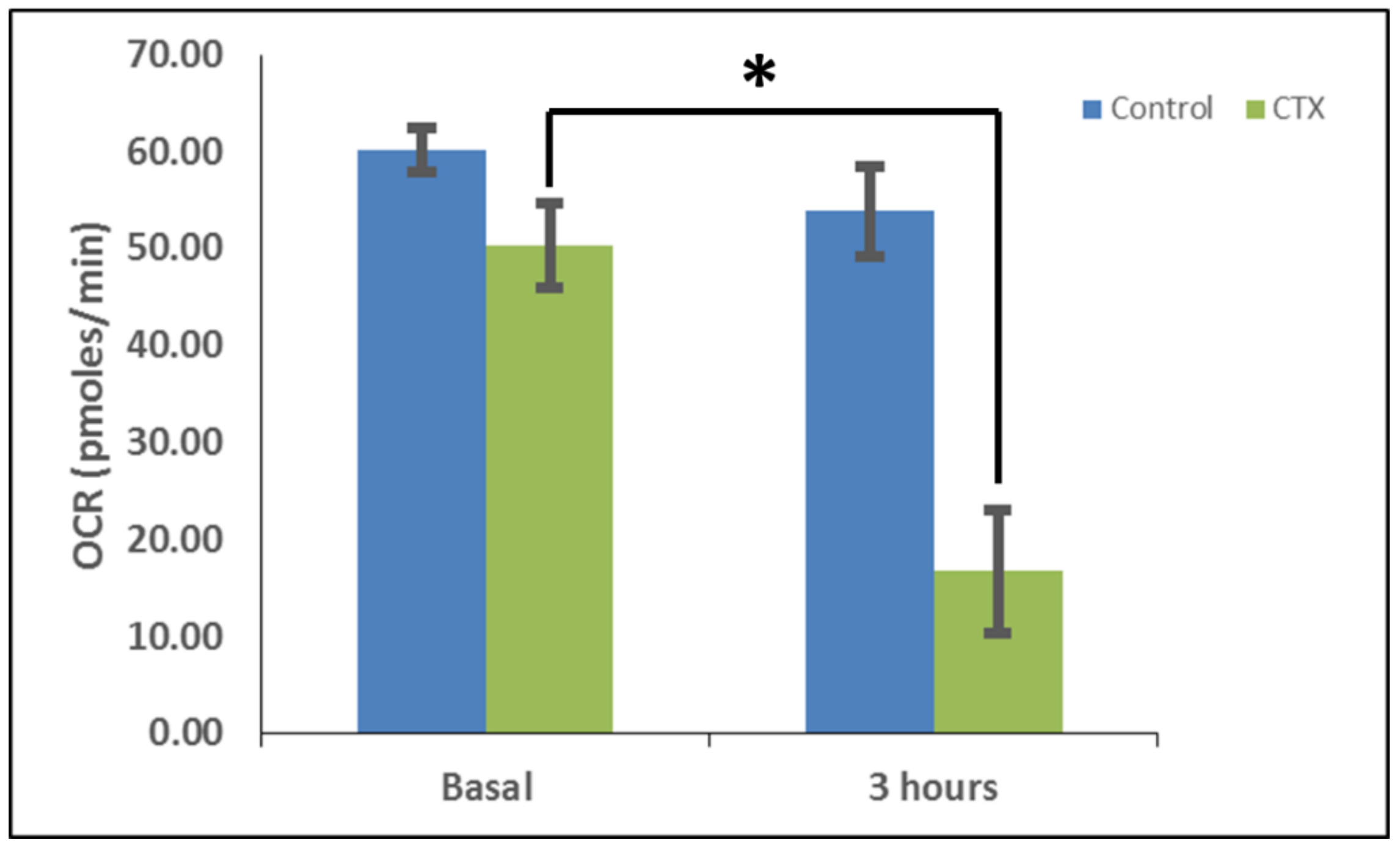
In order to measure the effects of cardiotoxin on mitochondrial respiration in neuroblastoma cells as a proxy of oxidative phosphorylation activity, a mitochondrial stress assay was performed by using an XF24e Extracellular Flux Analyzer (Seahorse Biosciences, Agilent Technologies) as previously described [
42], but with the following minor modifications. In brief, this assay was done by first plating SH-SY5Y cells at a density of 70,000 per well in complete medium (DMEM with 10% FBS) on a XF24 well cell culture microplate (Agilent Technologies). Cells were then exposed to cardiotoxin at the LD
50 (1.3 µM) in quadruplicate wells. Wells, B4 and C3, of the XF24 cell culture microplate were assigned as blanks (no cells) in order to assess background oxygen consumption rates. A calibration plate was also prepared by adding 1 mL of calibrant fluid (SeaHorse Bioscience, North Billerica, MA, USA) into the plate wells one day prior to performing the mitochondrial stress assay. On the day of the assay, the cells were washed with XF Media (XF Media (Seahorse), glucose (4.5 g/1 L), 1 mM sodium pyruvate, 1 mM glutamine). Oxygen consumption rates (OCRs) were analyzed at baseline and following the sequential addition of 1 μm oligomycin, 300 nm FCCP, and 1 μm rotenone/antimycin A to determine the ATP-linked OCRs, maximal OCRs, and mitochondrially-derived OCRs, respectively, as published previously [
42]. The final concentrations of oligomycin, FCCP, and rotenone/ antimycin-A were 1 μM each and were prepared in the XF Assay media in order to assess for ATP-dependent, maximal, and mitochondrial-specific oxygen consumption rates, respectively. The machine was programmed to expose the cells to cardiotoxin VII4 for 22 cycles (3 h) to measure the oxygen consumption rate (OCR). At the end of each assay, the OCRs and ECARs were normalized to the protein concentration (μg/mL) for their respective well.
5.8. Erythrosine Phosphorescence Quenching Studies
Unilamellar liposomes for erythrosine phosphorescence quenching assays were prepared as previously described [
21]. In brief, liposomes were incubated with 5 × 10
−6 M erythrosine for 20 h, followed by incubation with 10
−5 M ferrocene for 2 h at 4 °C. To remove unbound probes, the liposomes were chromatographed on a Sephadex G 50 column (1.8 × 50 cm) as previously published [
21]. For the liposomes used for phosphorescence assays, oxygen was enzymatically removed by glucose oxidase treatment. The analyzed liposome samples were contained in a square (10 mm) quartz chamber that was incubated at 18 °C. The erythrosine phosphorescence quenching by ferrocene in the presence or absence of defined cardiotoxin concentrations was detected with a filter specific for a wavelength greater than 700 nm following excitation with a pulsed laser apparatus (Chernogolovka, Russia). The lifetime of the excited state of erythrosine was estimated as the time dependence of the attenuation of the probe glow using semi-logarithmic coordinates. Samples for each data point were made and recorded in triplicate. The error of the erythrosine excited state lifetime was determined to be less than 5%.
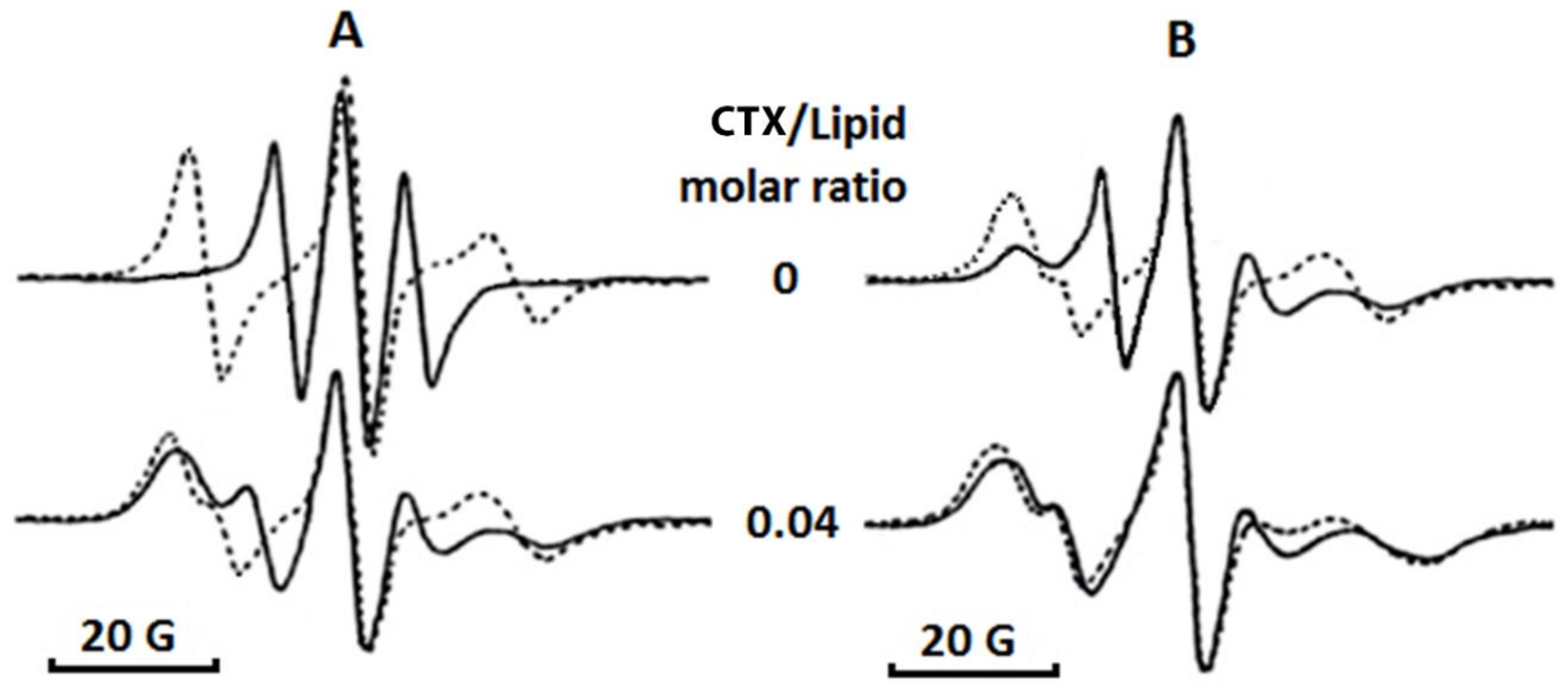
5.9. EPR Studies
For EPR studies, oriented multibilayer films were prepared as previously described [
21]. Briefly, 50 μL phospholipid suspension in buffer (10 mM Tris-HCl, pH 7.5, 0.1 M NaCl, 1 mM EDTA) was squeezed between two glass plates. The phospholipid concentrations in suspensions were prepared at 5 mM and they were made of either pure PC or PC + CL in molar ratios 9.5 and 0.5 or 8 and 2, respectively. Phospholipid suspensions contained a 10
−5 M 5-DSA spin probe and defined concentrations of cardiotoxin. EPR spectra of 5-DSA in multibilayer films were recorded with a Varian E-4 spectrometer (Palo Alto, CA USA) at modulation amplitudes not exceeding 2 × 10
−4 T and with a resonator input power not exceeding 20 mW at 18 °C. The analysis of the EPR spectra was done in terms of the
B/
C ratio [
21] and the
S parameter [
43].
B is defined as the intensity of the low-field component while
C is defined as the intensity of the central component of EPR spectra taken with respect to the magnetic field perpendicular to the bilayer normal. The formula used to calculate the
S parameter was previously published [
43]. Each sample was prepared and analyzed by EPR in three technical replicates for each of three independent samples. The means and standard deviations of these measurements were used as experimental data points. The variation between the triplicates was observed to be less than 5%.
5.10. 1H-NMR Studies
Unilamellar liposomes for 1H-NMR studies were prepared by drying phospholipids in chloroform via the continuous exposure of the sample to helium in vacuum for 1.5 h in order to form a lipid film. The film was then hydrated in a 2H2O buffer containing 10 mM Tris-HCl, pH 7.4, and 0.5 mM EDTA. The lipid suspension was sonicated for 15 min in helium media at 4 °C by employing an ultrasonic disperser, USDN-1 (St. Petersburg, Russia), at a frequency of 22 kHz. Unilamellar liposomes were centrifuged at 200 × g for 60 min to remove heavy phospholipid aggregates and then incubated in helium atmosphere for 15 h at 10 °C. Unilamellar liposomes were made of either pure PC or PC + CL in molar ratios of 9.5 and 0.5 or 8 and 2, respectively. The total concentration of phospholipids in unilamellar liposomes was 1.4 × 10−2 M. Cardiotoxin VII4 dissolved in a 2H2O-containing buffer was added directly into unilamellar liposomes at concentration 2.1 × 10−4 M. 1H-NMR spectra from unilamellar liposomes were recorded at 18 °C at an operating frequency of 200 MHz. The width of the 90° pulse was 8.7 μs, the relaxation delay was 50 μs, and the acquisition time for the free induction signal was 1 s. To split 1H NMR signals from the N+(CH3)3 groups of PC in the outer and inner leaflets of unilamellar liposomes, 10 μL of shift reagent (saturated K3Fe(CN)6 solution in 2H2O buffer) was added per 1 mL of liposomes. The measurements of the integral intensity of 1H NMR signals from the N+(CH3)3 groups of PC were done in triplicate readings. The variation between the triplicates was observed to be less than 6%.
5.11. Molecular Dynamics
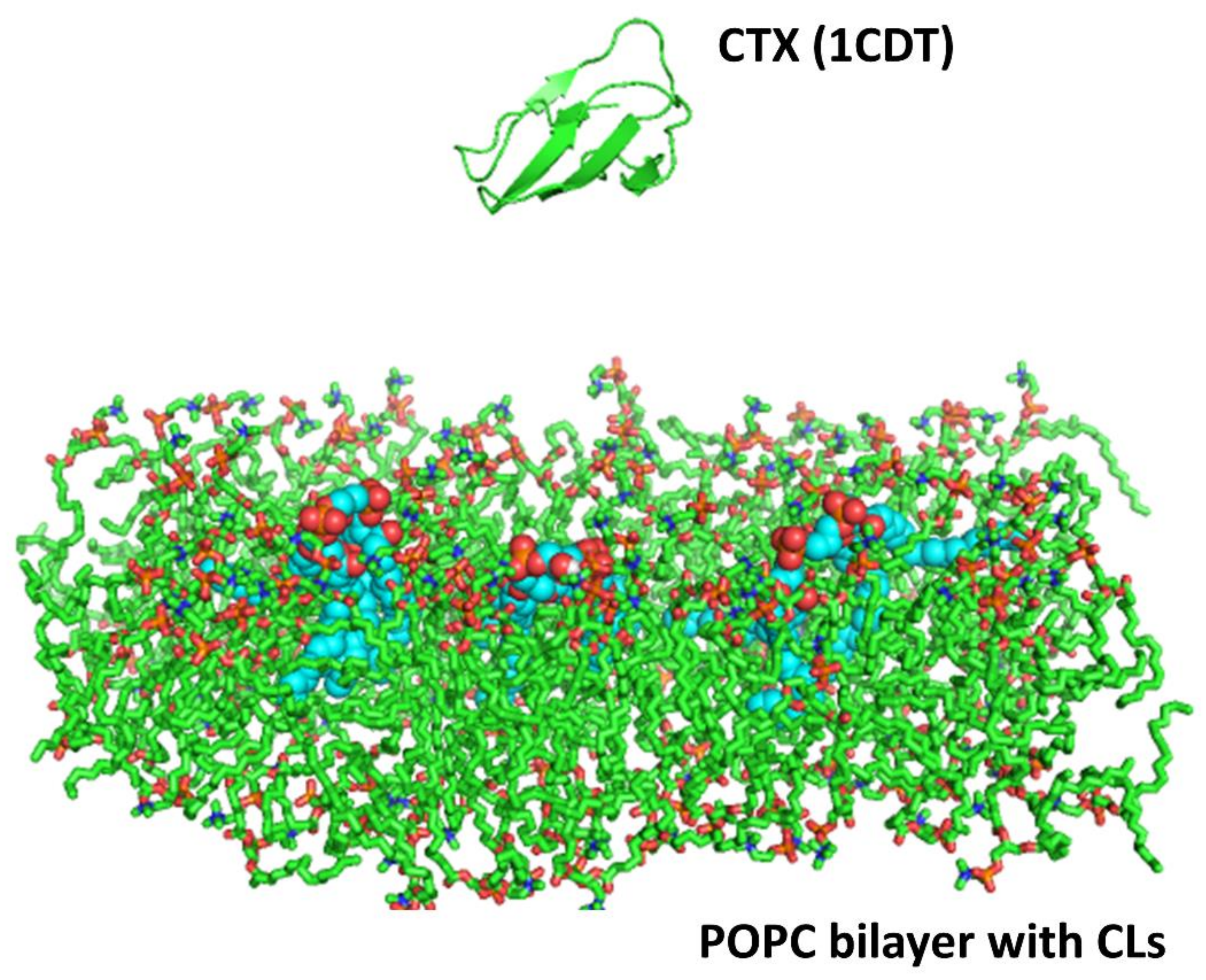
A homology model based on the solution NMR structure of cardiotoxin was built for cardiotoxin
Naja mossambica mossambica (PDB ID: 1CDT). In brief, the homology model was energy minimized due to the variability of NMR structures as previously published [
19]. The higher structural stability was determined by QMEAN scores, a scoring function for protein structures based on MD simulations and other protein models [
42,
43]. The processed protein template (cardiotoxin VII4) was then energy minimized using GROMACS version 4.5.1 with the GROMOS 53a6 force field parameters for proteins and lipids. The protein was then put into a system with a lipid bilayer with and without CL molecules and solvated.
Two simulations trajectories of 20 nanoseconds (ns) each were performed for both the cardiotoxin VII4 systems. The system consisted of the protein placed at a distance of 20 Å from a lipid bilayer made up of 128 molecules of 1-palmitoyl-2-oleoyl-phosphatidyl-choline (POPC) and three CL (CDL) molecules embedded in the bilayer, simulating the OMM. The cardiotoxin was placed about 20 Å away from the surface of the lipid bilayer, and the whole system was solvated with explicit SPC water molecules. Each simulation system was first energy minimized with 1000 steps of the steepest descent, followed by a short equilibration run of 200 picoseconds. During the equilibration, the protein backbone atoms were fixed and water and lipid molecules were allowed to relax. This was followed by 20 ns of the production run using GROMACS version 4.5.1 with GROMOS 53a6 force field parameters for the protein and lipids. The simulations were performed under constant NPT conditions, with an integration time step of 2 femtoseconds. The LINCS [
44] algorithm was used to constrain all bond lengths and the Particle Mesh Ewald method [
45] was used to compute the electrostatic interactions. The temperature was kept constant at T = 310 K by coupling to an external temperature bath with a coupling constant of 0.1 picosecond and a pressure coupling was employed to maintain a constant pressure of 1 bar. The force-field parameters for CDL was generated using PRODRG [
46]. In addition, two control simulations of 20 ns each were also performed for the cardiotoxin VII4 system with a bilayer consisting of only POPC lipids. The simulation protocols for these simulations were the same as those for toxin simulations with POPC and CDL molecules.
5.12. Molecular Docking
In order to identify potential phospholipid binding sites in cardiotoxin VII4, the phospholipid head groups of CL and PC or the complete molecules of PC and CL (head groups and alkyl chains) were docked with the solution NMR structures of cardiotoxin VII4 by using the AutoDockVina Version 4.2 program using a similar methodology as previously published [
47]. In brief, the PDB coordinates of CL were extracted from the bovine heart oxidoreductase crystal structure bound to CL (PDB ID# 1V54). The PDB coordinates of phosphatidylcholine (PC) were extracted from the structure of PITP complexed to DOPC (PDB ID# 1T27). The lipids were further edited to remove the alkyl chains using Avogadro as previously published [
48], and the overall charges were checked and energy minimized using AutoDock. The phospholipid “ligands”, which consisted of the phospholipid head groups and the complete molecules of PC and CL, contained rotatable bonds whereas the cytotoxins were kept as rigid molecules for each run. The overall molecular surface of cardiotoxin VII4 was considered for these docking studies (blind docking). A grid box was set up with the following dimensions: A center of x = 13.277; center of y = 25.025; center of z = –0.456; length of x = 36 Å; length of y = 46 Å; length of z = 36 Å. The setting for exhaustiveness was set up as 16, which gave us consistent results in at least three sets of docking for each ligand and cardiotoxin VII4 pair in this study. It is important to note that the grid box was large enough to do a “blind” docking by encompassing the entire surface of the cytotoxin VII4 and a phospholipid “ligand” for each molecular docking simulation. Following each Autodock run, the best nine docked conformations were analyzed for ionic, ion–polar, and hydrogen bond interactions between phospholipid polar head groups and charged and polar amino acids of cardiotoxin VII4 by using Python Molecular Viewer (MGL Tools, The Scripps Research Institute).
5.13. Statistics
Unless indicated otherwise, most results are expressed as mean ± S.E.M. from three independent experiments or a representative experiment. Data was analyzed by Student’s t test (two-tailed) for single comparisons. Multiple group comparisons were done by performing one-way ANOVA followed by Bonferroni-corrected Tukey’s test. P values less than 0.05 were considered statistically significant.

 and
and 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}