The Role of Toxins in the Pursuit for Novel Analgesics


Abstract
1. Introduction
2. Transient Receptor Potential Vanilloid 1 (TRPV1)
3. Transient Receptor Potential Ankyrin 1 (TRPA1)
4. Acid-Sensing Ion Channels (ASICs)
5. Voltage-Gated Sodium Channels
6. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Julius, D. TRP channels and pain. Annu. Rev. Cell Dev. Biol. 2013, 29, 355–384. [Google Scholar] [CrossRef] [PubMed]
- Julius, D.; Basbaum, A.I. Molecular mechanisms of nociception. Nature 2001, 413, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Mickle, A.D.; Shepherd, A.J.; Mohapatra, D.P.; Mickle, A.D.; Shepherd, A.J.; Mohapatra, D.P. Nociceptive TRP Channels: Sensory Detectors and Transducers in Multiple Pain Pathologies. Pharmaceuticals 2016, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Schaible, H.G. Peripheral and central mechanisms of pain generation. Handb. Exp. Pharmacol. 2007, 177, 3–28. [Google Scholar]
- Bohlen, C.J.; Julius, D. Receptor-targeting mechanisms of pain-causing toxins: How ow? Toxicon 2012, 60, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Binshtok, A.M. Mechanisms of Nociceptive Transduction and Transmission: A Machinery for Pain Sensation and Tools for Selective Analgesia. Int. Rev. Neurobiol. 2011, 97, 143–177. [Google Scholar] [PubMed]
- Rajapakse, D.; Liossi, C.; Howard, R.F. Presentation and management of chronic pain. Arch. Dis. Child. 2014, 99, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Hylands-White, N.; Duarte, R.V.; Raphael, J.H. An overview of treatment approaches for chronic pain management. Rheumatol. Int. 2017, 37, 29–42. [Google Scholar] [CrossRef] [PubMed]
- International Association for the Study of Pain. Clas-sification of chronic pain: Introduction Introduction. Pain 1986, 24, S3–S8. [Google Scholar]
- van Hecke, O.; Torrance, N.; Smith, B.H. Chronic pain epidemiology and its clinical relevance. Br. J. Anaesth. 2013, 111, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J.; Mannion, R.J. Neuropathic pain: Aetiology, symptoms, mechanisms, and management. Lancet 1999, 353, 1959–1964. [Google Scholar] [CrossRef]
- McGivern, J.G. Ziconotide: A review of its pharmacology and use in the treatment of pain. Neuropsychiatr. Dis. Treat. 2007, 3, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Prim. 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed]
- Patapoutian, A.; Tate, S.; Woolf, C.J. Transient receptor potential channels: Targeting pain at the source. Nat. Rev. Drug Discov. 2009, 8, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Laing, R.J.; Dhaka, A. ThermoTRPs and Pain. Neuroscientist 2016, 22, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Binder, A.; Baron, R. The Pharmacological Therapy of Chronic Neuropathic Pain. Dtsch. Arztebl. Int. 2016, 113, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Julius, D.; Nathans, J. Signaling by sensory receptors. Cold Spring Harb. Perspect. Biol. 2012, 4, a005991. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Szallasi, A. Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharmacol. Rev. 2014, 66, 676–814. [Google Scholar] [CrossRef] [PubMed]
- Weyer-Menkhoff, I.; Lötsch, J. Human pharmacological approaches to TRP-ion-channel-based analgesic drug development. Drug Discov. Today 2018, 23, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Trim, S.A.; Trim, C.M. Venom: The sharp end of pain therapeutics. Br. J. Pain 2013, 7, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.; Lingueglia, E. Pharmacology of acid-sensing ion channels—Physiological and therapeutical perspectives. Neuropharmacology 2015, 94, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Binshtok, A.M.; Bean, B.P.; Woolf, C.J. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature 2007, 449, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium Channels in Normal and Pathological Pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Jami, S.; Erickson, A.; Brierley, S.; Vetter, I. Pain-Causing Venom Peptides: Insights into Sensory Neuron Pharmacology. Toxins 2017, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Klint, J.K.; Senff, S.; Saez, N.J.; Seshadri, R.; Lau, H.Y.; Bende, N.S.; Undheim, E.A.B.; Rash, L.D.; Mobli, M.; King, G.F. Production of Recombinant Disulfide-Rich Venom Peptides for Structural and Functional Analysis via Expression in the Periplasm of E. coli. PLoS ONE 2013, 8, e63865. [Google Scholar] [CrossRef] [PubMed]
- Undheim, E.A.B.; Jenner, R.A.; King, G.F. Centipede venoms as a source of drug leads. Expert Opin. Drug Discov. 2016, 11, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M.; Cruz, L.J.; de Santos, V.; LeCheminant, G.W.; Griffin, D.; Zeikus, R.; McIntosh, J.M.; Galyean, R.; Varga, J.; Gray, W.R. Neuronal calcium channel antagonists. Discrimination between calcium channel subtypes using omega-conotoxin from Conus magus venom. Biochemistry 1987, 26, 2086–2090. [Google Scholar] [CrossRef] [PubMed]
- Kristipati, R.; Nádasdi, L.; Tarczy-Hornoch, K.; Lau, K.; Miljanich, G.P.; Ramachandran, J.; Bell, J.R. Characterization of the binding of omega-conopeptides to different classes of non-L-type neuronal calcium channels. Mol. Cell. Neurosci. 1994, 5, 219–228. [Google Scholar] [PubMed]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [PubMed]
- Tominaga, M.; Tominaga, T. Structure and function of TRPV1. Pflugers Arch. 2005, 451, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Szolcsányi, J.; Sándor, Z. Multisteric TRPV1 nocisensor: A target for analgesics. Trends Pharmacol. Sci. 2012, 33, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Appendino, G.; Nilius, B. Pharmacology of vanilloid transient receptor potential cation channels. Mol. Pharmacol. 2009, 75, 1262–1279. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, X.; Lespay-Rebolledo, C.; Brauchi, S. A structural view of ligand-dependent activation in thermoTRP channels. Front. Physiol. 2014, 5, 171. [Google Scholar] [CrossRef] [PubMed]
- Cao, E.; Cordero-Morales, J.F.; Liu, B.; Qin, F.; Julius, D. TRPV1 Channels Are Intrinsically Heat Sensitive and Negatively Regulated by Phosphoinositide Lipids. Neuron 2013, 77, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.D.; Alessandri-Haber, N. TRP channels: Targets for the relief of pain. Biochim. Biophys. Acta Mol. Basis Dis. 2007, 1772, 989–1003. [Google Scholar] [CrossRef] [PubMed]
- Jordt, S.E.; Tominaga, M.; Julius, D. Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc. Natl. Acad. Sci. USA 2000, 97, 8134–8139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Jara-Oseguera, A.; Chang, T.-H.; Bae, C.; Hanson, S.M.; Swartz, K.J. Heat activation is intrinsic to the pore domain of TRPV1. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Hazan, A.; Geron, M.; Steinberg, R.; Livni, L.; Matzner, H.; Priel, A. Activation of transient receptor potential vanilloid 1 by lipoxygenase metabolites depends on PKC phosphorylation. FASEB J. 2017, 31, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, V.; Thyagarajan, B.; Varnai, P.; Balla, A.; Balla, T.; Rohacs, T. Dual regulation of TRPV1 by phosphoinositides. J. Neurosci. 2007, 27, 7070–7080. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J.; Ma, Q. Nociceptors-noxious stimulus detectors. Neuron 2007, 55, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Bhave, G.; Zhu, W.; Wang, H.; Brasier, D.J.; Oxford, G.S.; Gereau IV, R.W. cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 2002, 35, 721–731. [Google Scholar] [CrossRef]
- Crandall, M.; Kwash, J.; Yu, W.; White, G. Activation of protein kinase C sensitizes human VR1 to capsaicin and to moderate decreases in pH at physiological temperatures in Xenopus oocytes. Pain 2002, 98, 109–117. [Google Scholar] [CrossRef]
- Szolcsányi, J. Forty years in capsaicin research for sensory pharmacology and physiology. Neuropeptides 2004, 38, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Fattori, V.; Hohmann, M.; Rossaneis, A.; Pinho-Ribeiro, F.; Verri, W. Capsaicin: Current Understanding of Its Mechanisms and Therapy of Pain and Other Pre-Clinical and Clinical Uses. Molecules 2016, 21, 844. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Leffler, A.; Malmberg, A.B.; Martin, W.J.; Trafton, J.; Petersen-Zeitz, K.R.; Koltzenburg, M.; Basbaum, A.I.; Julius, D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 2000, 288, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.B.; Gray, J.; Gunthorpe, M.J.; Hatcher, J.P.; Davey, P.T.; Overend, P.; Harries, M.H.; Latcham, J.; Clapham, C.; Atkinson, K.; et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 2000, 405, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Tisel, S.M.; Orestes, P.; Bhangoo, S.K.; Hoon, M.A. TRPV1-lineage neurons are required for thermal sensation. EMBO J. 2011, 30, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, V.; Rohacs, T. TRPV1: A Target for Rational Drug Design. Pharmaceuticals 2016, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Binder, A.; Wasner, G. Neuropathic pain: Diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010, 9, 807–819. [Google Scholar] [CrossRef]
- Immke, D.C.; Gavva, N.R. The TRPV1 receptor and nociception. Semin. Cell Dev. Biol. 2006, 17, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Lauria, G.; Morbin, M.; Lombardi, R.; Capobianco, R.; Camozzi, F.; Pareyson, D.; Manconi, M.; Geppetti, P. Expression of capsaicin receptor immunoreactivity in human peripheral nervous system and in painful neuropathies. J. Peripher. Nerv. Syst. 2006, 11, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Grundy, L.; Daly, D.M.; Chapple, C.; Grundy, D.; Chess-Williams, R. TRPV1 enhances the afferent response to P2X receptor activation in the mouse urinary bladder. Sci. Rep. 2018, 8, 197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Drzymalski, D.; Sun, L.; Xu, Q.; Jiao, C.; Wang, L.; Xie, S.; Qian, X.; Wu, H.; Xiao, F.; et al. Involvement of mGluR5 and TRPV1 in visceral nociception in a rat model of uterine cervical distension. Mol. Pain 2018, 14. [Google Scholar] [CrossRef] [PubMed]
- Andreev, Y.A.; Kozlov, S.A.; Korolkova, Y.V.; Dyachenko, I.A.; Bondarenko, D.A.; Skobtsov, D.I.; Murashev, A.N.; Kotova, P.D.; Rogachevskaja, O.A.; Kabanova, N.V.; et al. Polypeptide Modulators of TRPV1 Produce Analgesia without Hyperthermia. Mar. Drugs 2013, 11, 5100–5115. [Google Scholar] [CrossRef] [PubMed]
- Jara-Oseguera, A.; Simon, S.A.; Rosenbaum, T. TRPV1: On the road to pain relief. Curr. Mol. Pharmacol. 2008, 1, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Geron, M.; Hazan, A.; Priel, A. Animal toxins providing insights into TRPV1 activation mechanism. Toxins 2017, 9, 326. [Google Scholar] [CrossRef] [PubMed]
- Jordt, S.-E.; Julius, D. Molecular basis for species-specific sensitivity to “hot” chili peppers. Cell 2002, 108, 421–430. [Google Scholar] [CrossRef]
- Chou, M.Z.; Mtui, T.; Gao, Y.-D.; Kohler, M.; Middleton, R.E. Resiniferatoxin binds to the capsaicin receptor (TRPV1) near the extracellular side of the S4 transmembrane domain. Biochemistry 2004, 43, 2501–2511. [Google Scholar] [CrossRef] [PubMed]
- Raisinghani, M.; Pabbidi, R.M.; Premkumar, L.S. Activation of transient receptor potential vanilloid 1 (TRPV1) by resiniferatoxin. J. Physiol. 2005, 567, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Gavva, N.R.; Klionsky, L.; Qu, Y.; Shi, L.; Tamir, R.; Edenson, S.; Zhang, T.J.; Viswanadhan, V.N.; Toth, A.; Pearce, L.V.; et al. Molecular Determinants of Vanilloid Sensitivity in TRPV1. J. Biol. Chem. 2004, 279, 20283–20295. [Google Scholar] [CrossRef] [PubMed]
- Szallasi, A.; Blumberg, P.M. Resiniferatoxin, a phorbol-related diterpene, acts as an ultrapotent analog of capsaicin, the irritant constituent in red pepper. Neuroscience 1989, 30, 515–520. [Google Scholar] [CrossRef]
- Yang, F.; Xiao, X.; Lee, B.H.; Vu, S.; Yang, W.; Yarov-Yarovoy, V.; Zheng, J. The conformational wave in capsaicin activation of transient receptor potential vanilloid 1 ion channel. Nat. Commun. 2018, 9, 2879. [Google Scholar] [CrossRef] [PubMed]
- Elokely, K.; Velisetty, P.; Delemotte, L.; Palovcak, E.; Klein, M.L.; Rohacs, T.; Carnevale, V. Understanding TRPV1 activation by ligands: Insights from the binding modes of capsaicin and resiniferatoxin. Proc. Natl. Acad. Sci. USA 2016, 113, E137–E145. [Google Scholar] [CrossRef] [PubMed]
- Karai, L.; Brown, D.C.; Mannes, A.J.; Connelly, S.T.; Brown, J.; Gandal, M.; Wellisch, O.M.; Neubert, J.K.; Olah, Z.; Iadarola, M.J. Deletion of vanilloid receptor 1-expressing primary afferent neurons for pain control. J. Clin. Investig. 2004, 113, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.; Bates, B.D.; Keller, J.M.; Lopez, M.; Scholl, L.; Navarro, J.; Madian, N.; Haspel, G.; Nemenov, M.I.; Iadarola, M.J. Ablation of Rat TRPV1-Expressing Adelta/C-Fibers with Resiniferatoxin: Analysis of Withdrawal Behaviors, Recovery of Function and Molecular Correlates. Mol. Pain 2010, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- González-Cano, R.; Tejada, M.; Artacho-Cordón, A.; Nieto, F.; Entrena, J.; Wood, J.; Cendán, C.; González-Cano, R.; Tejada, M.Á.; Artacho-Cordón, A.; et al. Effects of Tetrodotoxin in Mouse Models of Visceral Pain. Mar. Drugs 2017, 15, 188. [Google Scholar] [CrossRef] [PubMed]
- Siemens, J.; Zhou, S.; Piskorowski, R.; Nikai, T.; Lumpkin, E. a; Basbaum, A.I.; King, D.; Julius, D. Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature 2006, 444, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Bohlen, C.J.; Priel, A.; Zhou, S.; King, D.; Siemens, J.; Julius, D. A bivalent tarantula toxin activates the capsaicin receptor, TRPV1, by targeting the outer pore domain. Cell 2010, 141, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Cao, E.; Julius, D.; Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 2016, 534, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Bae, C.; Anselmi, C.; Kalia, J.; Jara-Oseguera, A.; Schwieters, C.D.; Krepkiy, D.; Won Lee, C.; Kim, E.-H.; Kim, J. Il; Faraldo-Gómez, J.D.; et al. Structural insights into the mechanism of activation of the TRPV1 channel by a membrane-bound tarantula toxin. Elife 2016, 5, e11273. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Cao, E.; Liao, M.; Cheng, Y.; Julius, D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 2013, 504, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Geron, M.; Kumar, R.; Zhou, W.; Faraldo-Gómez, J.D.; Vásquez, V.; Priel, A. TRPV1 pore turret dictates distinct DkTx and capsaicin gating. Proc. Natl. Acad. Sci. USA 2018, 115, E11837–E11846. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yang, F.; Wei, N.; Hong, J.; Li, B.; Luo, L.; Rong, M.; Yarov-Yarovoy, V.; Zheng, J.; Wang, K.; et al. A pain-inducing centipede toxin targets the heat activation machinery of nociceptor TRPV1. Nat. Commun. 2015, 6, 8297. [Google Scholar] [CrossRef] [PubMed]
- Hakim, M.A.; Jiang, W.; Luo, L.; Li, B.; Yang, S.; Song, Y.; Lai, R. Scorpion Toxin, BmP01, Induces Pain by Targeting TRPV1 Channel. Toxins 2015, 7, 3671–3687. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yang, F.; Zhang, B.; Lee, B.H.; Li, B.; Luo, L.; Zheng, J.; Lai, R. A bimodal activation mechanism underlies scorpion toxin-induced pain. Sci. Adv. 2017, 3, e1700810. [Google Scholar] [CrossRef] [PubMed]
- Brederson, J.D.; Kym, P.R.; Szallasi, A. Targeting TRP channels for pain relief. Eur. J. Pharmacol. 2013, 716, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Bley, K. Topical capsaicin for pain management: Therapeutic potential and mechanisms of action of the new high-concentration capsaicin 8 patch. Br. J. Anaesth. 2011, 107, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, T.; Gordon-Shaag, A.; Munari, M.; Gordon, S.E. Ca2+/Calmodulin Modulates TRPV1 Activation by Capsaicin. J. Gen. Physiol. 2004, 123, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Koplas, P.A.; Rosenberg, R.L.; Oxford, G.S. The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. J. Neurosci. 1997, 17, 3525–3537. [Google Scholar] [CrossRef] [PubMed]
- Simone, D.A.; Nolano, M.; Johnson, T.; Wendelschafer-Crabb, G.; Kennedy, W.R. Intradermal injection of capsaicin in humans produces degeneration and subsequent reinnervation of epidermal nerve fibers: Correlation with sensory function. J. Neurosci. 1998, 18, 8947–8959. [Google Scholar] [CrossRef] [PubMed]
- Gerner, P.; Binshtok, A.M.; Wang, C.-F.; Hevelone, N.D.; Bean, B.P.; Woolf, C.J.; Wang, G.K. Capsaicin combined with local anesthetics preferentially prolongs sensory/nociceptive block in rat sciatic nerve. Anesthesiology 2008, 109, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, C.J.; Johnston, S.A.; Reynolds, L.R.; Al-Nadaf, S.; Budsberg, S.C. Efficacy of ABT-116, an antagonist of transient receptor potential vanilloid type 1, in providing analgesia for dogs with chemically induced synovitis. Am. J. Vet. Res. 2012, 73, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.Y.; Gavva, N.R. Therapeutic potential of vanilloid receptor TRPV1 agonists and antagonists as analgesics: Recent advances and setbacks. Brain Res. Rev. 2009, 60, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Uchytilova, E.; Spicarova, D.; Palecek, J. TRPV1 antagonist attenuates postoperative hypersensitivity by central and peripheral mechanisms. Mol. Pain 2014, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kort, M.E.; Kym, P.R. TRPV1 Antagonists: Clinical Setbacks and Prospects for Future Development. Prog. Med. Chem. 2012, 51, 57–70. [Google Scholar] [PubMed]
- Gavva, N.R.; Treanor, J.J.S.; Garami, A.; Fang, L.; Surapaneni, S.; Akrami, A.; Alvarez, F.; Bak, A.; Darling, M.; Gore, A.; et al. Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain 2008, 136, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Andreev, Y.A.; Kozlov, S.A.; Koshelev, S.G.; Ivanova, E.A.; Monastyrnaya, M.M.; Kozlovskaya, E.P.; Grishin, E. V Analgesic compound from sea anemone Heteractis crispa is the first polypeptide inhibitor of vanilloid receptor 1 (TRPV1). J. Biol. Chem. 2008, 283, 23914–23921. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, M.V.; Dorofeeva, N.A.; Komarova, M.S.; Korolkova, Y.V.; Andreev, Y.A.; Mosharova, I.V.; Grishin, E.V.; Tikhonov, D.B.; Kozlov, S.A. TRPV1 activation power can switch an action mode for its polypeptide ligands. PLoS ONE 2017, 12, e0177077. [Google Scholar] [CrossRef] [PubMed]
- Kitaguchi, T.; Swartz, K.J. An inhibitor of TRPV1 channels isolated from funnel Web spider venom. Biochemistry 2005, 44, 15544–15549. [Google Scholar] [CrossRef] [PubMed]
- Yaksh, T.L.; Farb, D.H.; Leeman, S.E.; Jessell, T.M. Intrathecal capsaicin depletes substance P in the rat spinal cord and produces prolonged thermal analgesia. Science 1979, 206, 481–483. [Google Scholar] [CrossRef] [PubMed]
- McGaraughty, S.; Chu, K.L.; Bitner, R.S.; Martino, B.; Kouhen, R. El; Han, P.; Nikkel, A.L.; Burgard, E.C.; Faltynek, C.R.; Jarvis, M.F. Capsaicin Infused Into the PAG Affects Rat Tail Flick Responses to Noxious Heat and Alters Neuronal Firing in the RVM. J. Neurophysiol. 2003, 90, 2702–2710. [Google Scholar] [CrossRef] [PubMed]
- Robbins, W.R.; Staats, P.S.; Levine, J.; Fields, H.L.; Allen, R.W.; Campbell, J.N.; Pappagallo, M. Treatment of intractable pain with topical large-dose capsaicin: Preliminary report. Anesth. Analg. 1998, 86, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Ellison, N.; Loprinzi, C.L.; Kugler, J.; Hatfield, A.K.; Miser, A.; Sloan, J.A.; Wender, D.B.; Rowland, K.M.; Molina, R.; Cascino, T.L.; et al. Phase III placebo-controlled trial of capsaicin cream in the management of surgical neuropathic pain in cancer patients. J. Clin. Oncol. 1997, 15, 2974–2980. [Google Scholar] [CrossRef] [PubMed]
- Zis, P.; Apsokardos, A.; Isaia, C.; Sykioti, P.; Vadalouca, A. Posttraumatic and postsurgical neuropathic pain responsive to treatment with capsaicin 8% topical patch. Pain Phys. 2014, 17, E213–E218. [Google Scholar]
- Kiani, J.; Sajedi, F.; Nasrollahi, S.A.; Esna-Ashari, F. A randomized clinical trial of efficacy and safety of the topical clonidine and capsaicin in the treatment of painful diabetic neuropathy. J. Res. Med. Sci. 2015, 20, 359–363. [Google Scholar] [PubMed]
- Blair, H.A. Capsaicin 8% Dermal Patch: A Review in Peripheral Neuropathic Pain. Drugs 2018, 78, 1489–1500. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.D.; Saeed, M.; Do, L.; Braz, J.; Basbaum, A.I.; Iadarola, M.J.; Wilson, D.M.; Dillon, W.P. CT-guided injection of a TRPV1 agonist around dorsal root ganglia decreases pain transmission in swine. Sci. Transl. Med. 2015, 7, 305ra145. [Google Scholar] [CrossRef] [PubMed]
- Iadarola, M.J.; Mannes, A.J. The vanilloid agonist resiniferatoxin for interventional-based pain control. Curr. Top. Med. Chem. 2011, 11, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Hoon, M.A. Ablation of TrpV1 neurons reveals their selective role in thermal pain sensation. Mol. Cell. Neurosci. 2010, 43, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Jeffry, J.A.; Yu, S.-Q.; Sikand, P.; Parihar, A.; Evans, M.S.; Premkumar, L.S. Selective Targeting of TRPV1 Expressing Sensory Nerve Terminals in the Spinal Cord for Long Lasting Analgesia. PLoS ONE 2009, 4, e7021. [Google Scholar] [CrossRef] [PubMed]
- Brown, D. Resiniferatoxin: The Evolution of the “Molecular Scalpel” for Chronic Pain Relief. Pharmaceuticals 2016, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Meents, J.E.; Ciotu, C.; Fischer, M.J.M.M. TRPA1—A molecular view. J. Neurophysiol. 2019, 121, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Zygmunt, P.M.; Högestätt, E.D. TRPA1. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2014; pp. 583–630. [Google Scholar]
- Gaudet, R. A primer on ankyrin repeat function in TRP channels and beyond. Mol. Biosyst. 2008, 4, 372. [Google Scholar] [CrossRef] [PubMed]
- Zayats, V.; Samad, A.; Minofar, B.; Roelofs, K.E.; Stockner, T.; Ettrich, R. Regulation of the transient receptor potential channel TRPA1 by its N-terminal ankyrin repeat domain. J. Mol. Model. 2013, 19, 4689–4700. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Morales, J.F.; Gracheva, E.O.; Julius, D. Cytoplasmic ankyrin repeats of transient receptor potential A1 (TRPA1) dictate sensitivity to thermal and chemical stimuli. Proc. Natl. Acad. Sci. USA 2011, 108, E1184–E1191. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, E.; Fernandes, M.; Keeble, J. The functions of TRPA1 and TRPV1: Moving away from sensory nerves. Br. J. Pharmacol. 2012, 166, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Fukuoka, T.; Obata, K.; Yamanaka, H.; Dai, Y.; Tokunaga, A.; Noguchi, K. Distinct expression of TRPM8, TRPA1, and TRPV1 mRNAs in rat primary afferent neurons with aδ/c-fibers and colocalization with trk receptors. J. Comp. Neurol. 2005, 493, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Mizuno, Y.; Kozai, D.; Yamamoto, S.; Kiyonaka, S.; Shibata, T.; Uchida, K.; Mori, Y. Molecular characterization of TRPA1 channel activation by cysteine-reactive inflammatory mediators. Channels 2008, 2, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, L.J.; Dubin, A.E.; Evans, M.J.; Marr, F.; Schultz, P.G.; Cravatt, B.F.; Patapoutian, A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 2007, 445, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A.; Kiselar, J.; Pumroy, R.A.; Han, S.; Moiseenkova-Bell, V.Y. Structural insights into the molecular mechanism of mouse TRPA1 activation and inhibition. J. Gen. Physiol. 2018, 150, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Karashima, Y.; Damann, N.; Prenen, J.; Talavera, K.; Segal, A.; Voets, T.; Nilius, B. Bimodal Action of Menthol on the Transient Receptor Potential Channel TRPA1. J. Neurosci. 2007, 27, 9874–9884. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Delling, M.; Jun, J.C.; Clapham, D.E. Oregano, thyme and clove-derived flavors and skin sensitizers activate specific TRP channels. Nat. Neurosci. 2006, 9, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.P.; Buber, M.T.; Yang, Q.; Cerne, R.; Cortés, R.Y.; Sprous, D.G.; Bryant, R.W. Thymol and related alkyl phenols activate the hTRPA1 channel. Br. J. Pharmacol. 2008, 153, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Jordt, S.-E.; Bautista, D.M.; Chuang, H.; McKemy, D.D.; Zygmunt, P.M.; Högestätt, E.D.; Meng, I.D.; Julius, D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 2004, 427, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.; Hwang, S.W. Polymodal Ligand Sensitivity of TRPA1 and Its Modes of Interactions: Figure 1. J. Gen. Physiol. 2009, 133, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Bautista, D.M.; Pellegrino, M.; Tsunozaki, M. TRPA1: A Gatekeeper for Inflammation. Annu. Rev. Physiol. 2013, 75, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Trevisani, M.; Siemens, J.; Materazzi, S.; Bautista, D.M.; Nassini, R.; Campi, B.; Imamachi, N.; Andre, E.; Patacchini, R.; Cottrell, G.S.; et al. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc. Natl. Acad. Sci. USA 2007, 104, 13519–13524. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.A.; Gentry, C.; Moss, S.; Bevan, S. Transient Receptor Potential A1 Is a Sensory Receptor for Multiple Products of Oxidative Stress. J. Neurosci. 2008, 28, 2485–2494. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Tai, Y.; Caceres, A.I.; Achanta, S.; Balakrishna, S.; Shao, X.; Fang, J.; Jordt, S.-E. Oxidized Phospholipid OxPAPC Activates TRPA1 and Contributes to Chronic Inflammatory Pain in Mice. PLoS ONE 2016, 11, e0165200. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, A.; Chapman, H.; Jalava, N.; Korjamo, T.; Saarnilehto, M.; Lindstedt, K.; Pertovaara, A. TRPA1: A Transducer and Amplifier of Pain and Inflammation. Basic Clin. Pharmacol. Toxicol. 2014, 114, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Koivisto, A.; Saarnilehto, M.; Chapman, H.; Kuokkanen, K.; Hao, B.; Huang, J.-L.; Wang, Y.-X.; Pertovaara, A. Spinal transient receptor potential ankyrin 1 channel contributes to central pain hypersensitivity in various pathophysiological conditions in the rat. Pain 2011, 152, 582–591. [Google Scholar] [CrossRef] [PubMed]
- McNamara, C.R.; Mandel-Brehm, J.; Bautista, D.M.; Siemens, J.; Deranian, K.L.; Zhao, M.; Hayward, N.J.; Chong, J.A.; Julius, D.; Moran, M.M.; et al. TRPA1 mediates formalin-induced pain. Proc. Natl. Acad. Sci. USA 2007, 104, 13525–13530. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.S.; La, J.-H.; Scheff, N.N.; Davis, B.M.; Albers, K.M.; Gebhart, G.F. TRPV1 and TRPA1 Antagonists Prevent the Transition of Acute to Chronic Inflammation and Pain in Chronic Pancreatitis. J. Neurosci. 2013, 33, 5603–5611. [Google Scholar] [CrossRef] [PubMed]
- Blackshaw, L.A. Transient receptor potential cation channels in visceral sensory pathways. Br. J. Pharmacol. 2014, 171, 2528–2536. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, T.K.; Altier, C. The role of TRPA1 in visceral inflammation and pain. Channels 2011, 5, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Kojima, R.; Nozawa, K.; Doihara, H.; Keto, Y.; Kaku, H.; Yokoyama, T.; Itou, H. Effects of novel TRPA1 receptor agonist ASP7663 in models of drug-induced constipation and visceral pain. Eur. J. Pharmacol. 2014, 723, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.M.S.; Lima-Júnior, R.C.P.; Bem, A.X.C.; Teixeira, C.G.; Grassi, L.S.; Medeiros, R.P.; Marques-Neto, R.D.; Callado, R.B.; Aragão, K.S.; Wong, D.V.T.; et al. Blockade of TRPA1 with HC-030031 attenuates visceral nociception by a mechanism independent of inflammatory resident cells, nitric oxide and the opioid system. Eur. J. Pain 2013, 17, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Cattaruzza, F.; Spreadbury, I.; Miranda-Morales, M.; Grady, E.F.; Vanner, S.; Bunnett, N.W. Transient receptor potential ankyrin-1 has a major role in mediating visceral pain in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G81–G91. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guo, C.-H.; Chowdhury, M.A.; Dai, T.-L.; Han, W. TRPA1 in the spinal dorsal horn is involved in post-inflammatory visceral hypersensitivity: In vivo study using TNBS-treated rat model. J. Pain Res. 2016, 9, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Brierley, S.M.; Hughes, P.A.; Page, A.J.; Kwan, K.Y.; Martin, C.M.; O’Donnell, T.A.; Cooper, N.J.; Harrington, A.M.; Adam, B.; Liebregts, T.; et al. The ion channel TRPA1 is required for normal mechanosensation and is modulated by algesic stimuli. Gastroenterology 2009, 137, 2084–2095. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Y.; Zuo, X.; Zhen, Y.; Yu, Y.; Gao, L. Transient receptor potential ankyrin-1 participates in visceral hyperalgesia following experimental colitis. Neurosci. Lett. 2008, 440, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Oshima, T.; Obata, K.; Sakurai, J.; Knowles, C.H.; Matsumoto, T.; Noguchi, K.; Miwa, H. Role of transient receptor potential A1 in gastric nociception. Digestion 2010, 82, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Andrade, E.L.; Meotti, F.C.; Calixto, J.B. TRPA1 antagonists as potential analgesic drugs. Pharmacol. Ther. 2012, 133, 189–204. [Google Scholar] [CrossRef] [PubMed]
- Eid, S.R.; Crown, E.D.; Moore, E.L.; Liang, H.A.; Choong, K.-C.; Dima, S.; Henze, D.A.; Kane, S.A.; Urban, M.O. HC-030031, a TRPA1 Selective Antagonist, Attenuates Inflammatory- and Neuropathy-Induced Mechanical Hypersensitivity. Mol. Pain 2008, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Preti, D.; Saponaro, G.; Szallasi, A. Transient receptor potential ankyrin 1 (TRPA1) antagonists. Pharm. Pat. Anal. 2015, 4, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Meotti, F.C.; Forner, S.; Lima-Garcia, J.F.; Viana, A.F.; Calixto, J.B. Antagonism of the transient receptor potential ankyrin 1 (TRPA1) attenuates hyperalgesia and urinary bladder overactivity in cyclophosphamide-induced haemorrhagic cystitis. Chem. Biol. Interact. 2013, 203, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, A.; Jalava, N.; Bratty, R.; Pertovaara, A. TRPA1 Antagonists for Pain Relief. Pharmaceuticals 2018, 11, 117. [Google Scholar] [CrossRef] [PubMed]
- Kistner, K.; Siklosi, N.; Babes, A.; Khalil, M.; Selescu, T.; Zimmermann, K.; Wirtz, S.; Becker, C.; Neurath, M.F.; Reeh, P.W.; et al. Systemic desensitization through TRPA1 channels by capsazepine and mustard oil—A novel strategy against inflammation and pain. Sci. Rep. 2016, 6, 28621. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.A.; Gentry, C.; Alenmyr, L.; Killander, D.; Lewis, S.E.; Andersson, A.; Bucher, B.; Galzi, J.-L.; Sterner, O.; Bevan, S.; et al. TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Δ9-tetrahydrocannabiorcol. Nat. Commun. 2011, 2, 551. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Hämäläinen, M.M.; Saarnilehto, M.; Koivisto, A.; Pertovaara, A. Attenuation of Mechanical Hypersensitivity by an Antagonist of the TRPA1 Ion Channel in Diabetic Animals. Anesthesiology 2009, 111, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Logashina, Y.A.; Mosharova, I.V.; Korolkova, Y.V.; Shelukhina, I.V.; Dyachenko, I.A.; Palikov, V.A.; Palikova, Y.A.; Murashev, A.N.; Kozlov, S.A.; Stensvåg, K.; et al. Peptide from Sea Anemone Metridium senile Affects Transient Receptor Potential Ankyrin-repeat 1 (TRPA1) Function and Produces Analgesic Effect. J. Biol. Chem. 2017, 292, 2992–3004. [Google Scholar] [CrossRef] [PubMed]
- Priest, B.T.; Blumenthal, K.M.; Smith, J.J.; Warren, V.A.; Smith, M.M. ProTx-I and ProTx-II: Gating modifiers of voltage-gated sodium channels. Toxicon 2007, 49, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Middleton, R.E.; Warren, V.A.; Kraus, R.L.; Hwang, J.C.; Liu, C.J.; Dai, G.; Brochu, R.M.; Kohler, M.G.; Gao, Y.-D.; Garsky, V.M.; et al. Two Tarantula Peptides Inhibit Activation of Multiple Sodium Channels. Biochemistry 2002, 41, 14734–14747. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.; Liu, B.; Cao, G.; Lipchik, A.M.M.; Perez, M.; Dekan, Z.; Mobli, M.; Daly, N.L.L.; Alewood, P.F.F.; Parker, L.L.L.; et al. A tarantula-venom peptide antagonizes the TRPA1 nociceptor ion channel by binding to the S1-S4 gating domain. Curr. Biol. 2014, 24, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Tonello, R.; Fusi, C.; Materazzi, S.; Marone, I.M.; De Logu, F.; Benemei, S.; Gonçalves, M.C.; Coppi, E.; Castro-Junior, C.J.; Gomez, M.V.; et al. The peptide Phα1β, from spider venom, acts as a TRPA1 channel antagonist with antinociceptive effects in mice. Br. J. Pharmacol. 2017, 174, 57–69. [Google Scholar] [CrossRef] [PubMed]
- de Souza, A.H.; Lima, M.C.; Drewes, C.C.; da Silva, J.F.; Torres, K.C.L.; Pereira, E.M.R.; de Castro, C.J.; Vieira, L.B.; Cordeiro, M.N.; Richardson, M.; et al. Antiallodynic effect and side effects of Phα1β, a neurotoxin from the spider Phoneutria nigriventer: Comparison with ω-conotoxin MVIIA and morphine. Toxicon 2011, 58, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.H.; Ferreira, J.; do Nascimento Cordeiro, M.; Vieira, L.B.; De Castro, C.J.; Trevisan, G.; Reis, H.; Souza, I.A.; Richardson, M.; Prado, M.A.M.; et al. Analgesic effect in rodents of native and recombinant Phα1β toxin, a high-voltage-activated calcium channel blocker isolated from armed spider venom. Pain 2008, 140, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Rigo, F.K.; Trevisan, G.; Rosa, F.; Dalmolin, G.D.; Otuki, M.F.; Cueto, A.P.; de Castro Junior, C.J.; Romano-Silva, M.A.; do Nascimento Cordeiro, M.; Richardson, M.; et al. Spider peptide Phα1β induces analgesic effect in a model of cancer pain. Cancer Sci. 2013, 104, 1226–1230. [Google Scholar] [CrossRef] [PubMed]
- Rigo, F.K.; Dalmolin, G.D.; Trevisan, G.; Tonello, R.; Silva, M.A.; Rossato, M.F.; Klafke, J.Z.; do Nascimento Cordeiro, M.; Castro Junior, C.J.; Montijo, D.; et al. Effect of ω-conotoxin MVIIA and Phα1β on paclitaxel-induced acute and chronic pain. Pharmacol. Biochem. Behav. 2013, 114–115, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Rigo, F.K.; Trevisan, G.; De Prá, S.D.-T.; Cordeiro, M.N.; Borges, M.H.; Silva, J.F.; Santa Cecilia, F.V.; de Souza, A.H.; de Oliveira Adamante, G.; Milioli, A.M.; et al. The spider toxin Phα1β recombinant possesses strong analgesic activity. Toxicon 2017, 133, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.B.M.; Sperotto, N.D.M.; Andrade, E.L.; Pereira, T.C.B.; Leite, C.E.; de Souza, A.H.; Bogo, M.R.; Morrone, F.B.; Gomez, M.V.; Campos, M.M. Spinal blockage of P/Q- or N-type voltage-gated calcium channels modulates functional and symptomatic changes related to haemorrhagic cystitis in mice. Br. J. Pharmacol. 2015, 172, 924–939. [Google Scholar] [CrossRef] [PubMed]
- Zurborg, S.; Yurgionas, B.; Jira, J.A.; Caspani, O.; Heppenstall, P.A. Direct activation of the ion channel TRPA1 by Ca2+. Nat. Neurosci. 2007, 10, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Chang, R.B.; Waters, H.N.; McKemy, D.D.; Liman, E.R. The Nociceptor Ion Channel TRPA1 Is Potentiated and Inactivated by Permeating Calcium Ions. J. Biol. Chem. 2008, 283, 32691–32703. [Google Scholar] [CrossRef] [PubMed]
- Logashina, Y.A.; Solstad, R.G.; Mineev, K.S.; Korolkova, Y.V.; Mosharova, I.V.; Dyachenko, I.A.; Palikov, V.A.; Palikova, Y.A.; Murashev, A.N.; Arseniev, A.S.; et al. New Disulfide-Stabilized Fold Provides Sea Anemone Peptide to Exhibit Both Antimicrobial and TRPA1 Potentiating Properties. Toxins 2017, 9, 154. [Google Scholar] [CrossRef] [PubMed]
- Bressan, E.; Touska, F.; Vetter, I.; Kistner, K.; Kichko, T.I.; Teixeira, N.B.; Picolo, G.; Cury, Y.; Lewis, R.J.; Fischer, M.J.M.; et al. Crotalphine desensitizes TRPA1 ion channels to alleviate inflammatory hyperalgesia. Pain 2016, 157, 2504–2516. [Google Scholar] [CrossRef] [PubMed]
- Machado, F.C.; Zambelli, V.O.; Fernandes, A.C.O.; Heimann, A.S.; Cury, Y.; Picolo, G. Peripheral interactions between cannabinoid and opioid systems contribute to the antinociceptive effect of crotalphine. Br. J. Pharmacol. 2014, 171, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, V.P.; Konno, K.; Chacur, M.; Sampaio, S.C.; Picolo, G.; Brigatte, P.; Zambelli, V.O.; Cury, Y. Crotalphine induces potent antinociception in neuropathic pain by acting at peripheral opioid receptors. Eur. J. Pharmacol. 2008, 594, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Konno, K.; Picolo, G.; Gutierrez, V.P.; Brigatte, P.; Zambelli, V.O.; Camargo, A.C.M.; Cury, Y. Crotalphine, a novel potent analgesic peptide from the venom of the South American rattlesnake Crotalus durissus terrificus. Peptides 2008, 29, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Brigatte, P.; Konno, K.; Gutierrez, V.P.; Sampaio, S.C.; Zambelli, V.O.; Picolo, G.; Curi, R.; Cury, Y. Peripheral kappa and delta opioid receptors are involved in the antinociceptive effect of crotalphine in a rat model of cancer pain. Pharmacol. Biochem. Behav. 2013, 109, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.; Schaefer, M. TRPA1 Is Differentially Modulated by the Amphipathic Molecules Trinitrophenol and Chlorpromazine. J. Biol. Chem. 2007, 282, 7145–7153. [Google Scholar] [CrossRef] [PubMed]
- Spassova, M.A.; Hewavitharana, T.; Xu, W.; Soboloff, J.; Gill, D.L. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc. Natl. Acad. Sci. USA 2006, 103, 16586–16591. [Google Scholar] [CrossRef] [PubMed]
- Bae, C.; Sachs, F.; Gottlieb, P.A. The Mechanosensitive Ion Channel Piezo1 Is Inhibited by the Peptide GsMTx4. Biochemistry 2011, 50, 6295–6300. [Google Scholar] [CrossRef] [PubMed]
- Bowman, C.L.; Gottlieb, P.A.; Suchyna, T.M.; Murphy, Y.K.; Sachs, F. Mechanosensitive ion channels and the peptide inhibitor GsMTx-4: History, properties, mechanisms and pharmacology. Toxicon 2007, 49, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Gnanasambandam, R.; Ghatak, C.; Yasmann, A.; Nishizawa, K.; Sachs, F.; Ladokhin, A.S.; Sukharev, S.I.; Suchyna, T.M. GsMTx4: Mechanism of Inhibiting Mechanosensitive Ion Channels. Biophys. J. 2017, 112, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, M.; Nishizawa, K. Molecular Dynamics Simulations of a Stretch-Activated Channel Inhibitor GsMTx4 with Lipid Membranes: Two Binding Modes and Effects of Lipid Structure. Biophys. J. 2007, 92, 4233–4243. [Google Scholar] [CrossRef] [PubMed]
- Kwan, K.Y.; Allchorne, A.J.; Vollrath, M.A.; Christensen, A.P.; Zhang, D.-S.; Woolf, C.J.; Corey, D.P. TRPA1 Contributes to Cold, Mechanical, and Chemical Nociception but Is Not Essential for Hair-Cell Transduction. Neuron 2006, 50, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Story, G.M.; Gereau, R.W. Numbing the Senses: Role of TRPA1 in Mechanical and Cold Sensation. Neuron 2006, 50, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Rosa, F.; Trevisan, G.; Rigo, F.K.; Tonello, R.; Andrade, E.L.; do Nascimento Cordeiro, M.; Calixto, J.B.; Gomez, M.V.; Ferreira, J. Phα1β, a peptide from the venom of the spider Phoneutria nigriventer shows antinociceptive effects after continuous infusion in a neuropathic pain model in rats. Anesth. Analg. 2014, 119, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, R.; Champigny, G.; Bassilana, F.; Heurteaux, C.; Lazdunski, M. A proton-gated cation channel involved in acid-sensing. Nature 1997, 386, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Cristofori-Armstrong, B.; Rash, L.D. Acid-sensing ion channel (ASIC) structure and function: Insights from spider, snake and sea anemone venoms. Neuropharmacology 2017, 127, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.; Diochot, S.; Salinas, M.; Deval, E.; Noël, J.; Lingueglia, E. Venom toxins in the exploration of molecular, physiological and pathophysiological functions of acid-sensing ion channels. Toxicon 2013, 75, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, E.B.; Kawate, T.; Gouaux, E. Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature 2009, 460, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Jasti, J.; Furukawa, H.; Gonzales, E.B.; Gouaux, E. Structure of acid-sensing ion channel 1 at 1.9 Å resolution and low pH. Nature 2007, 449, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Deval, E. Acid-Sensing Ion Channels and nociception in the peripheral and central nervous systems. Neuropharmacology 2015, 94, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Holzer, P. Acid-sensing ion channels in gastrointestinal function. Neuropharmacology 2015, 94, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Deval, E.; Gasull, X.; Noël, J.; Salinas, M.; Baron, A.; Diochot, S.; Lingueglia, E. Acid-Sensing Ion Channels (ASICs): Pharmacology and implication in pain. Pharmacol. Ther. 2010, 128, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-G.; Yu, Y.; Zhang, Z.-D.; Cao, H.; Xu, T.-L. ASIC3 Channels Integrate Agmatine and Multiple Inflammatory Signals through the Nonproton Ligand Sensing Domain. Mol. Pain 2010, 6, 88. [Google Scholar] [CrossRef] [PubMed]
- Deval, E.; Noël, J.; Lay, N.; Alloui, A.; Diochot, S.; Friend, V.; Jodar, M.; Lazdunski, M.; Lingueglia, E. ASIC3, a sensor of acidic and primary inflammatory pain. EMBO J. 2008, 27, 3047–3055. [Google Scholar] [CrossRef] [PubMed]
- Qadri, Y.J.; Rooj, A.K.; Fuller, C.M. ENaCs and ASICs as therapeutic targets. Am. J. Physiol. Physiol. 2012, 302, C943–C965. [Google Scholar] [CrossRef] [PubMed]
- Dorofeeva, N.A.; Barygin, O.I.; Staruschenko, A.; Bolshakov, K.V.; Magazanik, L.G. Mechanisms of non-steroid anti-inflammatory drugs action on ASICs expressed in hippocampal interneurons. J. Neurochem. 2008, 106, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Voilley, N.; de Weille, J.; Mamet, J.; Lazdunski, M. Nonsteroid anti-inflammatory drugs inhibit both the activity and the inflammation-induced expression of acid-sensing ion channels in nociceptors. J. Neurosci. 2001, 21, 8026–8033. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Chu, X.; Maysami, S.; Li, M.; Si, H.; Cottrell, J.E.; Simon, R.P.; Xiong, Z. Inhibition of Acid Sensing Ion Channel Currents by Lidocaine in Cultured Mouse Cortical Neurons. Anesth. Anal. 2011, 112, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Li, X.; Wang, G.; Fei, J.; Meng, T.; Zhang, X.; Yu, J.; Yu, J.; Li, J. Inhibition of acid-sensing ion channel currents by propofol in rat dorsal root ganglion neurons. Clin. Exp. Pharmacol. Physiol. 2014, 41, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Escoubas, P.; De Weille, J.R.; Lecoq, A.; Diochot, S.; Waldmann, R.; Champigny, G.; Moinier, D.; Ménez, A.; Lazdunski, M. Isolation of a tarantula toxin specific for a class of proton-gated Na+ channels. J. Biol. Chem. 2000, 275, 25116–25121. [Google Scholar] [CrossRef] [PubMed]
- Saez, N.J.; Mobli, M.; Bieri, M.; Chassagnon, I.R.; Malde, A.K.; Gamsjaeger, R.; Mark, A.E.; Gooley, P.R.; Rash, L.D.; King, G.F. A dynamic pharmacophore drives the interaction between Psalmotoxin-1 and the putative drug target acid-sensing ion channel 1a. Mol. Pharmacol. 2011, 80, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, T.W.; Lee, K.G.; Gormley, M.G.; Askwith, C.C. Heteromeric Acid-Sensing Ion Channels (ASICs) Composed of ASIC2b and ASIC1a Display Novel Channel Properties and Contribute to Acidosis-Induced Neuronal Death. J. Neurosci. 2011, 31, 9723–9734. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kalbacher, H.; Gründer, S. Interaction of acid-sensing ion channel (ASIC) 1 with the tarantula toxin psalmotoxin 1 is state dependent. J. Gen. Physiol. 2006, 127, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Baconguis, I.; Gouaux, E. Structural plasticity and dynamic selectivity of acid-sensing ion channel-spider toxin complexes. Nature 2012, 489, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.J.P.P.; Benz, J.; Stohler, P.; Tetaz, T.; Joseph, C.; Huber, S.; Schmid, G.; Hügin, D.; Pflimlin, P.; Trube, G.; et al. Structure of the acid-sensing ion channel 1 in complex with the gating modifier Psalmotoxin 1. Nat. Commun. 2012, 3, 936. [Google Scholar] [CrossRef] [PubMed]
- Mazzuca, M.; Heurteaux, C.; Alloui, A.; Diochot, S.; Baron, A.; Voilley, N.; Blondeau, N.; Escoubas, P.; Gélot, A.; Cupo, A.; et al. A tarantula peptide against pain via ASIC1a channels and opioid mechanisms. Nat. Neurosci. 2007, 10, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Matricon, J.; Gelot, A.; Etienne, M.; Lazdunski, M.; Muller, E.; Ardid, D. Spinal cord plasticity and acid-sensing ion channels involvement in a rodent model of irritable bowel syndrome. Eur. J. Pain 2011, 15, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Baron, A.; Salinas, M.; Douguet, D.; Scarzello, S.; Dabert-Gay, A.-S.; Debayle, D.; Friend, V.V.V.; Alloui, A.; Lazdunski, M.; et al. Black mamba venom peptides target acid-sensing ion channels to abolish pain. Nature 2012, 490, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Er, S.Y.; Cristofori-Armstrong, B.; Escoubas, P.; Rash, L.D. Discovery and molecular interaction studies of a highly stable, tarantula peptide modulator of acid-sensing ion channel 1. Neuropharmacology 2017, 127, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Chassagnon, I.R.; McCarthy, C.A.; Chin, Y.K.-Y.; Pineda, S.S.; Keramidas, A.; Mobli, M.; Pham, V.; De Silva, T.M.; Lynch, J.W.; Widdop, R.E.; et al. Potent neuroprotection after stroke afforded by a double-knot spider-venom peptide that inhibits acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. USA 2017, 114, 3750–3755. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Baron, A.; Rash, L.D.; Deval, E.; Escoubas, P.; Scarzello, S.; Salinas, M.; Lazdunski, M. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. EMBO J. 2004, 23, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.E.; Cristofori-Armstrong, B.; Anangi, R.; Rosengren, K.J.; Lau, C.H.Y.; Mobli, M.; Brust, A.; Alewood, P.F.; King, G.F.; Rash, L.D. Understanding the Molecular Basis of Toxin Promiscuity: The Analgesic Sea Anemone Peptide APETx2 Interacts with Acid-Sensing Ion Channel 3 and hERG Channels via Overlapping Pharmacophores. J. Med. Chem. 2014, 57, 9195–9203. [Google Scholar] [CrossRef] [PubMed]
- Peigneur, S.; Béress, L.; Möller, C.; Marí, F.; Forssmann, W.-G.; Tytgat, J. A natural point mutation changes both target selectivity and mechanism of action of sea anemone toxins. FASEB J. 2012, 26, 5141–5151. [Google Scholar] [CrossRef] [PubMed]
- Rahman, T.; Smith, E.S.J. In silico assessment of interaction of sea anemone toxin APETx2 and acid sensing ion channel 3. Biochem. Biophys. Res. Commun. 2014, 450, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Deval, E.; Noël, J.; Gasull, X.; Delaunay, A.; Alloui, A.; Friend, V.; Eschalier, A.; Lazdunski, M.; Lingueglia, E. Acid-sensing ion channels in postoperative pain. J. Neurosci. 2011, 31, 6059–6066. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, M.; Kolker, S.J.; Sluka, K.A. Acid-Sensing Ion Channel 3 Expression in Mouse Knee Joint Afferents and Effects of Carrageenan-Induced Arthritis. J. Pain 2009, 10, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.C.W.; Otsuka, E.; Wagstrom, E.; Jensen, C.S.; Price, M.P.; Gebhart, G.F. Short-Term Sensitization of Colon Mechanoreceptors Is Associated With Long-Term Hypersensitivity to Colon Distention in the Mouse. Gastroenterology 2007, 133, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Mourier, G.; Salinas, M.; Kessler, P.; Stura, E.A.; Leblanc, M.; Tepshi, L.; Besson, T.; Diochot, S.; Baron, A.; Douguet, D.; et al. Mambalgin-1 Pain-relieving Peptide, Stepwise Solid-phase Synthesis, Crystal Structure, and Functional Domain for Acid-sensing Ion Channel 1a Inhibition. J. Biol. Chem. 2016, 291, 2616–2629. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Alloui, A.; Rodrigues, P.; Dauvois, M.; Friend, V.; Aissouni, Y.; Eschalier, A.; Lingueglia, E.; Baron, A. Analgesic effects of mambalgin peptide inhibitors of acid-sensing ion channels in inflammatory and neuropathic pain. Pain 2016, 157, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Salinas, M.; Besson, T.; Delettre, Q.; Diochot, S.; Boulakirba, S.; Douguet, D.; Lingueglia, E. Binding site and inhibitory mechanism of the mambalgin-2 pain-relieving peptide on acid-sensing ion channel 1a. J. Biol. Chem. 2014, 289, 13363–13373. [Google Scholar] [CrossRef] [PubMed]
- Bohlen, C.J.; Chesler, A.T.; Sharif-Naeini, R.; Medzihradszky, K.F.; Zhou, S.; King, D.; Sánchez, E.E.; Burlingame, A.L.; Basbaum, A.I.; Julius, D. A heteromeric Texas coral snake toxin targets acid-sensing ion channels to produce pain. Nature 2011, 479, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Baconguis, I.; Bohlen, C.J.; Goehring, A.; Julius, D.; Gouaux, E. X-Ray Structure of Acid-Sensing Ion Channel 1–Snake Toxin Complex Reveals Open State of a Na+-Selective Channel. Cell 2014, 156, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Izumi, M.; Ikeuchi, M.; Ji, Q.; Tani, T. Local ASIC3 modulates pain and disease progression in a rat model of osteoarthritis. J. Biomed. Sci. 2012, 19, 77. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, J.; Spencer, R.H.; Garsky, V.M.; Liang, A.; Leitl, M.D.; Cato, M.J.; Cook, S.P.; Kane, S.; Urban, M.O. Reversal of acid-induced and inflammatory pain by the selective ASIC3 inhibitor, APETx2. Br. J. Pharmacol. 2010, 161, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Structure and function of voltage-gated sodium channels at atomic resolution. Exp. Physiol. 2014, 99, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.L. Resurgence of Sodium Channel Research. Annu. Rev. Physiol. 2001, 63, 871–894. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ou, S.-W.; Wang, Y.-J. Distribution and function of voltage-gated sodium channels in the nervous system. Channels 2017, 11, 534–554. [Google Scholar] [CrossRef] [PubMed]
- de Lera Ruiz, M.; Kraus, R.L. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 2015, 58, 7093–7118. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.N.; Boorman, J.P.; Okuse, K.; Baker, M.D. Voltage-gated sodium channels and pain pathways. J. Neurobiol. 2004, 61, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.R.; Sheets, P.L.; Waxman, S.G. The roles of sodium channels in nociception: Implications for mechanisms of pain. Pain 2007, 131, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.; Tang, L.; Madge, D.J.; Stevens, E.B. The role of sodium channels in neuropathic pain. Semin. Cell Dev. Biol. 2006, 17, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Nassar, M.A.; Stirling, L.C.; Forlani, G.; Baker, M.D.; Matthews, E.A.; Dickenson, A.H.; Wood, J.N. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc. Natl. Acad. Sci. USA 2004, 101, 12706–12711. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Y.; MacFarlane, J.; MacDonald, M.; Thompson, J.; Dube, M.-P.; Mattice, M.; Fraser, R.; Young, C.; Hossain, S.; Pape, T.; et al. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin. Genet. 2007, 71, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Dahllund, L.; Eriksson, A.B.; Hellgren, D.; Karlsson, U.; Lund, P.-E.; Meijer, I.A.; Meury, L.; Mills, T.; Moody, A.; et al. A stop codon mutation in SCN9A causes lack of pain sensation. Hum. Mol. Genet. 2007, 16, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. From genes to pain: Nav1.7 and human pain disorders. Trends Neurosci. 2007, 30, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Rush, A.M.; Cummins, T.R.; Hisama, F.M.; Novella, S.; Tyrrell, L.; Marshall, L.; Waxman, S.G. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 2005, 128, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Fertleman, C.R.; Baker, M.D.; Parker, K.A.; Moffatt, S.; Elmslie, F.V.; Abrahamsen, B.; Ostman, J.; Klugbauer, N.; Wood, J.N.; Gardiner, R.M.; et al. SCN9A Mutations in Paroxysmal Extreme Pain Disorder: Allelic Variants Underlie Distinct Channel Defects and Phenotypes. Neuron 2006, 52, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Y.; Li, S.; Xu, Z.; Li, H.; Ma, L.; Fan, J.; Bu, D.; Liu, B.; Fan, Z.; et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J. Med. Genet. 2004, 41, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Faber, C.G.; Hoeijmakers, J.G.J.; Ahn, H.-S.; Cheng, X.; Han, C.; Choi, J.-S.; Estacion, M.; Lauria, G.; Vanhoutte, E.K.; Gerrits, M.M.; et al. Gain of function NaV1.7 mutations in idiopathic small fiber neuropathy. Ann. Neurol. 2012, 71, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Hoeijmakers, J.G.J.; Ahn, H.-S.; Zhao, P.; Shah, P.; Lauria, G.; Gerrits, M.M.; te Morsche, R.H.M.; Dib-Hajj, S.D.; Drenth, J.P.H.; et al. Nav1.7-related small fiber neuropathy: Impaired slow-inactivation and DRG neuron hyperexcitability. Neurology 2012, 78, 1635–1643. [Google Scholar] [CrossRef] [PubMed]
- Faber, C.G.; Lauria, G.; Merkies, I.S.J.; Cheng, X.; Han, C.; Ahn, H.-S.; Persson, A.-K.; Hoeijmakers, J.G.J.; Gerrits, M.M.; Pierro, T.; et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 19444–19449. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Han, C.; Estacion, M.; Vasylyev, D.; Hoeijmakers, J.G.J.; Gerrits, M.M.; Tyrrell, L.; Lauria, G.; Faber, C.G.; Dib-Hajj, S.D.; et al. PROPANE Study Group Gain-of-function mutations in sodium channel NaV1.9 in painful neuropathy. Brain 2014, 137, 1627–1642. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yang, Y.; Zhao, P.; Gerrits, M.M.; Hoeijmakers, J.G.J.; Bekelaar, K.; Merkies, I.S.J.; Faber, C.G.; Dib-Hajj, S.D.; Waxman, S.G. Small-Fiber Neuropathy Nav1.8 Mutation Shifts Activation to Hyperpolarized Potentials and Increases Excitability of Dorsal Root Ganglion Neurons. J. Neurosci. 2013, 33, 14087–14097. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Vasylyev, D.; Macala, L.J.; Gerrits, M.M.; Hoeijmakers, J.G.J.; Bekelaar, K.J.; Dib-Hajj, S.D.; Faber, C.G.; Merkies, I.S.J.; Waxman, S.G. The G1662S NaV1.8 mutation in small fibre neuropathy: Impaired inactivation underlying DRG neuron hyperexcitability. J. Neurol. Neurosurg. Psychiatry 2014, 85, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wood, J.N. The roles of sodium channels in nociception: Implications for mechanisms of neuropathic pain. Pain Med. 2011, 12 (Suppl. 3), S93–S99. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Y.P.; Price, N.; Namdari, R.; Cohen, C.J.; Lamers, M.H.; Winters, C.; Price, J.; Young, C.E.; Verschoof, H.; Sherrington, R.; et al. Treatment of Nav1.7-mediated pain in inherited erythromelalgia using a novel sodium channel blocker. Pain 2012, 153, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Fozzard, H.A.; Sheets, M.F.; Hanck, D.A. The Sodium Channel as a Target for Local Anesthetic Drugs. Front. Pharmacol. 2011, 2, 68. [Google Scholar] [CrossRef] [PubMed]
- Hockley, J.R.F.; Winchester, W.J.; Bulmer, D.C. The voltage-gated sodium channel NaV 1.9 in visceral pain. Neurogastroenterol. Motil. 2016, 28, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Inserra, M.C.; Israel, M.R.; Caldwell, A.; Castro, J.; Deuis, J.R.; Harrington, A.M.; Keramidas, A.; Garcia-Caraballo, S.; Maddern, J.; Erickson, A.; et al. Multiple sodium channel isoforms mediate the pathological effects of Pacific ciguatoxin-1. Sci. Rep. 2017, 7, 42810. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.-H.; Zhou, Y.-L.; Xu, G.-Y. Targeting voltage-gated sodium channels for treatment for chronic visceral pain. World J. Gastroenterol. 2011, 17, 2357–2364. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.; Melgar, S. Lack of colonic-inflammation-induced acute visceral hypersensitivity to colorectal distension in Na(v)1.9 knockout mice. Eur. J. Pain 2008, 12, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Hillsley, K.; Lin, J.-H.; Stanisz, A.; Grundy, D.; Aerssens, J.; Peeters, P.J.; Moechars, D.; Coulie, B.; Stead, R.H. Dissecting the role of sodium currents in visceral sensory neurons in a model of chronic hyperexcitability using Nav1.8 and Nav1.9 null mice. J. Physiol. 2006, 576, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Laird, J.M.A.; Souslova, V.; Wood, J.N.; Cervero, F. Deficits in visceral pain and referred hyperalgesia in Nav1.8 (SNS/PN3)-null mice. J. Neurosci. 2002, 22, 8352–8356. [Google Scholar] [CrossRef] [PubMed]
- Samad, O.A.; Tan, A.M.; Cheng, X.; Foster, E.; Dib-Hajj, S.D.; Waxman, S.G. Virus-mediated shRNA Knockdown of Nav1.3 in Rat Dorsal Root Ganglion Attenuates Nerve Injury-induced Neuropathic Pain. Mol. Ther. 2013, 21, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Mattei, C. Tetrodotoxin, a Candidate Drug for Nav1.1-Induced Mechanical Pain? Mar. Drugs 2018, 16, 72. [Google Scholar] [CrossRef] [PubMed]
- Osteen, J.D.; Herzig, V.; Gilchrist, J.; Emrick, J.J.; Zhang, C.; Wang, X.; Castro, J.; Garcia-Caraballo, S.; Grundy, L.; Rychkov, G.Y.; et al. Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature 2016, 534, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Salvatierra, J.; Castro, J.; Erickson, A.; Li, Q.; Braz, J.; Gilchrist, J.; Grundy, L.; Rychkov, G.Y.; Deiteren, A.; Rais, R.; et al. NaV1.1 inhibition can reduce visceral hypersensitivity. JCI Insight 2018, 3, 121000. [Google Scholar] [CrossRef] [PubMed]
- Lindia, J.A.; Köhler, M.G.; Martin, W.J.; Abbadie, C. Relationship between sodium channel NaV1.3 expression and neuropathic pain behavior in rats. Pain 2005, 117, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Strong, J.A.; Ye, L.; Mao, J.-X.; Zhang, J.-M. Knockdown of sodium channel NaV1.6 blocks mechanical pain and abnormal bursting activity of afferent neurons in inflamed sensory ganglia. Pain 2013, 154, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Strong, J.A.; Zhang, J.-M. Local knockdown of the NaV1.6 sodium channel reduces pain behaviors, sensory neuron excitability, and sympathetic sprouting in rat models of neuropathic pain. Neuroscience 2015, 291, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Tytgat, J. Voltage-gated sodium channel modulation by scorpion α-toxins. Toxicon 2007, 49, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Deuis, J.R.; Mueller, A.; Israel, M.R.; Vetter, I. The pharmacology of voltage-gated sodium channel activators. Neuropharmacology 2017, 127, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.C.; Lewis, R.J. Sodium channels and pain: From toxins to therapies. Br. J. Pharmacol. 2018, 175, 2138–2157. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, J.; Olivera, B.M.; Bosmans, F. Animal Toxins Influence Voltage-Gated Sodium Channel Function. Handb. Exp. Pharmacol. 2014, 221, 203–229. [Google Scholar] [PubMed]
- Cestèle, S.; Catterall, W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar] [CrossRef]
- Stevens, M.; Peigneur, S.; Tytgat, J. Neurotoxins and Their Binding Areas on Voltage-Gated Sodium Channels. Front. Pharmacol. 2011, 2, 71. [Google Scholar] [CrossRef] [PubMed]
- Fozzard, H.A.; Lipkind, G.M. The Tetrodotoxin Binding Site Is within the Outer Vestibule of the Sodium Channel. Mar. Drugs 2010, 8, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Li, Z.; Jiang, Y.; Pan, X.; Wu, J.; Cristofori-Armstrong, B.; Smith, J.J.; Chin, Y.K.Y.; Lei, J.; Zhou, Q.; et al. Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science 2018, 362, eaau2596. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Cestèle, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.H.Y.; King, G.F.; Mobli, M. Molecular basis of the interaction between gating modifier spider toxins and the voltage sensor of voltage-gated ion channels. Sci. Rep. 2016, 6, 34333. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Martin-Eauclaire, M.-F.; Swartz, K.J. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 2008, 456, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yarov-Yarovoy, V.; Kahn, R.; Gordon, D.; Gurevitz, M.; Scheuer, T.; Catterall, W.A. Mapping the receptor site for -scorpion toxins on a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. USA 2011, 108, 15426–15431. [Google Scholar] [CrossRef] [PubMed]
- Campos, F.V.; Chanda, B.; Beirão, P.S.L.; Bezanilla, F. α-Scorpion Toxin Impairs a Conformational Change that Leads to Fast Inactivation of Muscle Sodium Channels. J. Gen. Physiol. 2008, 132, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.C.; Qu, Y.; Tanada, T.N.; Scheuer, T.; Catterall, W.A. Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3–S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J. Biol. Chem. 1996, 271, 15950–15962. [Google Scholar] [CrossRef] [PubMed]
- Cestèle, S.; Qu, Y.; Rogers, J.C.; Rochat, H.; Scheuer, T.; Catterall, W.A. Voltage sensor-trapping: Enhanced activation of sodium channels by beta-scorpion toxin bound to the S3–S4 loop in domain II. Neuron 1998, 21, 919–931. [Google Scholar] [CrossRef]
- Cestèle, S.; Scheuer, T.; Mantegazza, M.; Rochat, H.; Catterall, W.A. Neutralization of gating charges in domain II of the sodium channel alpha subunit enhances voltage-sensor trapping by a beta-scorpion toxin. J. Gen. Physiol. 2001, 118, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Leipold, E.; Borges, A.; Heinemann, S.H. Scorpion β-toxin interference with NaV channel voltage sensor gives rise to excitatory and depressant modes. J. Gen. Physiol. 2012, 139, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Nieto, F.R.; Cobos, E.J.; Tejada, M.Á.; Sánchez-Fernández, C.; González-Cano, R.; Cendán, C.M. Tetrodotoxin (TTX) as a therapeutic agent for pain. Mar. Drugs 2012, 10, 281–305. [Google Scholar] [CrossRef] [PubMed]
- Lago, J.; Rodríguez, L.P.; Blanco, L.; Vieites, J.M.; Cabado, A.G. Tetrodotoxin, an Extremely Potent Marine Neurotoxin: Distribution, Toxicity, Origin and Therapeutical Uses. Mar. Drugs 2015, 13, 6384–6406. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Black, J.A.; Waxman, S.G. Voltage-gated sodium channels: Therapeutic targets for pain. Pain Med. 2009, 10, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Wickenden, A.D.; Chaplan, S.R. Sodium channel blockers for the treatment of neuropathic pain. Neurotherapeutics 2009, 6, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.S.; Park, S.K.; Chung, K.; Chung, J.M. Low dose of tetrodotoxin reduces neuropathic pain behaviors in an animal model. Brain Res. 2000, 871, 98–103. [Google Scholar] [CrossRef]
- Nieto, F.R.; Entrena, J.M.; Cendán, C.M.; Pozo, E. Del; Vela, J.M.; Baeyens, J.M. Tetrodotoxin inhibits the development and expression of neuropathic pain induced by paclitaxel in mice. Pain 2008, 137, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, P.; Levine, J.D. Antihyperalgesic effect of tetrodotoxin in rat models of persistent muscle pain. Neuroscience 2015, 311, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Beloeil, H.; Ababneh, Z.; Chung, R.; Zurakowski, D.; Mulkern, R.V.; Berde, C.B. Effects of bupivacaine and tetrodotoxin on carrageenan-induced hind paw inflammation in rats (Part 1): Hyperalgesia, edema, and systemic cytokines. Anesthesiology 2006, 105, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Salas, M.M.; McIntyre, M.K.; Petz, L.N.; Korz, W.; Wong, D.; Clifford, J.L. Tetrodotoxin suppresses thermal hyperalgesia and mechanical allodynia in a rat full thickness thermal injury pain model. Neurosci. Lett. 2015, 607, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Marcil, J.; Walczak, J.-S.; Guindon, J.; Ngoc, A.H.; Lu, S.; Beaulieu, P. Antinociceptive effects of tetrodotoxin (TTX) in rodents. Br. J. Anaesth. 2006, 96, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Hagen, N.A.; Fisher, K.M.; Lapointe, B.; du Souich, P.; Chary, S.; Moulin, D.; Sellers, E.; Ngoc, A.H.; Canadian Tetrodotoxin Study Group. An open-label, multi-dose efficacy and safety study of intramuscular tetrodotoxin in patients with severe cancer-related pain. J. Pain Symptom Manag. 2007, 34, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Hagen, N.A.; Cantin, L.; Constant, J.; Haller, T.; Blaise, G.; Ong-Lam, M.; du Souich, P.; Korz, W.; Lapointe, B. Tetrodotoxin for Moderate to Severe Cancer-Related Pain: A Multicentre, Randomized, Double-Blind, Placebo-Controlled, Parallel-Design Trial. Pain Res. Manag. 2017, 2017, 7212713. [Google Scholar] [CrossRef] [PubMed]
- England, S.; de Groot, M.J. Subtype-selective targeting of voltage-gated sodium channels. Br. J. Pharmacol. 2009, 158, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Blumenthal, K.; Jackson, J.O.; Liang, S.; Cummins, T.R. The tarantula toxins ProTx-II and huwentoxin-IV differentially interact with human Nav1.7 voltage sensors to inhibit channel activation and inactivation. Mol. Pharmacol. 2010, 78, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, S.; Kraus, R.L.; Scheuer, T.; Catterall, W.A. Inhibition of sodium channel gating by trapping the domain II voltage sensor with protoxin II. Mol. Pharmacol. 2008, 73, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Schmalhofer, W.A.; Calhoun, J.; Burrows, R.; Bailey, T.; Kohler, M.G.; Weinglass, A.B.; Kaczorowski, G.J.; Garcia, M.L.; Koltzenburg, M.; Priest, B.T. ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol. Pharmacol. 2008, 74, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Cummins, T.R.; Alphy, S.; Blumenthal, K.M. Molecular interactions of the gating modifier toxin ProTx-II with NaV 1.5: Implied existence of a novel toxin binding site coupled to activation. J. Biol. Chem. 2007, 282, 12687–12697. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.-I.; Sekino, S.; Ikegami, M.; Ikeda, H.; Kamei, J. Antihyperalgesic effects of ProTx-II, a Nav1.7 antagonist, and A803467, a Nav1.8 antagonist, in diabetic mice. J. Exp. Pharmacol. 2015, 7, 11–16. [Google Scholar] [PubMed]
- Flinspach, M.; Xu, Q.; Piekarz, A.D.; Fellows, R.; Hagan, R.; Gibbs, A.; Liu, Y.; Neff, R.A.; Freedman, J.; Eckert, W.A.; et al. Insensitivity to pain induced by a potent selective closed-state Nav1.7 inhibitor. Sci. Rep. 2017, 7, 39662. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, E.; Cassulini, R.R.; Silva, D.F.; Clement, H.; Schiavon, E.; Zamudio, F.Z.; Odell, G.; Arcangeli, A.; Clare, J.J.; Alagón, A.; et al. Target promiscuity and heterogeneous effects of tarantula venom peptides affecting Na+ and K+ ion channels. J. Biol. Chem. 2010, 285, 4130–4142. [Google Scholar] [CrossRef] [PubMed]
- Saez, N.J.; Senff, S.; Jensen, J.E.; Er, S.Y.; Herzig, V.; Rash, L.D.; King, G.F. Spider-venom peptides as therapeutics. Toxins 2010, 2, 2851–2871. [Google Scholar] [CrossRef] [PubMed]
- Clement, H.; Odell, G.; Zamudio, F.Z.; Redaelli, E.; Wanke, E.; Alagón, A.; Possani, L.D. Isolation and characterization of a novel toxin from the venom of the spider Grammostola rosea that blocks sodium channels. Toxicon 2007, 50, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Lampe, R.A. Analgesic Peptides From Venom of Grammostola Spatulata and Use Thereof. U.S. Patent 5877026A, 2 March 1999. [Google Scholar]
- Revell, J.D.; Lund, P.-E.; Linley, J.E.; Metcalfe, J.; Burmeister, N.; Sridharan, S.; Jones, C.; Jermutus, L.; Bednarek, M.A. Potency optimization of Huwentoxin-IV on hNav1.7: A neurotoxin TTX-S sodium-channel antagonist from the venom of the Chinese bird-eating spider Selenocosmia huwena. Peptides 2013, 44, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Moyer, B.D.; Murray, J.K.; Ligutti, J.; Andrews, K.; Favreau, P.; Jordan, J.B.; Lee, J.H.; Liu, D.; Long, J.; Sham, K.; et al. Pharmacological characterization of potent and selective NaV1.7 inhibitors engineered from Chilobrachys jingzhao tarantula venom peptide JzTx-V. PLoS ONE 2018, 13, e0196791. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.K.; Ligutti, J.; Liu, D.; Zou, A.; Poppe, L.; Li, H.; Andrews, K.L.; Moyer, B.D.; McDonough, S.I.; Favreau, P.; et al. Engineering potent and selective analogues of GpTx-1, a tarantula venom peptide antagonist of the Na(V)1.7 sodium channel. J. Med. Chem. 2015, 58, 2299–2314. [Google Scholar] [CrossRef] [PubMed]
- Rahnama, S.; Deuis, J.R.; Cardoso, F.C.; Ramanujam, V.; Lewis, R.J.; Rash, L.D.; King, G.F.; Vetter, I.; Mobli, M. The structure, dynamics and selectivity profile of a NaV1.7 potency-optimised huwentoxin-IV variant. PLoS ONE 2017, 12, e0173551. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Li, P.; Chen, B.; Huang, J.; Lai, R.; Liu, J.; Rong, M. Selective Closed-State Nav1.7 Blocker JZTX-34 Exhibits Analgesic Effects against Pain. Toxins 2018, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Peng, D.; Huang, B.; Yang, Q.; Zhang, Q.; Chen, M.; Rong, M.; Liu, Z. Discovery of a Novel Nav1.7 Inhibitor From Cyriopagopus albostriatus Venom With Potent Analgesic Efficacy. Front. Pharmacol. 2018, 9, 1158. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xiao, Y.; Kang, D.; Liu, J.; Li, Y.; Undheim, E.A.B.; Klint, J.K.; Rong, M.; Lai, R.; King, G.F. Discovery of a selective NaV1.7 inhibitor from centipede venom with analgesic efficacy exceeding morphine in rodent pain models. Proc. Natl. Acad. Sci. USA 2013, 110, 17534–17539. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.Y.; Cristofori-Armstrong, B.; Undheim, E.A.B.; King, G.F.; Rash, L.D. Three Peptide Modulators of the Human Voltage-Gated Sodium Channel 1.7, an Important Analgesic Target, from the Venom of an Australian Tarantula. Toxins 2015, 7, 2494–2513. [Google Scholar] [CrossRef] [PubMed]
- Klint, J.K.; Smith, J.J.; Vetter, I.; Rupasinghe, D.B.; Er, S.Y.; Senff, S.; Herzig, V.; Mobli, M.; Lewis, R.J.; Bosmans, F.; et al. Seven novel modulators of the analgesic target NaV 1.7 uncovered using a high-throughput venom-based discovery approach. Br. J. Pharmacol. 2015, 172, 2445–2458. [Google Scholar] [CrossRef] [PubMed]
- Deuis, J.R.; Dekan, Z.; Wingerd, J.S.; Smith, J.J.; Munasinghe, N.R.; Bhola, R.F.; Imlach, W.L.; Herzig, V.; Armstrong, D.A.; Rosengren, K.J.; et al. Pharmacological characterisation of the highly NaV1.7 selective spider venom peptide Pn3a. Sci. Rep. 2017, 7, 40883. [Google Scholar] [CrossRef] [PubMed]
- Cherki, R.S.; Kolb, E.; Langut, Y.; Tsveyer, L.; Bajayo, N.; Meir, A. Two tarantula venom peptides as potent and differential Na(V) channels blockers. Toxicon 2014, 77, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Bulaj, G.; Zhang, M.-M.; Green, B.R.; Fiedler, B.; Layer, R.T.; Wei, S.; Nielsen, J.S.; Low, S.J.; Klein, B.D.; Wagstaff, J.D.; et al. Synthetic muO-conotoxin MrVIB blocks TTX-resistant sodium channel NaV1.8 and has a long-lasting analgesic activity. Biochemistry 2006, 45, 7404–7414. [Google Scholar] [CrossRef] [PubMed]
- Ekberg, J.; Jayamanne, A.; Vaughan, C.W.; Aslan, S.; Thomas, L.; Mould, J.; Drinkwater, R.; Baker, M.D.; Abrahamsen, B.; Wood, J.N.; et al. muO-conotoxin MrVIB selectively blocks Nav1.8 sensory neuron specific sodium channels and chronic pain behavior without motor deficits. Proc. Natl. Acad. Sci. USA 2006, 103, 17030–17035. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.O.; Peigneur, S.; Diniz, M.R.V.; Tytgat, J.; Beirão, P.S.L. Inhibitory effect of the recombinant Phoneutria nigriventer Tx1 toxin on voltage-gated sodium channels. Biochimie 2012, 94, 2756–2763. [Google Scholar] [CrossRef] [PubMed]
- Leipold, E.; DeBie, H.; Zorn, S.; Borges, A.; Olivera, B.M.; Terlau, H.; Heinemann, S.H. muO conotoxins inhibit NaV channels by interfering with their voltage sensors in domain-2. Channels 2007, 1, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Knapp, O.; Nevin, S.T.; Yasuda, T.; Lawrence, N.; Lewis, R.J.; Adams, D.J. Biophysical properties of Na(v) 1.8/Na(v) 1.2 chimeras and inhibition by µO-conotoxin MrVIB. Br. J. Pharmacol. 2012, 166, 2148–2160. [Google Scholar] [CrossRef] [PubMed]
- Wingerd, J.S.; Mozar, C.A.; Ussing, C.A.; Murali, S.S.; Chin, Y.K.-Y.; Cristofori-Armstrong, B.; Durek, T.; Gilchrist, J.; Vaughan, C.W.; Bosmans, F.; et al. The tarantula toxin β/δ-TRTX-Pre1a highlights the importance of the S1-S2 voltage-sensor region for sodium channel subtype selectivity. Sci. Rep. 2017, 7, 974. [Google Scholar] [CrossRef] [PubMed]
- Jensen, H.S.; Grunnet, M.; Bastlund, J.F. Therapeutic potential of Na(V)1.1 activators. Trends Pharmacol. Sci. 2014, 35, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Zhu, Y.; La, J.-H.; Wills, Z.P.; Gebhart, G.F. Experimental and computational evidence for an essential role of NaV1.6 in spike initiation at stretch-sensitive colorectal afferent endings. J. Neurophysiol. 2015, 113, 2618–2634. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; McLellan, A.T. Opioid Abuse in Chronic Pain--Misconceptions and Mitigation Strategies. N. Engl. J. Med. 2016, 374, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.; Schäfer, M.; Machelska, H. Attacking pain at its source: New perspectives on opioids. Nat. Med. 2003, 9, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2003, 2, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L. Toxins and drug discovery. Toxicon 2014, 92, 193–200. [Google Scholar] [CrossRef] [PubMed]
- de Souza, J.M.; Goncalves, B.D.C.; Gomez, M.V.; Vieira, L.B.; Ribeiro, F.M. Animal Toxins as Therapeutic Tools to Treat Neurodegenerative Diseases. Front. Pharmacol. 2018, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Lewis, R.J. Use of venom peptides to probe ion channel structure and function. J. Biol. Chem. 2010, 285, 13315–13320. [Google Scholar] [CrossRef] [PubMed]
- Kalia, J.; Milescu, M.; Salvatierra, J.; Wagner, J.; Klint, J.K.; King, G.F.; Olivera, B.M.; Bosmans, F. From foe to friend: Using animal toxins to investigate ion channel function. J. Mol. Biol. 2015, 427, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Hamad, M.K.; He, K.; Abdulrazeq, H.F.; Mustafa, A.M.; Luceri, R.; Kamal, N.; Ali, M.; Nakhla, J.; Herzallah, M.M.; Mammis, A. Potential Uses of Isolated Toxin Peptides in Neuropathic Pain Relief: A Literature Review. World Neurosurg. 2018, 113, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Renukuntla, J.; Vadlapudi, A.D.; Patel, A.; Boddu, S.H.S.; Mitra, A.K. Approaches for enhancing oral bioavailability of peptides and proteins. Int. J. Pharm. 2013, 447, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Borchardt, R.T. Optimizing oral absorption of peptides using prodrug strategies. J. Control. Release 1999, 62, 231–238. [Google Scholar] [CrossRef]
- White, T.R.; Renzelman, C.M.; Rand, A.C.; Rezai, T.; McEwen, C.M.; Gelev, V.M.; Turner, R.A.; Linington, R.G.; Leung, S.S.F.; Kalgutkar, A.S.; et al. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat. Chem. Biol. 2011, 7, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Linde, Y.; Ovadia, O.; Safrai, E.; Xiang, Z.; Portillo, F.P.; Shalev, D.E.; Haskell-Luevano, C.; Hoffman, A.; Gilon, C. Structure-activity relationship and metabolic stability studies of backbone cyclization and N-methylation of melanocortin peptides. Biopolymers 2008, 90, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Rezai, T.; Yu, B.; Millhauser, G.L.; Jacobson, M.P.; Lokey, R.S. Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers. J. Am. Chem. Soc. 2006, 128, 2510–2511. [Google Scholar] [CrossRef] [PubMed]
- Brandelli, A. Nanostructures as promising tools for delivery of antimicrobial peptides. Mini Rev. Med. Chem. 2012, 12, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.C.; Kumari, A.; Yadav, R. Development of peptide and protein nanotherapeutics by nanoencapsulation and nanobioconjugation. Peptides 2011, 32, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pei, Y.; Zhang, X.; Gu, Z.; Zhou, Z.; Yuan, W.; Zhou, J.; Zhu, J.; Gao, X. PEGylated PLGA nanoparticles as protein carriers: Synthesis, preparation and biodistribution in rats. J. Control. Release 2001, 71, 203–211. [Google Scholar] [CrossRef]
- Gulbake, A.; Jain, S.K. Chitosan: A potential polymer for colon-specific drug delivery system. Expert Opin. Drug Deliv. 2012, 9, 713–729. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Shah, T.; Amin, A. Polysaccharides: A targeting strategy for colonic drug delivery. Expert Opin. Drug Deliv. 2011, 8, 779–796. [Google Scholar] [CrossRef] [PubMed]


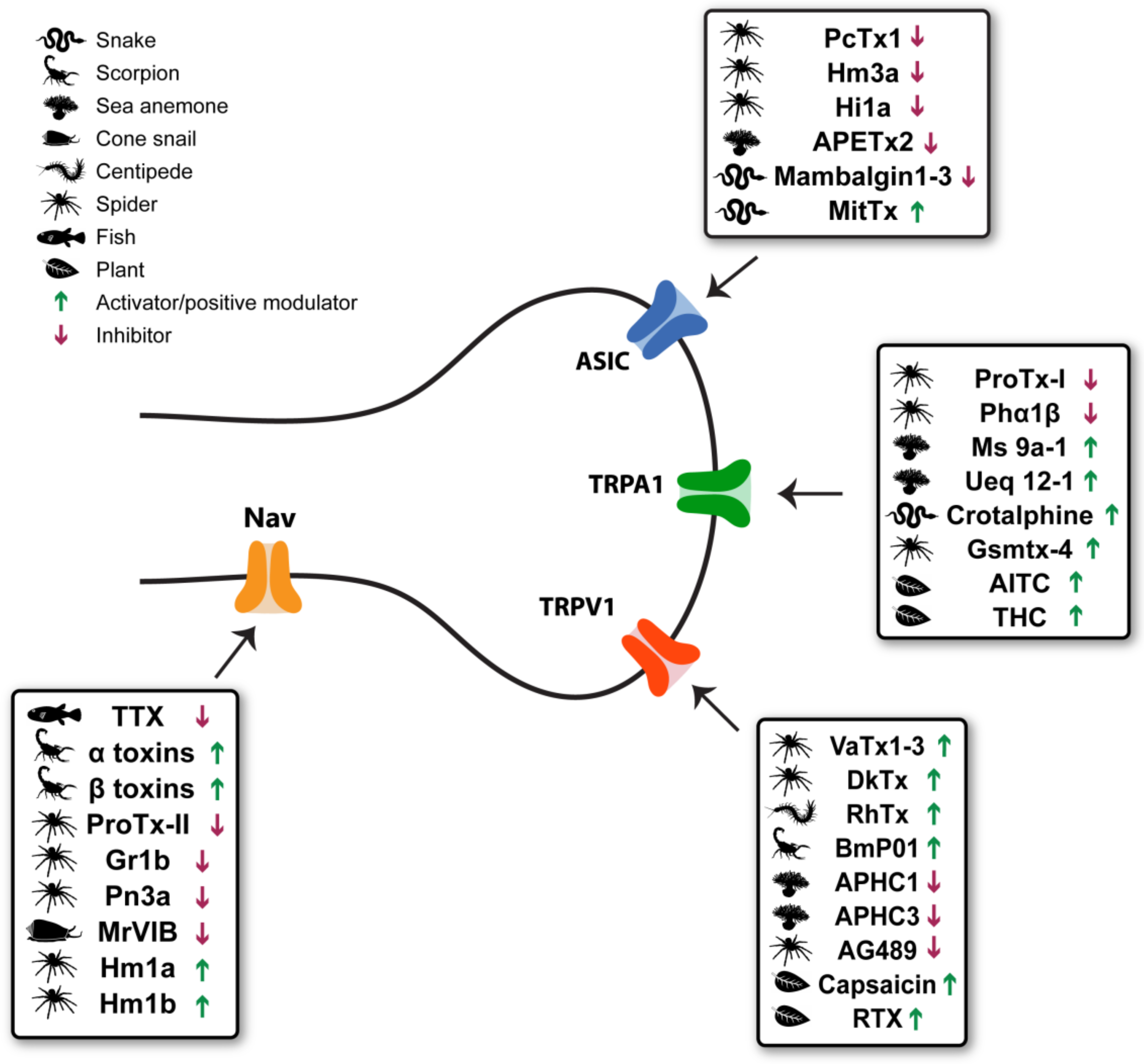{kind=link}
| Toxin | Nociceptive Effect | Pain Model |
|---|---|---|
| Capsaicin Capsicum family | Analgesia (following pain) | Acute (rats) [56,92,93]. Neuropathic pain (humans) [94,95,96,97,98]. |
| RTX Euphorbia resinifera | Analgesia (following pain) | Acute (pigs, mice and rats) [99,100,101,102]. Inflammatory (dogs, rats and mice) [65,101,102]. Cancer-related pain (humans and dogs) [65,103]. |
| VaTx1-3 Psalmopoeus cambridgei | Pain | Acute (mice) [68]. |
| DkTx Ornithoctonus huwena | NA | - |
| RhTx Scolopendra subspinipes mutilans | Pain | Acute (mice) [75]. |
| BmP01 Mesobuthus martensii | Pain | Acute (mice) [76]. |
| APHC1,3 Heteractis crispa | Analgesia | Acute (mice) [55]. Inflammatory (mice) [55] |
| AG489 Agelenopsis aperta | NA | - |
| Toxin | Nociceptive Effect | Pain Model |
|---|---|---|
| ProTx-I Thrixopelma pruriens | NA | - |
| Phα1β Phoneutria nigriventer | Analgesia | Acute [148,150]. Inflammatory (mice) [148,150,153]. Neuropathic (rats) [148,150,153,171]. Post-operative pain (mice) [149]. Cancer-related pain (mice and rats) [151,152,153]. |
| Ms 9a-1 metridium senile | Analgesia | Acute and inflammatory (mice) [144]. |
| Ueq 12-1 metridium senile | Analgesia | Acute and inflammatory (mice) [144,157]. |
| Crotalphine Crotalus durissus terrificus | Analgesia | Acute (mice) [161]. Inflammatory (rats) [158,159,161]. Neuropathic (rats) [160]. Cancer-related pain (rats) [162]. |
| Gsmtx-4 Grammostola spatulata | NA | - |
| Toxin | Nociceptive Effect | Pain Model |
|---|---|---|
| PcTx1 Psalmopoeus cambridgei | Analgesia | Acute (mice) [193]. Inflammatory (mice) [193]. Neuropathic (mice and rats) [193]. Visceral (rats) [194]. |
| Hi1a Hadronyche infensa | NA | - |
| Hm3a Heteroscodra maculata | NA | - |
| APETx2 Anthopleura elegantissima | Analgesia | Inflammatory (rats) [181,210,211]. Post-operative pain, rats [202]. |
| Mambalgin1-3 Dendroaspis polylepis Dendroaspis angusticeps | Analgesia | Acute (mice) [195]. Inflammatory (mice) [195]. |
| MitTx Micrurus tener tener | Pain | Acute (mice) [208]. |
| Toxin | Nociceptive Effect | Pain Model |
|---|---|---|
| Tetrodotoxin Tetraodontidae | Analgesia | Inflammatory (mice and rats) [273,274,276]. Neuropathic (mice and rats) [271,273,275,276]. Visceral (mice and rats) [67,276]. Cancer-related pain (mice and humans) [272,273,277,278]. |
| ProTx-II Thrixopelma pruriens | Analgesia | Acute and inflammatory (rats) [285]. Diabetic neuropathic pain (mice) [284]. |
| β-TRTX-Gr1b Grammostola rosea | Analgesia | Acute and inflammatory (rats) [289]. |
| μ-theraphotoxin-Pn3a Pamphobeteus nigricolor | Analgesia(only when co-administrated with opioids) | Acute and inflammatory (mice and rats) [299]. |
| μO-conotoxin MrVIB Conus marmoreus | Analgesia | Acute (rats) [301]. Inflammatory (rats) [302]. Post-operative pain (rats) [301]. Neuropathic (rats) [302]. |
| δ-theraphotoxin-Hm1a δ-theraphotoxin-Hm1b Heteroscodra maculata | Pain | Pain and mechanical hypersensitivity (mice) [245]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maatuf, Y.; Geron, M.; Priel, A. The Role of Toxins in the Pursuit for Novel Analgesics. Toxins 2019, 11, 131. https://doi.org/10.3390/toxins11020131
Maatuf Y, Geron M, Priel A. The Role of Toxins in the Pursuit for Novel Analgesics. Toxins. 2019; 11(2):131. https://doi.org/10.3390/toxins11020131
Chicago/Turabian StyleMaatuf, Yossi, Matan Geron, and Avi Priel. 2019. "The Role of Toxins in the Pursuit for Novel Analgesics" Toxins 11, no. 2: 131. https://doi.org/10.3390/toxins11020131
APA StyleMaatuf, Y., Geron, M., & Priel, A. (2019). The Role of Toxins in the Pursuit for Novel Analgesics. Toxins, 11(2), 131. https://doi.org/10.3390/toxins11020131