1. Introduction
The genes encoding the subtilase cytotoxin (SubAB) are present in various Shiga toxin-producing
Escherichia coli (STEC) strains [
1,
2,
3]. SubAB was originally found and characterized in the
E. coli O113:H21 strain 98NK2, which was isolated from a patient suffering from hemolytic uremic syndrome (HUS) [
4,
5]. Several virulence profiling studies showed that
subAB genes are often present in sheep and wild ruminant STEC isolates. [
3,
6,
7]. In addition, such STEC did not contain the locus of enterocyte effacement (LEE) [
8,
9,
10]. To our knowledge,
subAB genes are always detected in Shiga toxin (
stx)-positive strains. Therefore, it was hypothesized that the pathogenic function of SubAB is linked to that of Stx [
5,
11]. However, animal studies gave first insights into the pathogenesis by intoxication with only SubAB [
5,
11,
12,
13]. For example, Seyahian et al. [
13] showed that SubAB1 injection in sublethal doses causes severe damages in kidneys, hearts, and livers in rats, which can contribute to HUS pathogenesis. Comparable damages were already described for mice and were linked to HUS symptoms observed in humans [
5,
11]. Therefore, SubAB may be considered as a pathogenicity factor, which contributes to the virulence of STEC.
The genes encoding SubAB1 are located on the virulence plasmid pO113 [
5]. In addition to the plasmid-located
subAB1 genes, the chromosomal variant genes
subAB2-1,
subAB2-2, and
subAB2-3 were described [
5,
6,
14,
15], and further
subAB variant genes were suggested [
16].
SubAB is composed of an enzymatically active A-subunit (SubA) and five B-subunits (SubB), the latter mediating the binding of the toxin to its target cells. Byres et al. [
17] identified
N-glycolylneuraminic acid (Neu5Gc), which is α2,3-linked to a glycan receptor, as a binding target of the B-pentamer. However, latest studies revealed that SubAB may be able to bind to a variety of glycans on the cell surface. In particular,
N-glycans seem to be the main binding partners, and
O-glycans play only a minor role in target cell recognition by SubAB [
18]. Upon binding, the holo-toxin is retrogradely transported to the endoplasmic reticulum, where the A-subunit binds to the endoplasmic Hsp70 chaperone BiP/GRP78 [
19]. SubA specifically cleaves BiP at the L–L motif between its ATPase and substrate-binding domain, causing an unfold protein (UPR) stress response that ultimately leads to apoptosis of the target cell [
19,
20].
The assembly of AB
5 toxins seems to be a crucial step for their functionality. Some members of this family are released from the cell by specific secretion systems as holo-toxin complexes [
21,
22]. Thus, it was assumed that Stx and SubAB also form stable complexes before binding to their targets. However, Pellino et al. [
23] showed that preassembly of Stx1 and Stx2 is not required for successful intoxication. They proposed that binding of the B-subunits to their receptor on the target cell could be a template for the formation of holo-toxin complexes. Funk et al. [
24] demonstrated that the separately expressed A- and B-subunits of different SubAB variants are able to form functional homo- and hetero-complexes with high cytotoxic activity. Currently, it is not known whether SubAB subunits assemble in solution to form stable toxin complexes.
The aim of the current study was to analyze the capability of recombinant SubAB subunits to form homo- and hetero-complexes in vitro. Therefore, effective purification protocols were established, followed by analyzing the activity of the purified proteins by cytotoxicity assays on HeLa cells. Subsequently, the separately expressed and purified recombinant SubAB subunits were analyzed biochemically using far-ultraviolet (UV) circular dichroism (CD) spectroscopy and size-exclusion chromatography (SEC). The homo- and hetero-complex formations were characterized with analytical ultracentrifugation (aUC). If possible, dissociation constants Kd were determined by isothermal titration calorimetry (ITC). In addition, we performed flow cytometry (FACS) analysis to further elucidate binding of SubAB complexes to target cells when no stable toxin complexes were detectable in solution.
3. Discussion
In this study, we demonstrated, for the first time, that toxin complex formations of SubAB variants have different characteristics. Stable complexes not only assembled in solution, but most likely also on the surface of the target cells. Furthermore, our results indicate that a stable pentamer of B-subunits is mandatory for the AB5 complex formation in solution.
AB
5 toxins are structurally characterized by an A-subunit which is predominantly composed of α-helices [
30]. In contrast, the pentameric B-subunits typically contain a higher amount of β-sheets [
31]. Crystal structure analyses of the functional SubAB holo-toxin complex by Le Nours et al. [
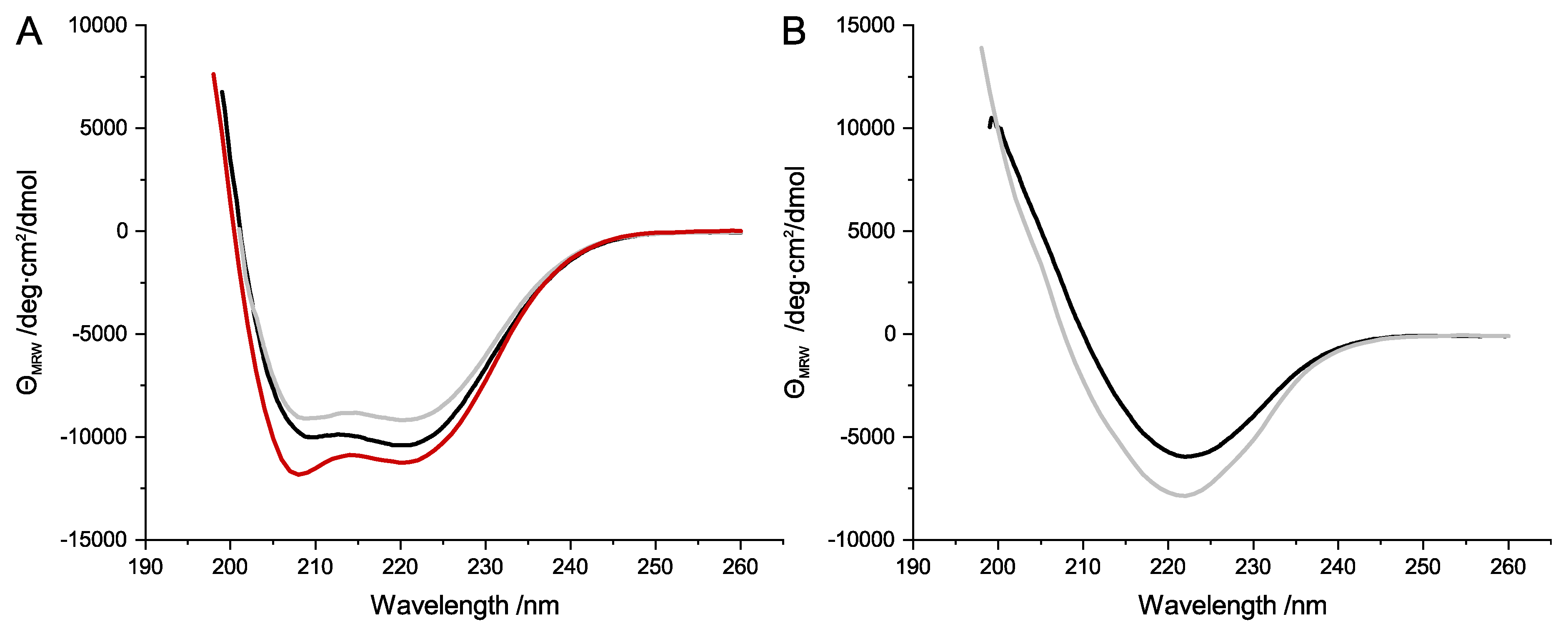
32] showed that this fundamental distribution of secondary structure throughout the protein complex is true for SubAB1-His. In the current study, secondary structure analyses of the separately expressed and purified SubAB subunits revealed that SubA1-His, SubA2-2-His, and SubA2-2 showed typical α-helical signals in the far-UV CD spectra (
Figure 3A). The His-tagged B-subunits also showed the expected pattern of proteins composed of β-sheets (
Figure 3B).
SubA variants und SubB variants demonstrated different thermal stabilities. SubA subunits denatured irreversibly at approximately 65 °C, and SubB subunits denatured irreversibly at about 80 °C. The thermal stabilities indicated that the subunits used in this study were natively folded and stable at the temperatures that were applied throughout the experiments to characterize their complex formation capabilities. In addition, the His-tag had no observable effect on the secondary structure, thermal stability, and oligomerization of SubA2-2.
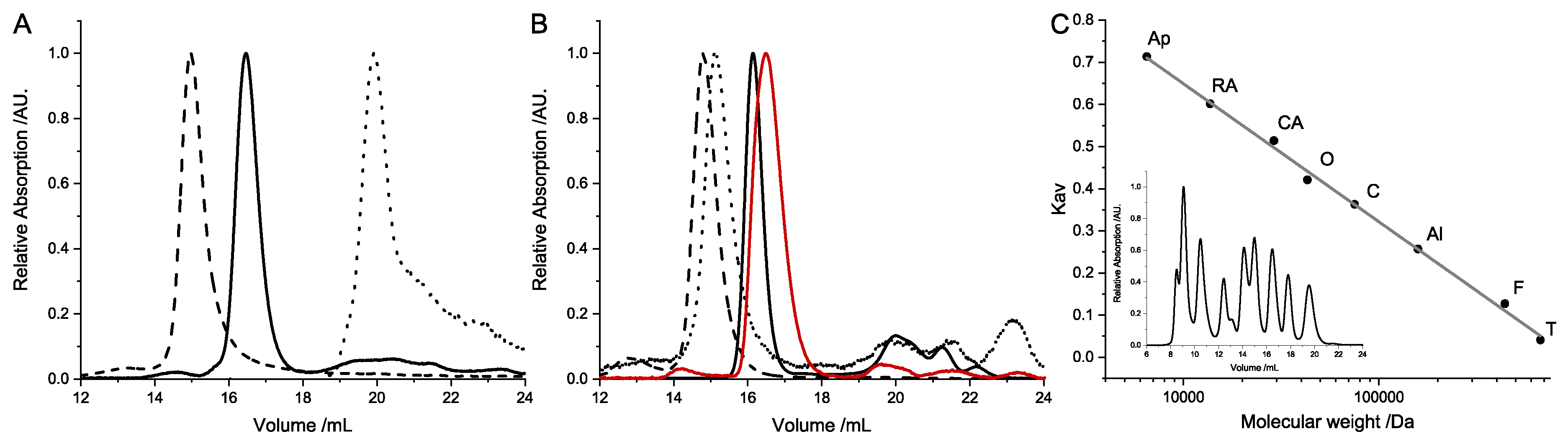
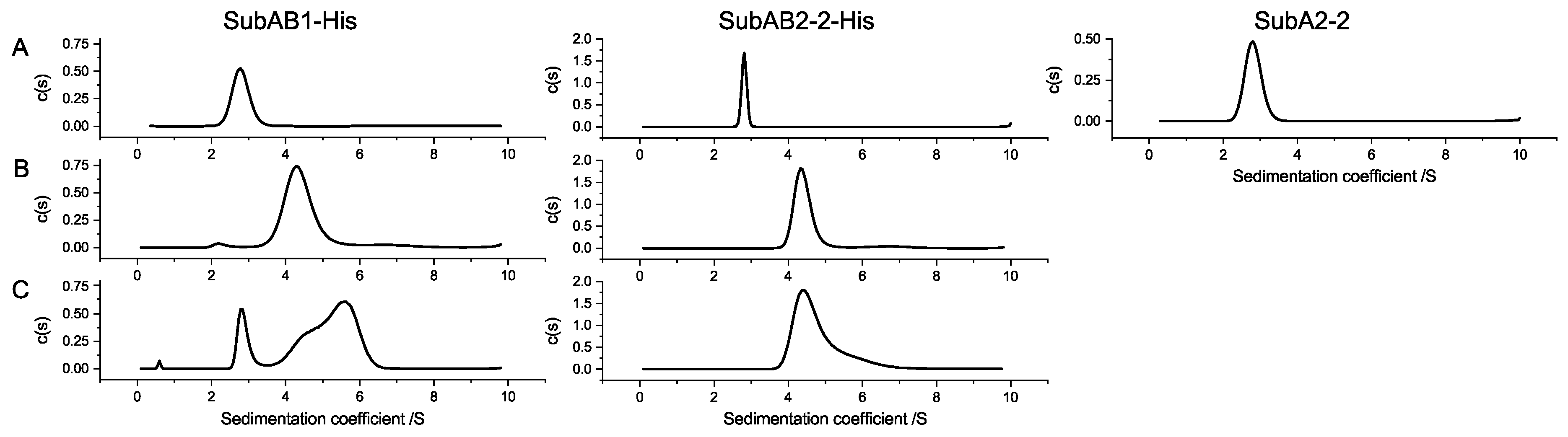
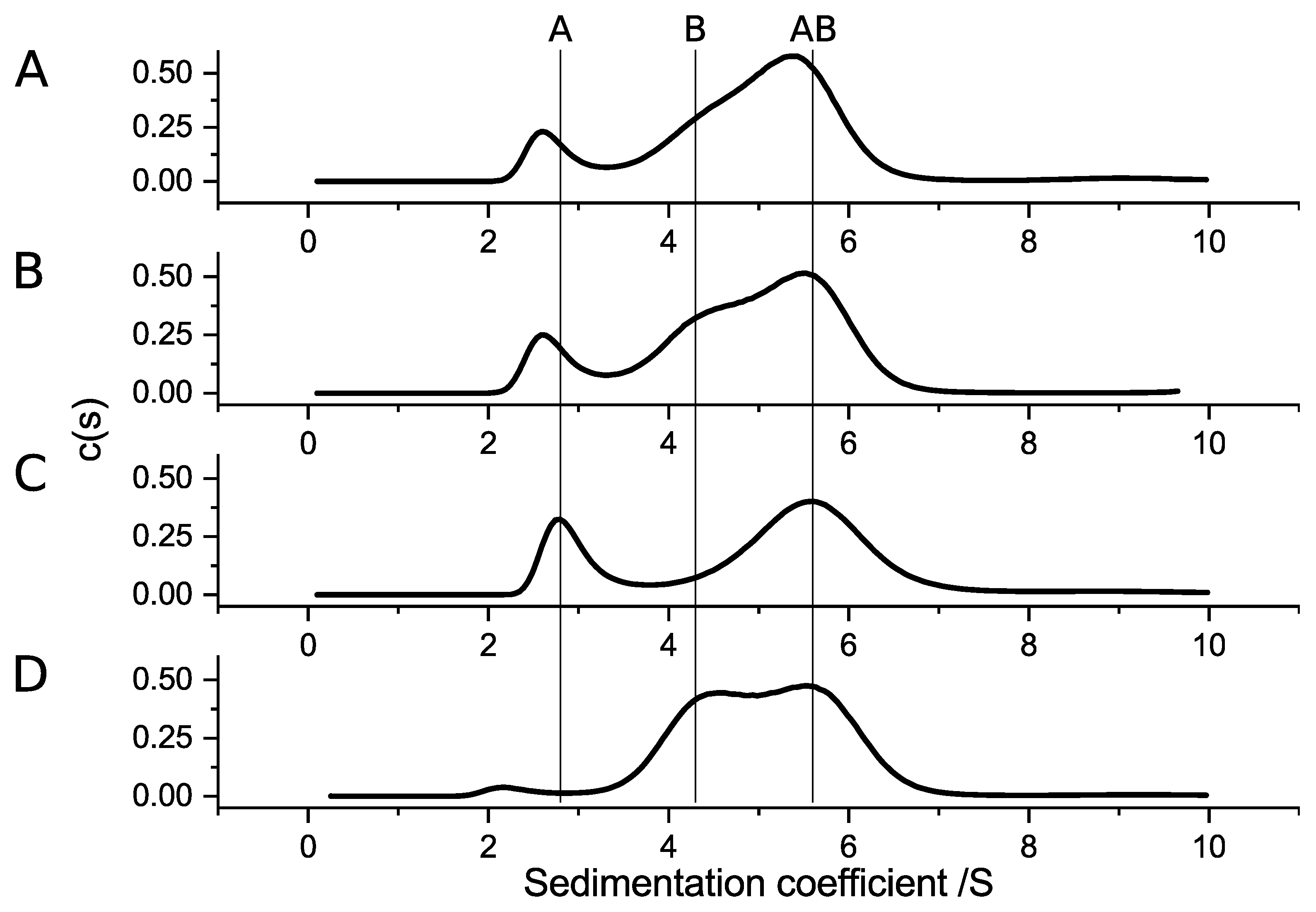
The separated aUC analyses of SubAB1-His subunits resulted in two distinct species, sedimenting with S-values which are comparable to the molecular sizes that were obtained from SEC analysis. This shows that SubA1-His is a monomer and SubB1-His a pentamer. The mixture of both subunits resulted in a new sedimentation species with an S-value and estimated molecular weight that correspond to the formation of an AB
5 complex in solution (
Figure 5 SubAB1-His, left column). The corresponding experiment for SubAB2-2-His showed comparable results for the single runs, but the results of the mixture of SubAB2-2-His did not reveal the formation of a peak with lager sedimentation coefficient, which would indicate an interaction species. Moreover, there was a clearly tailed peak observable at 4.8 S, which can be interpreted as the free B-subunits showing transient interactions with the A-subunit; however, no clear complex formation was observed for SubAB1-His (
Figure 5 SubAB2-2-His, middle column) [
29]. Possible reasons for the different complex-forming abilities could be the amino-acid composition of the variants. As mentioned earlier, the overall identity of the amino-acid sequence between the SubA and SubB subunits is 94.5% and 92.4%, respectively, which corresponds to 18 different amino acids in SubA2-2-His compared to SubA1-His and nine differences for SubB2-2-His compared to SubB1-His. No particular region was identifiable in the proteins where the changes in the sequence pile up. In contrast, the alterations were rather evenly distributed throughout the whole protein. In total, 13 amino acids throughout SubA1 were identified to be part of the interaction regions of the toxin complex [
32]. None of them were different in SubA2-2-His. The same was true for the eight amino acids of the B-subunits, which were identified to be part of the interface between A- and B-subunits. However, in very close proximity to the AB interface, which is located between amino acids 91 and 94 in SubB, the proline at position 96 is altered to a leucine in SubB2-2 (amino-acid count includes the signal peptide). These two amino acids do not discriminate in their chemical properties but in their size. Based on this finding, we suspect that the missing ability of SubAB2-2-His to form stable toxin complexes in solution is caused by the leucine at position 96.
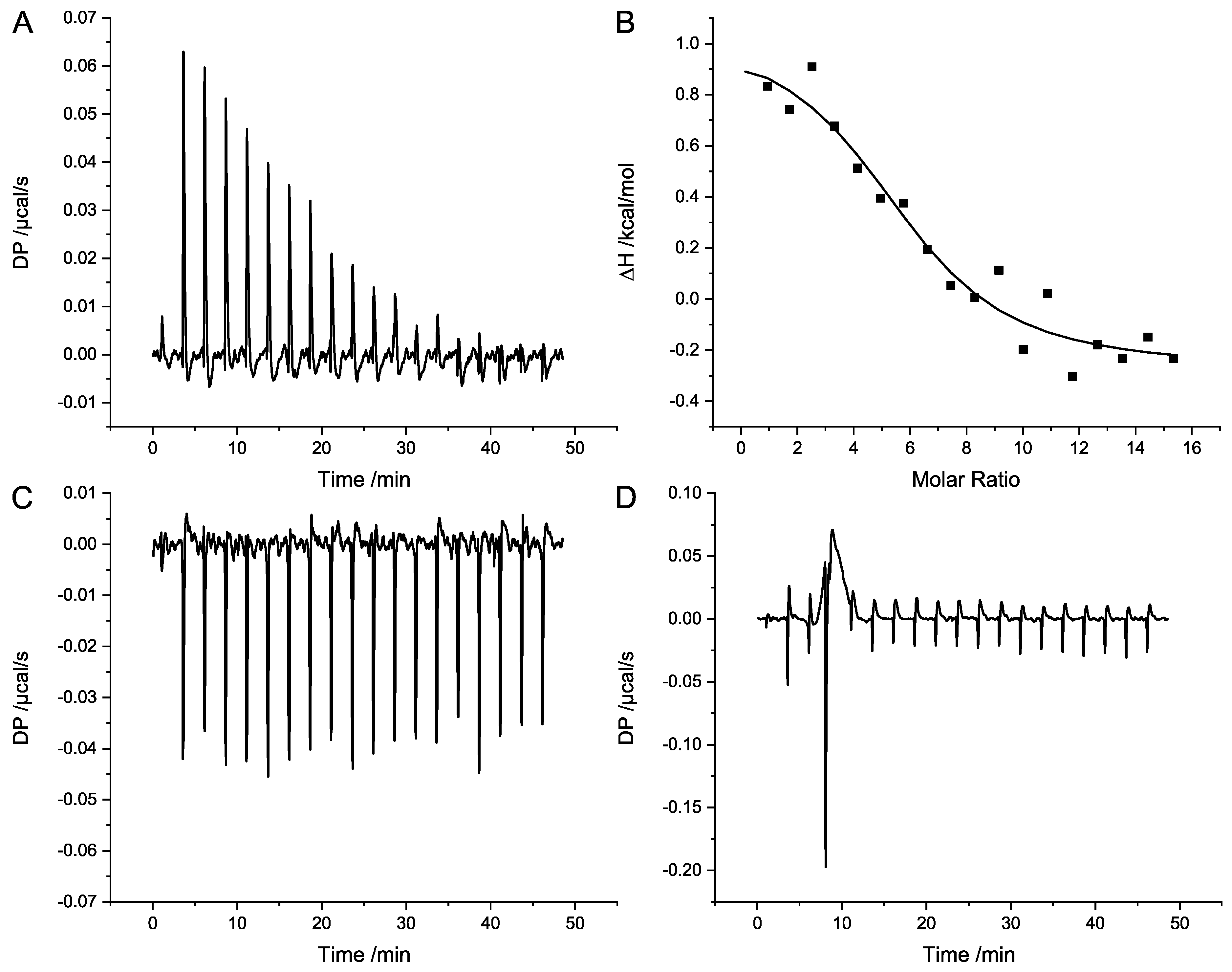
The analyses of complex formation of SubA1-His and SubB1-His with ITC revealed a dissociation constant in the lower micromolar range. This confirms the observation that this variant is able to form homo-complexes in solution. The corresponding experiments for SubAB2-2-His did not result in any binding signal given the applied condition, leading to the assumption that the SubAB2-2-His is not able to form stable complexes in solutions, or, if so, then only with a much larger dissociation constant. As described earlier, it is thought that AB
5 toxins are assembled in complexes to be able to bind to their target cells and mediate their toxicity. Nevertheless, Pellino et al. [
23] showed that the preassembly of the AB
5 toxin complex is not necessary for intoxication by Stx. Furthermore, they showed that the assembly occurs at the receptor interface. Taking this new model for the functionality of AB
5 toxins into account, our results led to the hypothesis that SubAB2-2-His forms a stable and functional toxin complex on the surface of the target cells.
To prove this suggestion, flow cytometry experiments were performed to understand the binding behavior of the separate SubAB2-2-His subunits. Pre-incubation resulted in a clear increase in the fluorescence for *SubA2-2-His + SubB2-2-His (
Figure 7), which could be interpreted as better binding and could be attributed to the formation of AB
5 complexes prior to the binding event. This is also supported by the observation made by the aUC experiments, which suggests the formation of transient SubAB2-2-His complexes (
Figure 5 SubAB2-2-His, middle column). Contrarily, the results for SubA2-2-His + *SubB2-2-His showed that there is no positive observable effect on the binding. The observed reduction in fluorescence could be a consequence of the AB
5 complex formation and binding to HeLa cells, as changes in the surrounding of fluorescent dyes tend to affect their signal [
33,
34]. In the cell-bound state of the complex, the fluorophores that are linked to *SubB2-2-His are sandwiched between the cell wall and the associated A-subunit. This constellation could shield the fluorophores and result in a decrease of the signal. Thus, this indicates that the formation of transient toxin complexes during the pre-incubation facilitates the binding to HeLa-cells and that stable SubAB2-2-His complexes can be formed at the target cell.
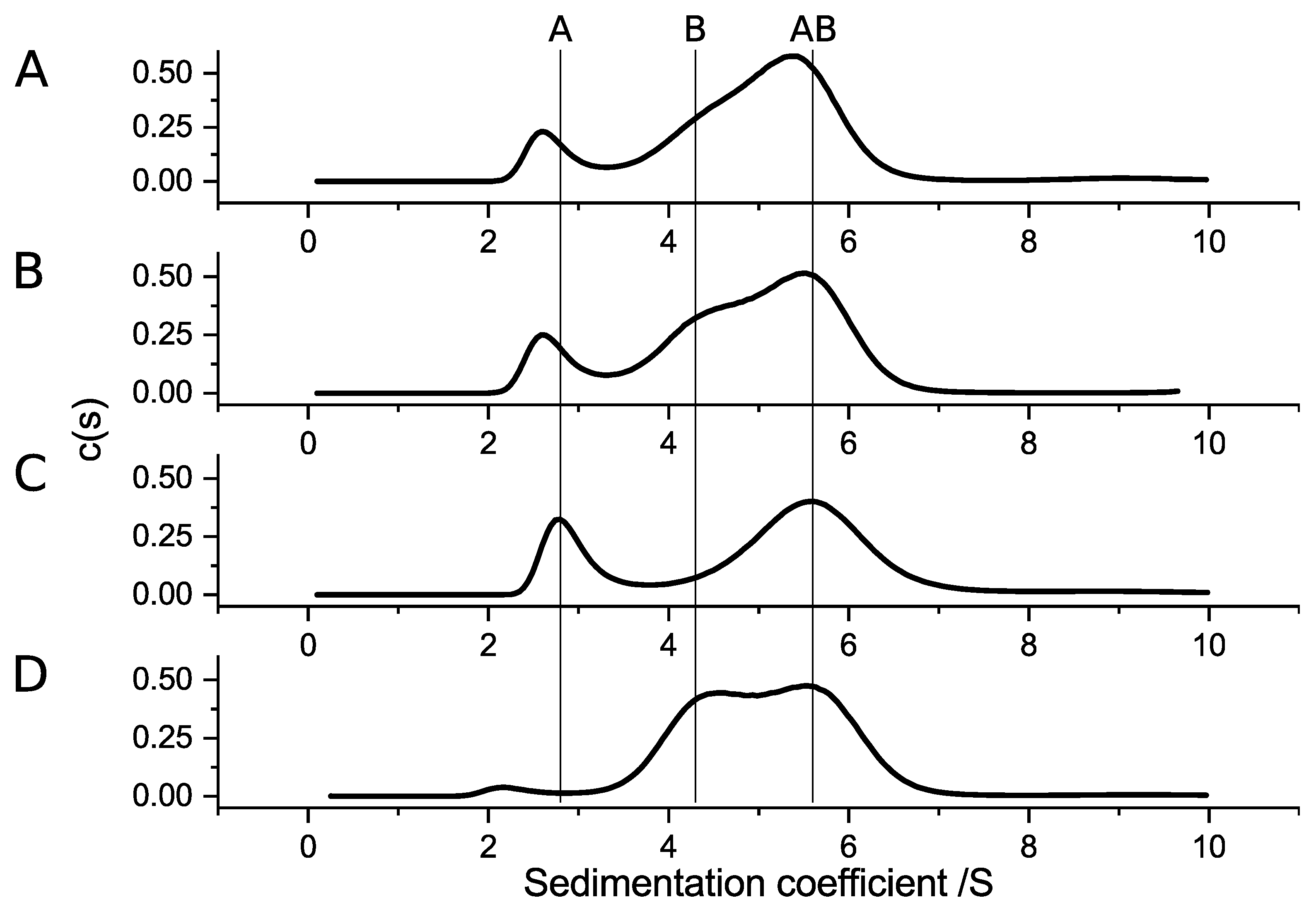
Furthermore, Funk et al. [
24] showed, in cell culture-based assays, that SubAB subunits of different variants successfully intoxicated Vero cells. This suggests that the formation of hetero-complexes within the SubAB family is possible. Here, we investigated if the subunits of SubAB1-His, SubAB2-2-His, and SubA2-2 were able to form hetero-complexes (
Figure 8). All combinations showed clear complex formation. Based on the results obtained in the previous experiments, we expected complex formation of all A-subunits with SubB1-His, because, so far, stable complexes were observed only for SubAB1-His. Furthermore, we assumed that SubB2-2-His is not able to form stable complexes in solution due to its amino-acid composition. However,
Figure 8 shows different behaviors of the subunits. Specifically, while the complex formation of SubA2-2-His with SubB1-His and SubA2-2 with SubB1-His matched our expectations, this was not true for the complexes observed for SubA1-His with SubB2-2-His and for SubA2-2 with SubB2-2-His. Based on the size of the peaks corresponding to the AB
5 hetero-complexes, samples containing SubB1-His as a B-subunit formed larger amounts of complexes compared to the mixtures with SubB2-2-His. In addition, SubA2-2 bound much better to the B-subunits (
Figure 8C,D) than SubA2-2-His. This indicates that the His-tag linked to the toxin subunits has an effect on the biochemical properties.
Conclusively, we characterized the AB5 toxin complex assembly of different SubAB variants using aUC and ITC experiments. In our experimental settings, readily assembled B pentamers were mandatory for the complex formation. Furthermore, in comparison to other SubAB variants, SubAB1-His revealed the most stable complexes in solution. This is most likely attributed to the SubB1-His subunit, as other SubA variants also formed stable complexes with this subunit. SubAB2-2-His tended to form transient complexes in solution, which simplifies the binding to target cells. Nevertheless, stable complexes of this variant were not observed in solution, leading to our hypothesis that the cell surface is the main location of stable complex formation for SubAB2-2-His. However, further studies are required to fully understand the different properties of SubAB variants and their complex formations. In addition, it remains unclear how SubAB2-2 forms functional complexes which lead to a comparable intoxication of HeLa cells, as observed for SubAB2-2-His; thus, this requires further investigation.
4. Materials and Methods
4.1. Cloning, Expression, and Purification of SubAB Variant Subunits
4.1.1. Cloning of SubA2-2
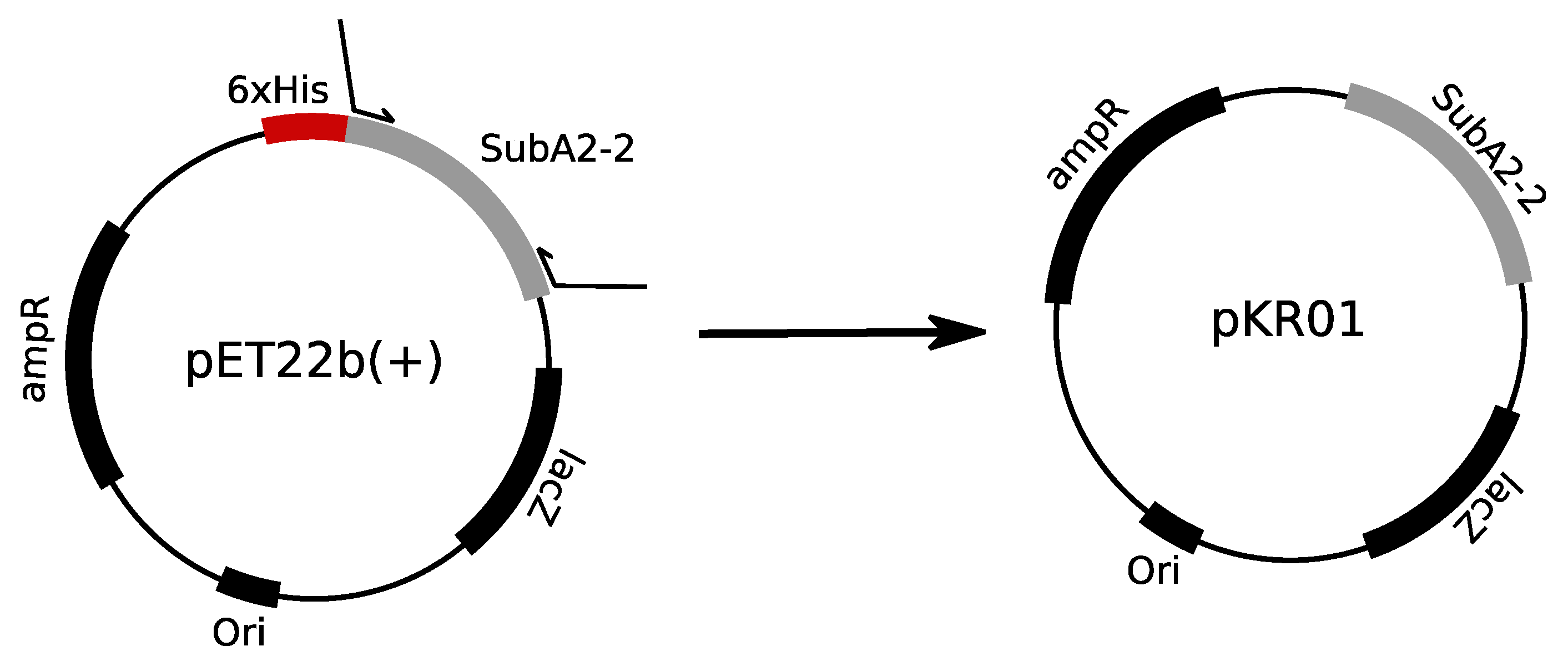
For expression of His-tag free SubA2-2, plasmids pKR01 was constructed. Therefore, the
subA insert of the expression plasmids described by [
24] were amplified in standard 50-µL PCR reactions using the Phusion High Fidelity Polymerase (Thermo Fisher Scientific, Waltham, MA, USA) with the primer pairs listed in
Table 3 (Eurofins Scientific, Luxemburg, Luxemburg) according to the manufacturer’s manual to ensure that the sequences of the His-tagged and untagged subunits used in the previous and current study were identical. Prior to digestion with the restriction enzymes
XbaI and
XhoI (both Thermo Fisher Scientific, Waltham, MA, USA), the insert was purified with the Wizard
® SV Gel and PCR clean-up kit (Promega, Fitchburg, WI, USA) according to the manufacturer’s recommendations (Handbook 2009, Promega, Fitchburg, WI, USA). Digestion of the insert and the pET16b(+) vector (Novagen, Darmstaft, Germany) was performed as recommended by the web tool DoubleDigest calculator of Thermo Scientific. After inactivation of the restriction enzymes at 80 °C for 15 min, the insert was again purified with the Wizard
® SV Gel and PCR clean-up kit. The digested vector was loaded on a 1% (
w/
v) agarose gel, separated at 120 V with 90 min run time, and subsequently extracted with the Wizard
® SV Gel and PCR clean-up kit. Ligation was performed with a 1:5 molar ratio of vector to insert—applying 20 ng of vector—and the T4 Ligase (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturers’ manual (Manual, 2014, Thermo Fisher Scientific, Waltham, MA, USA). The ligated DNA was transformed in chemical competent
E. coli XL-1 Blue; positive clones were identified via colony PCR (using Primer pET16b_seq_for and pET16b_seq_rev,
Table 3). A colony PCR was conducted in a total reaction volume of 20 µL containing 2 µL of template, 200 µmol/L dNTPs, 200 nM of each primer, 0.5 U of
Taq polymerase, and 1× ThermoPol buffer. The templates were generated by suspending one colony in 20 µL of sterile water and incubation at 95 °C for 10 min of the cell suspension. Positive clones were verified by Sanger sequencing using the same primers as for the colony PCR after plasmid preparation (QIAprep Spin Mini Prep kit, Handbook, July 2006, Qiagen, Venlo, Netherlands). Sanger sequencing was performed with a CEQ™8000 automated sequencer (Beckman Coulter, Brea, CA, USA) as described previously [
24]. The cloning strategy is depicted in
Figure 9. Correct expression plasmids were transformed in
E. coli C41 (DE3) to deliberately allow expression of the subunit.
4.1.2. Expression and Cell Lysis of SubAB Variants
Expression of SubA2-2 subunit was performed in an auto-induction medium ZYM-5052 according to Studier [
25]. The volume of a typical expression culture was 400 mL, which was composed of 200 mL of 2× ZY (2% (
w/v) tryptone, 1% (
w/v) yeast extract, pH 7.0), 8 mL of 50× M (1.25 mol/L Na
2HPO
4, 1.25 mol/L KH
2PO
4, 2.5 mol/L NH
4Cl, 0.25 mol/L Na
2SO
4), 8 mL of 50× 5052 (2.5% (
w/v) glycerol, 0.25% (
w/v)
d-(+)-glucose, 1.0% (
w/v) lactose), 0.8 mL 1 mol/L MgSO
4, and 150 µg/mL ampicillin. The expression culture was inoculated 1:50 with an overnight culture. Prior to inoculation, the overnight culture was pelleted (5000×
g, 5 min, room temperature (RT)) and the cells were resuspended in fresh medium. Growth of the cultures took place at 37 °C with shaking at 180 rpm until an OD
600 nm of about 1.5 was reached and the auto-induced expression started. Subsequently, the culture was incubated for protein expression overnight at 20 °C and 180 rpm shaking. Expression was stopped by incubating on ice for 20 min. After that, the expression was harvested by centrifugation at 4000×
g, 15 min, 4 °C. To remove residual medium components, cell pellets were washed in 30 mL of washing buffer (50 mmol/L Tris/HCl pH 7.5, 100 mmol/L NaCl) and then stored at −20 °C until purification. Expression of SubAB-His subunits was performed as described by [
24].
Cell lysis of all expressions was performed in the first buffer using the subsequent purification. The cell pellets were resuspended in 5 mL of buffer/1 g cell pellet and supplemented with 1 mg/mL lysozyme. After an incubation of 1 h on ice, the cells were disrupted by sonication in 12 cycles with 10 s on time and 20 s off time, with an amplitude of 70% and a microtip at a Sonifier® SFX150 Cell disruptor (Branson Ultrasonics Corporation, Danbury, CT, USA). The lysate was cleared by centrifugation at 30,000× g, 4 °C for 45 min and later used for purification.
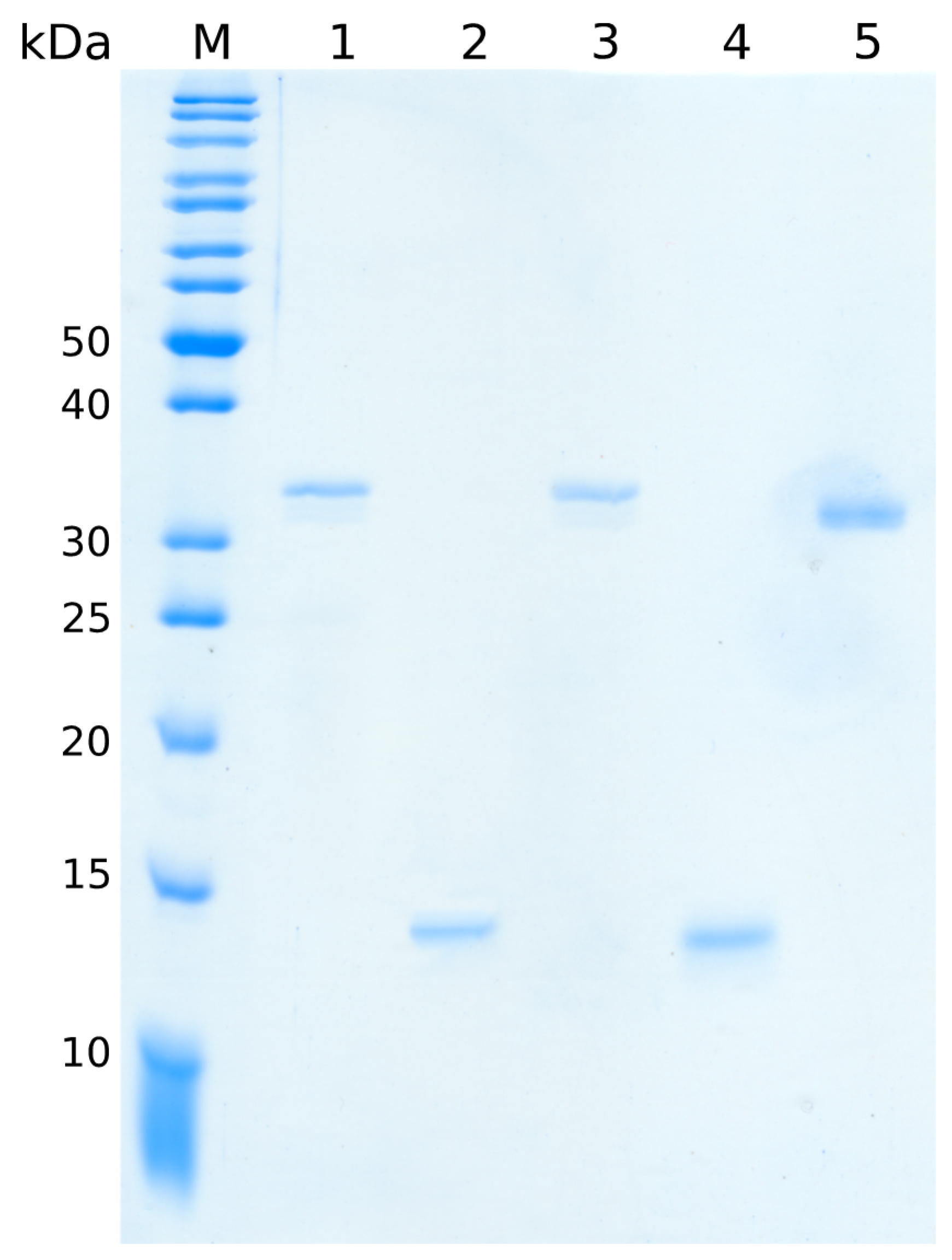
4.1.3. Purification of Recombinant Subtilase Subunits
SubA-His subunits were purified with gravity flow His-columns according to the protocol by Funk et al. [
24]. For the removal of imidazole and remaining impurities of the sample, the SubA-His subunits were run on a Superdex75pg gel filtration column in standard PBS supplemented with 10% glycerol to increase the protein stability during storage (GE Healthcare, Chicago, IL, USA).
The SubA2-2 subunit was purified with a two-column approach on an ÄKTA pure system (GE Healthcare, Chicago, IL, USA) consisting of the cation exchange SPff column and a gel filtration Superdex75pg column (GE Healthcare Chicago, IL, USA). The cleared lysate was loaded via a 50-mL superloop on the first column equilibrated in SP1A buffer (50 mmol/L Tris/HCl pH 7.5, 5 mmol/L EDTA). After a washing step with five column volumes (CVs) of buffer A, elution was performed with a linear gradient against SP1 B buffer (50 mmol/L Tris/HCl pH 7.5, 5 mmol/L EDTA, 1 mol/L NaCl) and 2-mL fractionation. Protein-containing elution fractions were analyzed with 12.5% (
v/v) SDS-PAGE (180 V, 35 mA per gel, 80 min), and all SubA2-2 positive fractions were pooled for polishing on a Superdex75pg column. Gel filtration was performed in standard PBS with 10% (
v/v) glycerol. After identification by SDS-PAGE and pooling of the target protein-containing fraction, the pool was concentrated with Amicon Ultra-15 spin columns cut off at 10 kDa (Merck Milllipore, Burlington, MA, USA), aliquoted, and stored at −70 °C. Similar to the His-tagged A subunit, all His-tagged B-subunits were purified following the protocol by [
24] with an additional gel filtration step on a Superdex75pg in PBS + 10% (
v/v) glycerol.
4.1.4. Concentration Detection
For all experiments, the concentration of the analyzed proteins was determined spectroscopically by measuring the absorption at 280 nm using a Nanodrop2000 device (Thermo Fisher Scientific, Waltham, MA, USA). The length of all subunits, molecular size, and corresponding extinction coefficient were calculated with ExPasy/Protparam and are given in
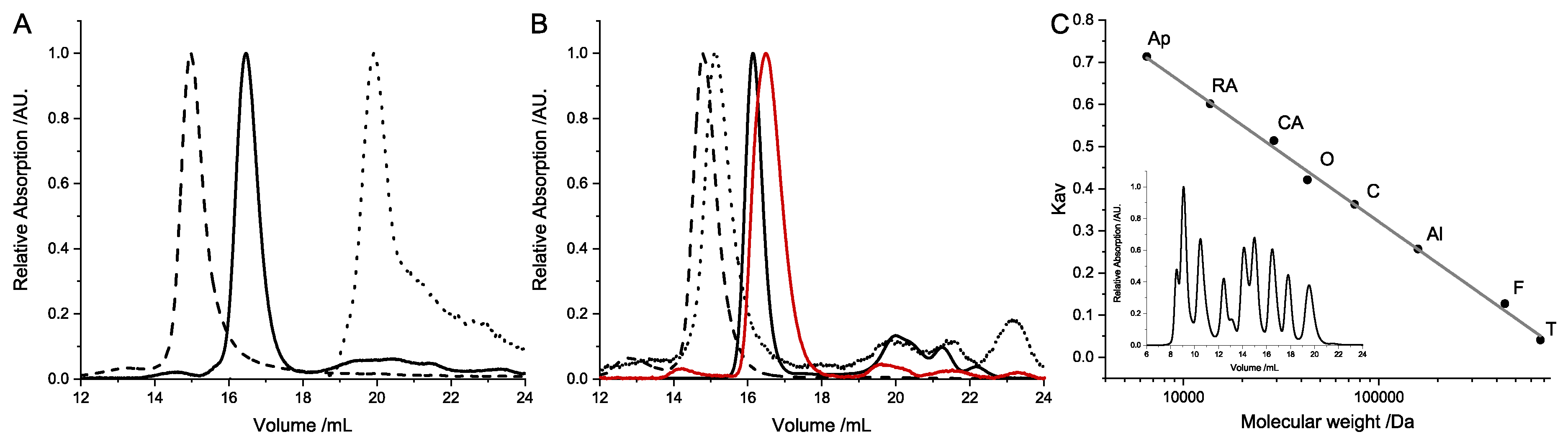
Table 4.
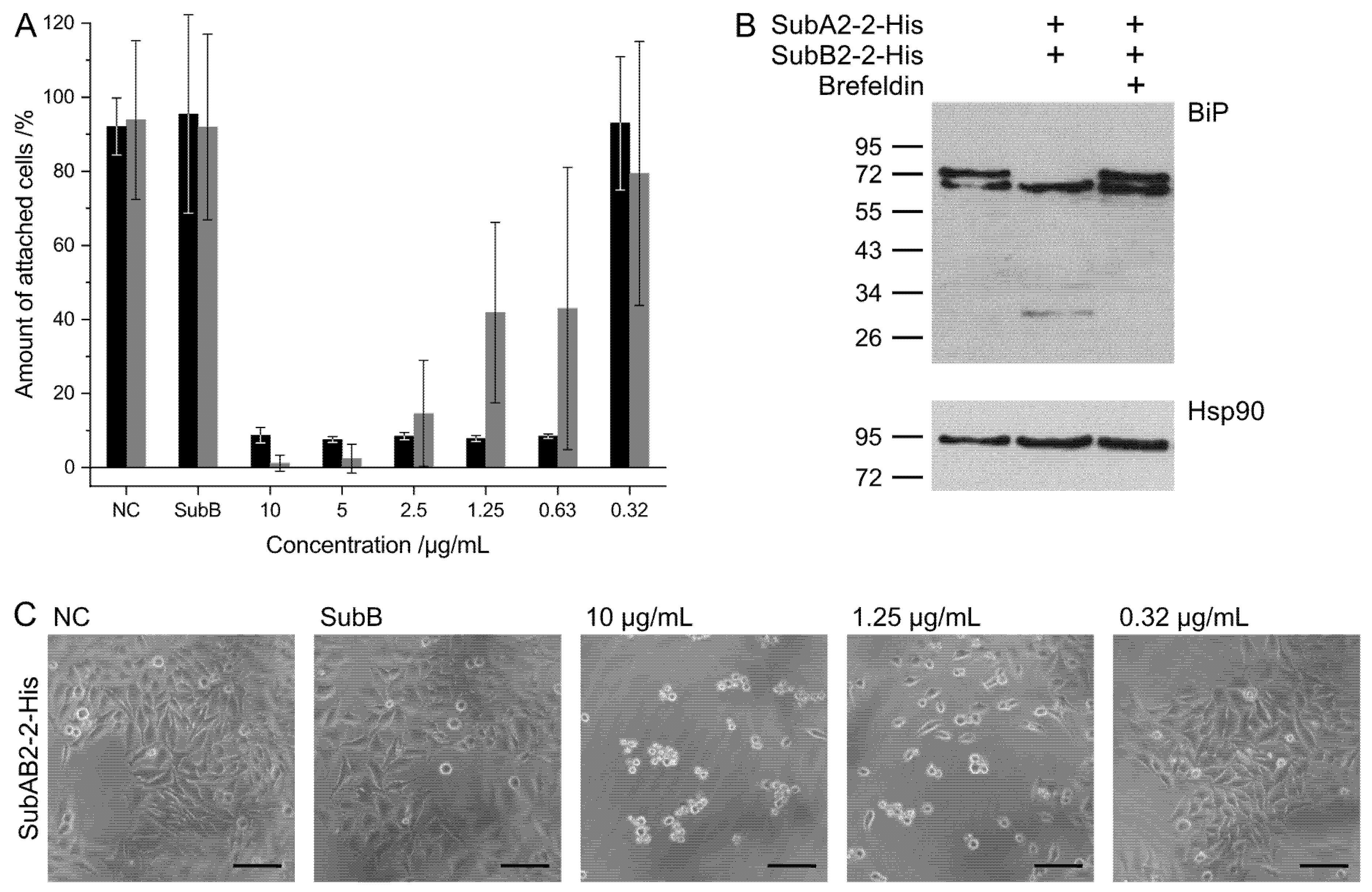
4.2. Cytotoxicity Assay
The determination of the cytotoxicity of the respective SubAB complexes and separate subunits, assays with HeLa cells were performed as described elsewhere [
24]. The detection of the assays was either performed as batch detection using crystal violet staining or direct counting of rounded and unaltered cells after 72 h of incubation with the applied toxins. For intoxication, the separate SubAB subunits were mixed in a 1:5 molar ratio, and total protein concentrations from 10 µg/mL to 0.32 µg/mL were transferred to the epithelial cells. This ratio was chosen to simulate the addition of AB
5 holo-toxin complexes. For calculation of the average and standard deviation for cytotoxicity, three independent experiments (biological replicates) with three technical replicates each were performed.
4.3. Detection of BiP Cleavage
Verification of BiP cleavage in the intoxicated HeLa cells was performed with 1 × 106 cells in 12.5 mL, seeded in 12-well plates distributed to 8 × 104 cell per well. After overnight growth, the cells were washed one time with PBS and incubated with serum-free MEM or 10 µmol/L Brefeldin A (Selleckchem, Munich, Germany) in serum-free MEM for 30 min. Subsequently, the toxin was added, and the cells were further incubated for 4 h. In total, 10 µg/mL toxin was added to the cells containing the A- and B-subunit in a 1:5 molar ratio. After another washing step with PBS, cells were detached by adding 2.5× Laemmli sample buffer containing DTT and mechanically removed from the surface. Cells were transferred in reaction tubes to be incubated at 95 °C for 10 min. Then, 40 µL of each sample was applied to a 12.5% SDS PAGE. Following this, the proteins were blotted onto a nitrocellulose membrane, and BiP was detected using anti-GRP78/BiP rat (1:1000, Santa Cruz Biotechnology, Dallas, TX, USA) and anti-rat-HRP (1:2500, Cell Signaling Technology, Danvers, MA, USA). The detection was performed by enhanced chemiluminescence (ECL, Millipore, Burlington, MA, USA). For loading control, Hsp90 was detected after stripping of the blot using anti-Hsp90-mouse (1:1000, Santa Cruz, Dallas, TX, USA) and mBP (1:2500, Santa Cruz, Dallas, TX, USA).
4.4. Size-Exclusion Chromatography
Size-exclusion chromatography was performed in standard measurement buffer (50 mmol/L Tris/HCl pH 7.5, 150 mmol/L NaCl, 10 mmol/L MgCl2) on a Superdex200increase 10/30 GL column (GE Healthcare, Chicago, IL, USA). For an estimation of the molecular size and oligomerization of the separate subunits, the column was calibrated with blue dextran, aldolase, ovalbumin, conalbumin, carbonic anhydrase, and ribonuclease A (GE Healthcare HMW/LMW calibration kit, GE Healthcare, Chicago, IL, USA). The molecular weight was calculated by the determination of the Kav value. All measurements were performed at least thrice.
4.5. Analytical Ultracentrifugation
Oligomerization and the formation of AB
5 toxin complexes were analyzed by analytical ultracentrifugation (aUC). For each variant, the A- and B-subunits were analyzed independently and in a 1:5 molar mixtures to detect the formation of complexes. All measurements were performed in standard measurement buffer. The sedimentation of the samples was monitored in an Optima-XL-I Beckman centrifuge at 42,000 rpm with absorption spectra (Beckman Coulter, Brea, CA, USA). Data analysis was performed with SEDFIT v. 15.01b (2015) by Peter Schuck [
27]. Viscosity and density of the solvent were calculated with SEDNTERP v.1.09 (2006) by Hayes, Laue, and Philo [
35].
4.6. CD Spectroscopy
Experiments were performed at a Jasco J715 CD Spectrometer with a PTC-348 WI Peltier Unit using default settings (Jasco, Pfungstadt, Germany). Prior to the measurements, all samples were dialyzed in standard PBS buffer overnight. After centrifugation at maximal speed in an Eppendorf centrifuge, the concentration was detected with a Nanodrop2000 (Thermo Fisher Scientific, Waltham, MA, USA) and 200 µg/mL samples were prepared for measurement. Far-UV spectra were measured from 260 nm to 195 nm at 25 °C, and temperature transitions, monitored at 220 nm for the A-subunits and 228 nm and 218 nm for B-subunits, were recorded from 25 °C to 95 °C. Far-UV CD spectra were recorded with 100 nm/min of continuous scanning, 0.1 nm of data pitch, 4 s of response time, 1 nm of band width, and 10 accumulations. Raw data were recorded in millidegrees, and the signal was converted to the mean residue ellipticity (deg∙cm2/dmol). Temperature transitions were recorded with 1 °C/min of heating rate, 0.1 °C of data pitch, 1 s of response time, and 1 nm of band width. To evaluate the melting or denaturing temperature, all transition measurements were fitted with a simple logistic fit using OriginPro2019 from OriginLab (Northampton, MA, USA).
4.7. Isothermal Titration Calorimetry
To analyze the complex formation in vitro, isothermal titration experiments were performed using a Malvern PEAKCal device. All samples were prepared by dialysis overnight against the standard measuring buffer and concentrated with Amicon spinfilters (Merck Millipore, Burlington, MA, USA) to adjust concentrations. The standard assay settings were 750 rpm for stirring, with 5 µW used as the reference power; after 60 s of initial delay, the injection process started with 0.4 µL and an injection duration of 0.8 s. The first injection was followed by 18 injections with 2 µL and a duration of 4 s with 150 s of spacing. For all titrations, 300 µL of 4 µmol/L SubA was titrated with 20 µL of 320 µmol/L SubB solution in 19 steps at 25 °C. Evaluation of the titrations was performed with the MicroCal evaluation software from Malvern (Worcestershire, UK).
4.8. Flow Cytometry
To investigate cell surface binding, purified proteins were labeled with DyLightTM488 NHS ester (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacture’s protocol (Handbook 2011). After the labeling reaction, unbound dye was removed using Zeba Spin Desalting Columns (Thermo Fisher Scientific, Waltham, MA, USA). Activity of labeled components was verified via intoxication assays on HeLa cells and was comparable to the unlabeled variants. The labeled compounds were indicated by asterisk *.
HeLa cells were detached from culture plates using 25 mmol/L EDTA in PBS for 30 min at 37 °C and washed with PBS twice. Cells were aliquoted to 2 × 105 cells/tube, and binding was performed with the indicated amount of labeled proteins for 2 min in PBS on ice. For the binding of pre-incubated toxins, the two protein subunits were pre-incubated together for 30 min in PBS on ice prior the addition to HeLa cells. After toxin binding, cells were washed with PBS once and subjected to flow cytometric analysis using the BD FACSCelesta (BD Biosciences, Franklin Lakes, NJ, USA). As a negative control, untreated Hela cells were used. Data were analyzed using Flowing software (version 2.5.1) by Perttu Terho.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}