Hadrurid Scorpion Toxins: Evolutionary Conservation and Selective Pressures

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
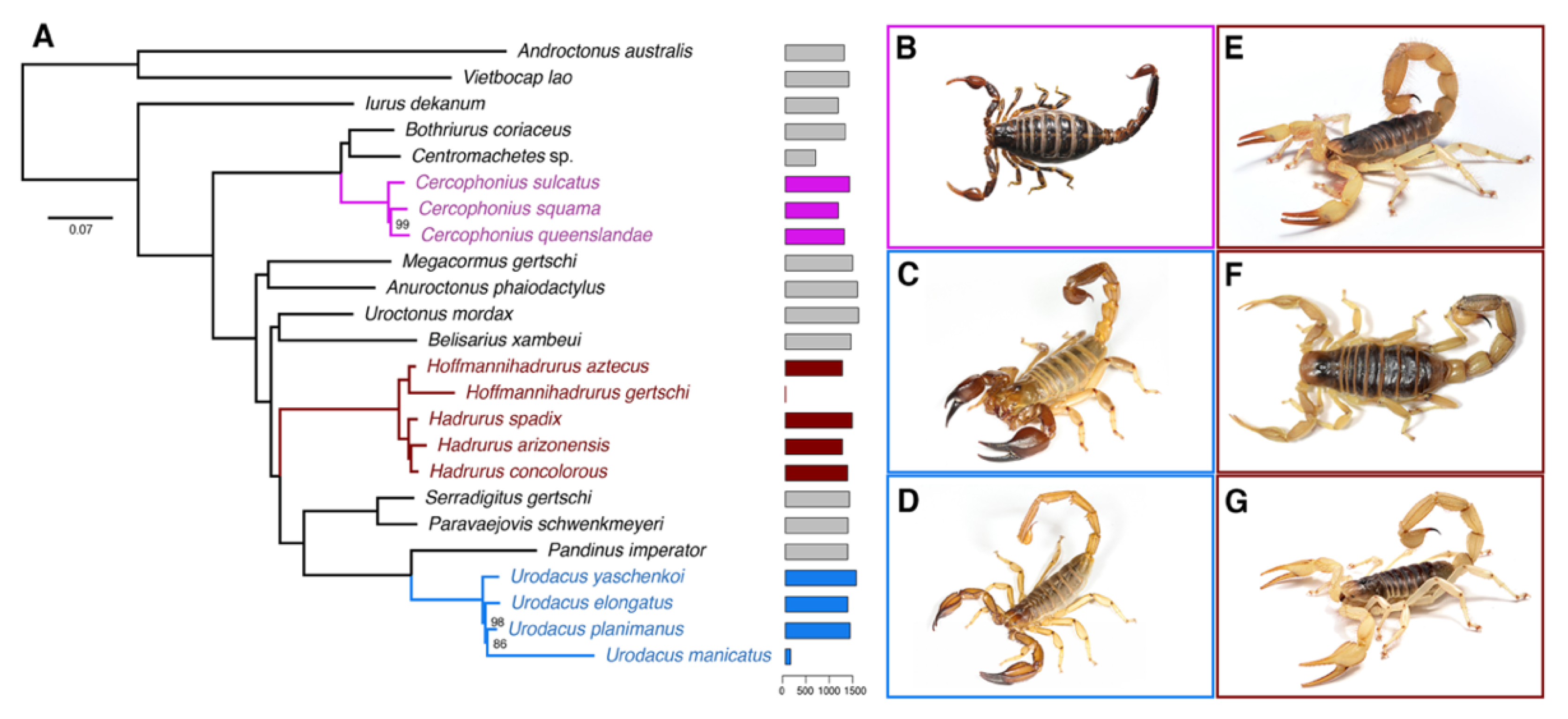
2.1. Hadrurid Phylogenomic Tree and Divergence Time Estimate
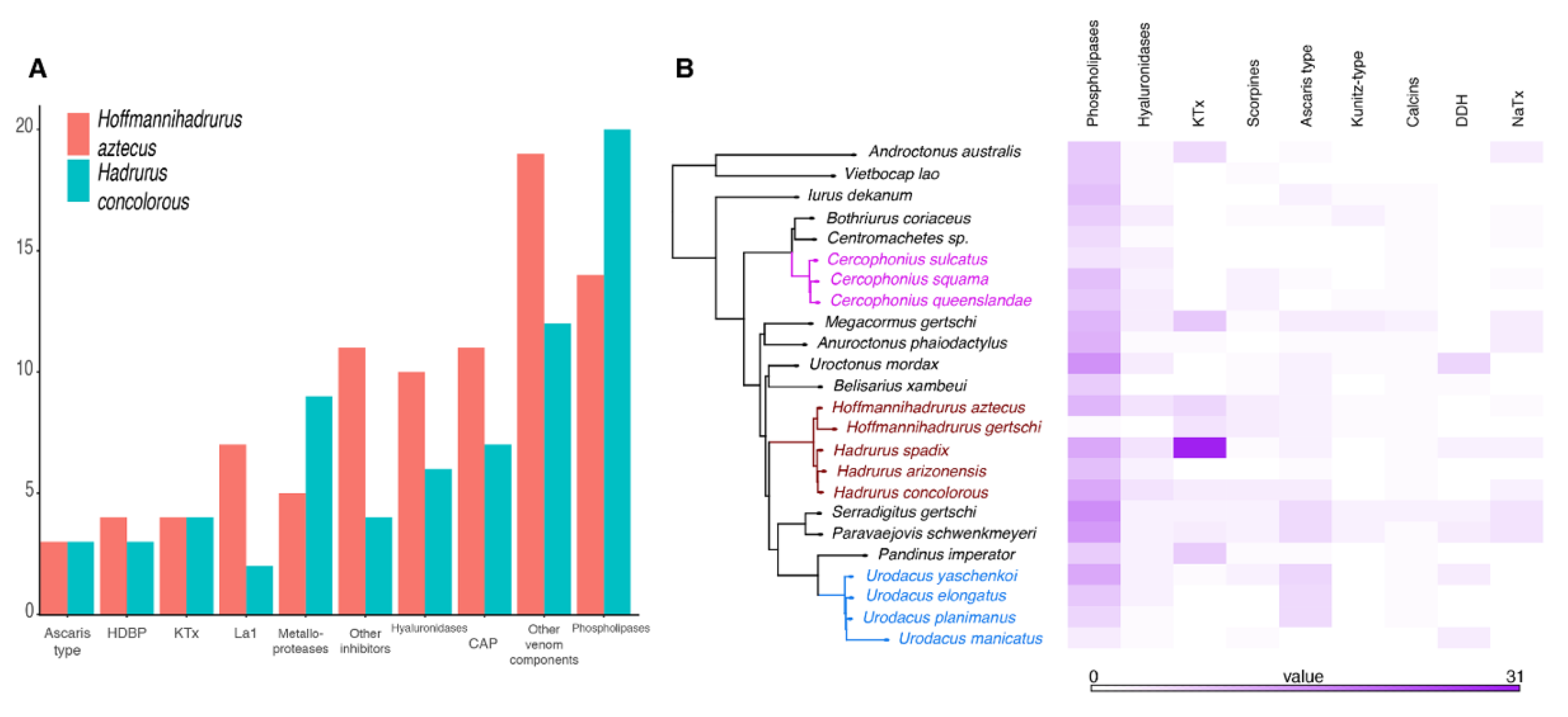
2.2. The Repertoire of Venom-Specific Transcripts in the H. concolorous and H. aztecus Libraries
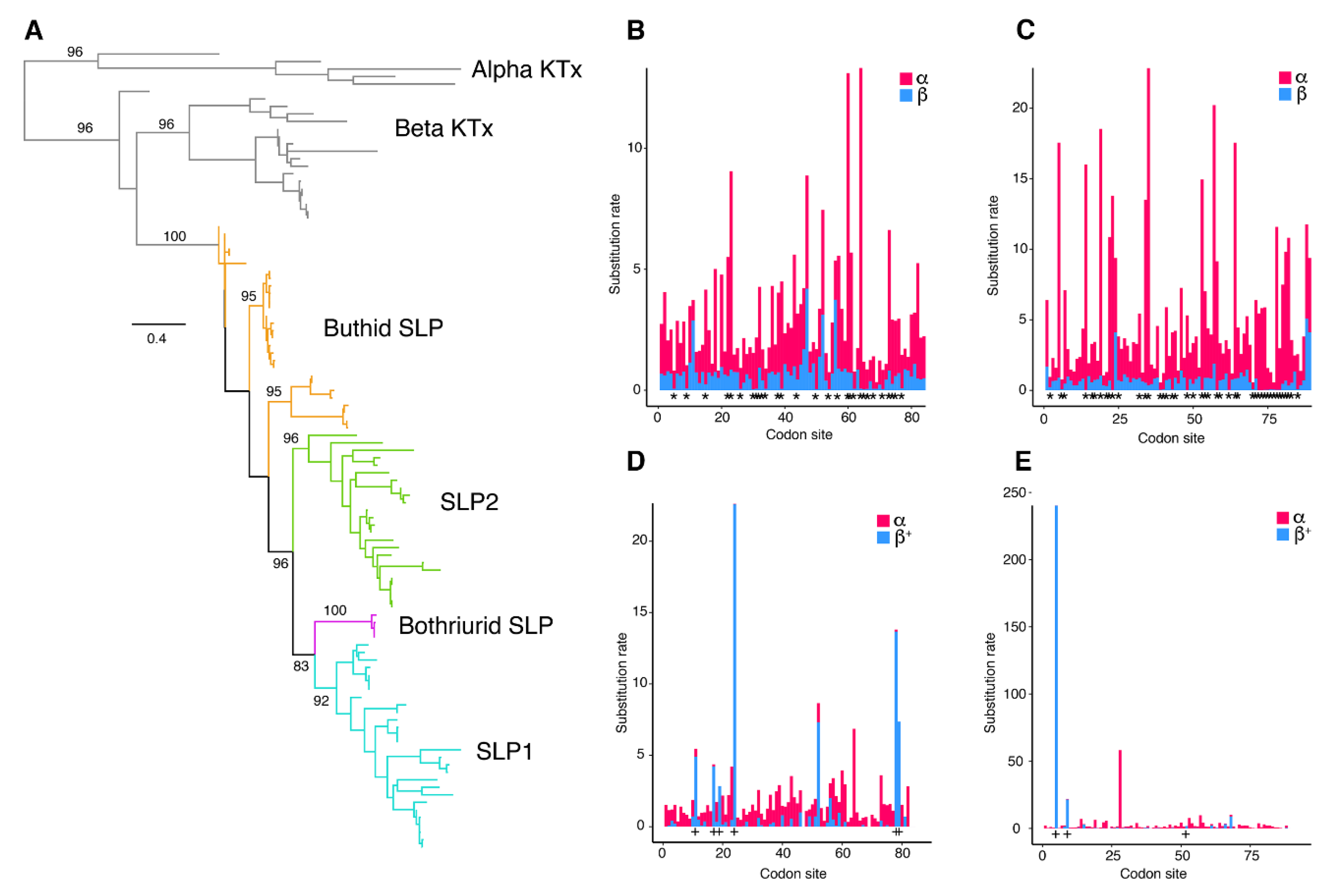
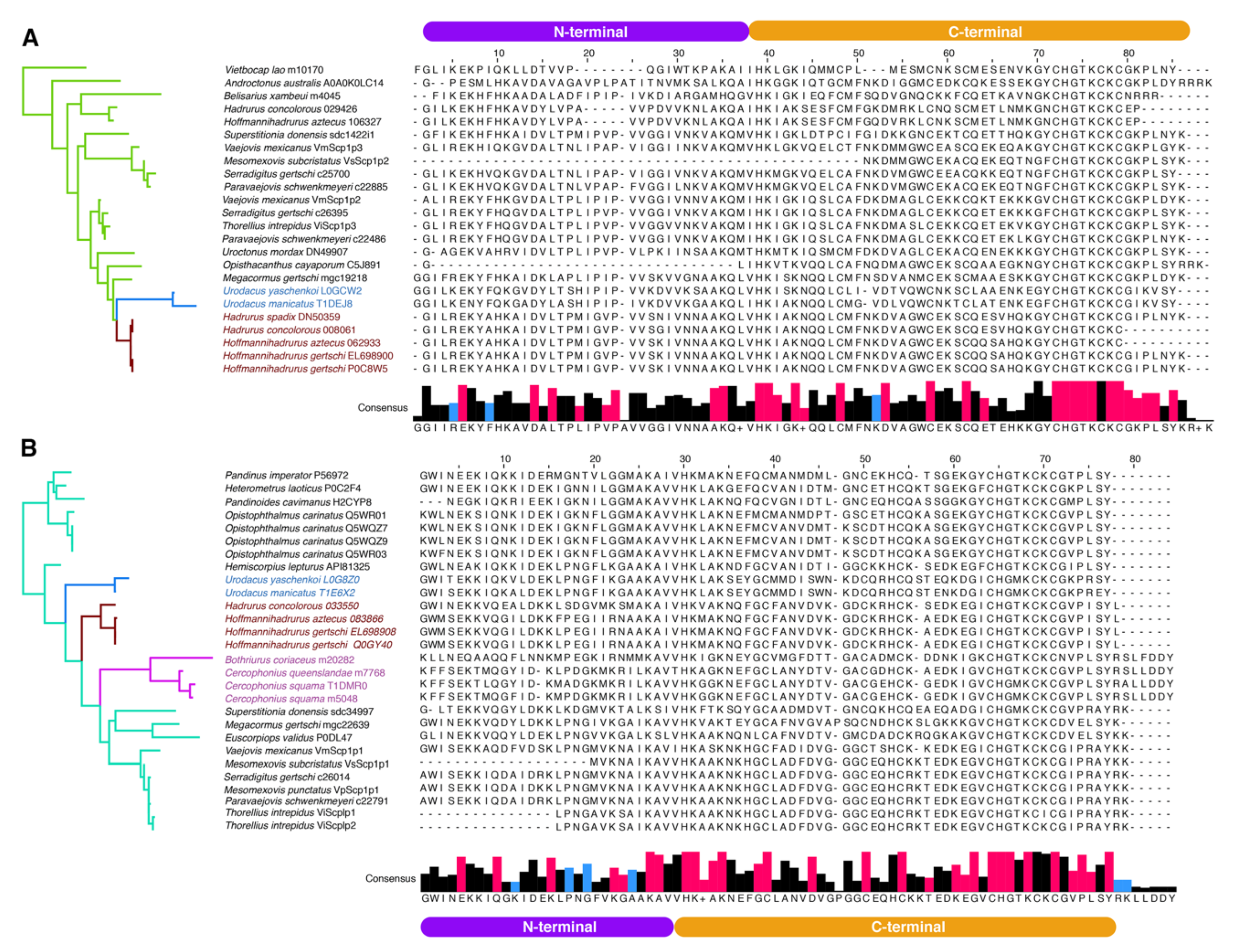
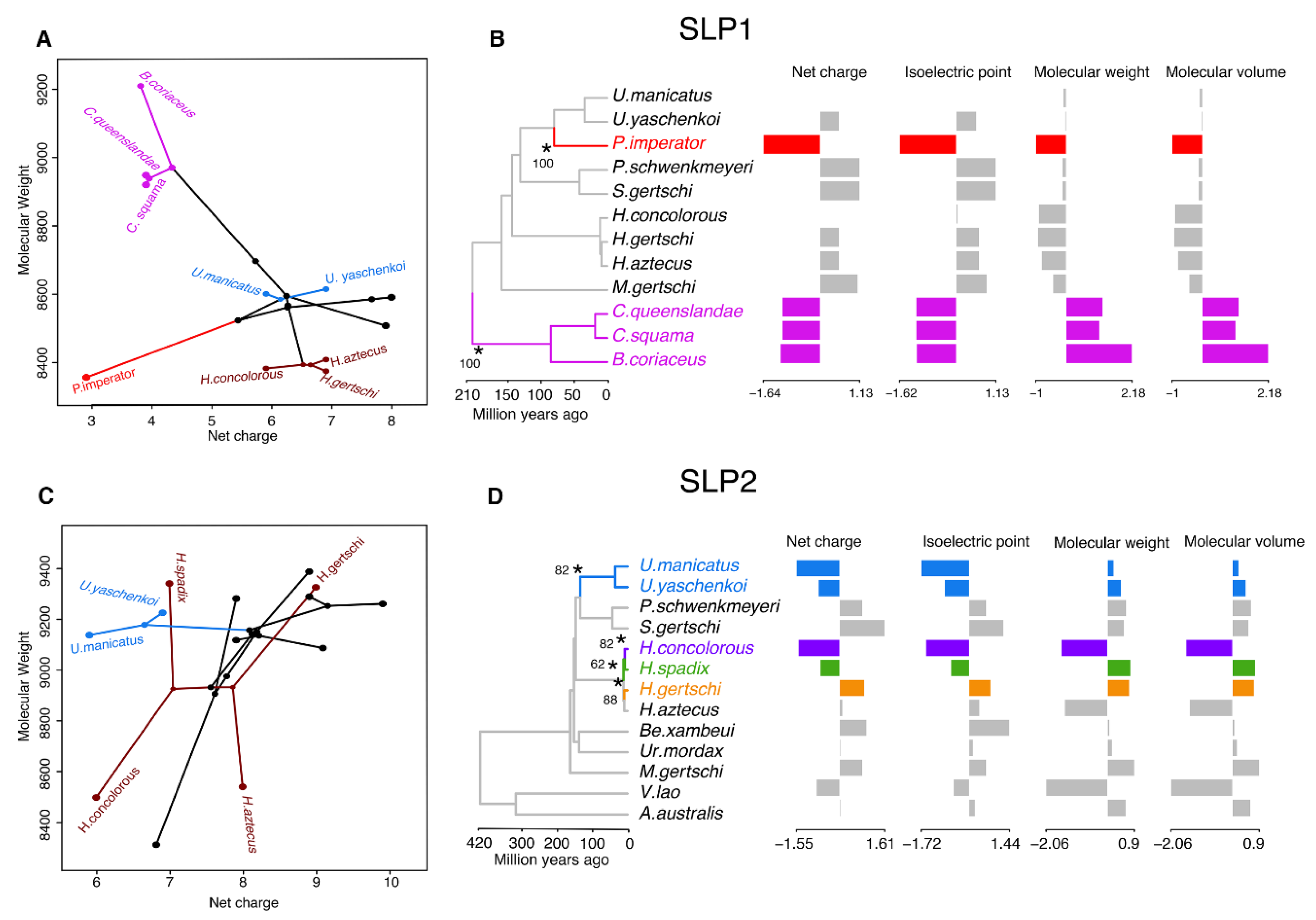
2.3. Molecular Evolution of Scorpine-Like Peptides (SLP)
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Specimen Collection and RNA Sequencing
5.2. Orthology Inference and Phylogenetic Methods
5.3. Venom Transcriptome Analyses of Hadrurus Concolorous and Hoffmannihadrurus Aztecus
5.4. Gene Tree Analysis and Molecular Evolution of Scorpines
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fry, B.G.; Roelants, K.; Champagne, D.E.; Scheib, H.; Tyndall, J.D.A.; King, G.F.; Nevalainen, T.J.; Norman, J.A.; Lewis, R.J.; Norton, R.S.; et al. The Toxicogenomic Multiverse: Convergent Recruitment of Proteins Into Animal Venoms. Annu. Rev. Genom. Hum. Genet. 2009, 10, 483–511. [Google Scholar] [CrossRef]
- Nelsen, D.R.; Nisani, Z.; Cooper, A.M.; Fox, G.A.; Gren, E.C.K.; Corbit, A.G.; Hayes, W.K. Poisons, toxungens, and venoms: Redefining and classifying toxic biological secretions and the organisms that employ them. Biol. Rev. 2013, 89, 450–465. [Google Scholar] [CrossRef]
- Morgenstern, D.; King, G.F. The venom optimization hypothesis revisited. Toxicon 2013, 63, 120–128. [Google Scholar] [CrossRef]
- Casewell, N.R.; Wüster, W.; Vonk, F.J.; Harrison, R.A.; Fry, B.G. Complex cocktails: The evolutionary novelty of venoms. Trends Ecol. Evol. 2013, 28, 219–229. [Google Scholar] [CrossRef]
- King, G.F.; Hardy, M.C. Spider-Venom Peptides: Structure, Pharmacology, and Potential for Control of Insect Pests. Annu. Rev. Entomol. 2013, 58, 475–496. [Google Scholar] [CrossRef]
- Torres, A.F.C.; Huang, C.; Chong, C.-M.; Leung, S.W.; Prieto-da-Silva, Á.R.B.; Havt, A.; Quinet, Y.P.; Martins, A.M.C.; Lee, S.M.Y.; Rádis-Baptista, G. Transcriptome Analysis in Venom Gland of the Predatory Giant Ant Dinoponera quadriceps: Insights into the Polypeptide Toxin Arsenal of Hymenopterans. PLoS ONE 2014, 9, e87556. [Google Scholar] [CrossRef]
- Asgari, S.; Rivers, D.B. Venom Proteins from Endoparasitoid Wasps and Their Role in Host-Parasite Interactions. Annu. Rev. Entomol. 2011, 56, 313–335. [Google Scholar] [CrossRef]
- Undheim, E.A.B.; King, G.F. On the venom system of centipedes (Chilopoda), a neglected group of venomous animals. Toxicon 2011, 57, 512–524. [Google Scholar] [CrossRef]
- Undheim, E.A.B.; Jones, A.; Clauser, K.R.; Holland, J.W.; Pineda, S.S.; King, G.F.; Fry, B.G. Clawing through Evolution: Toxin Diversification and Convergence in the Ancient Lineage Chilopoda (Centipedes). Mol. Biol. Evol. 2014, 31, 2124–2148. [Google Scholar] [CrossRef]
- Undheim, E.; Fry, B.; King, G. Centipede Venom: Recent Discoveries and Current State of Knowledge. Toxins 2015, 7, 679–704. [Google Scholar] [CrossRef]
- von Reumont, B.M.; Blanke, A.; Richter, S.; Alvarez, F.; Bleidorn, C.; Jenner, R.A. The First Venomous Crustacean Revealed by Transcriptomics and Functional Morphology: Remipede Venom Glands Express a Unique Toxin Cocktail Dominated by Enzymes and a Neurotoxin. Mol. Biol. Evol. 2013, 31, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnakone, P.; Possani, L.D.; Schwartz, E.F.; Rodriguez de la Vega, R.C. Scorpion Venoms; Springer: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Gopalakrishnakone, P.; Corzo, G.; de Lima, M.E.; Diego-Garcia, E. Spider Venoms; Springer: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Possani, L.D.; Becerril, B.; Delepierre, M.; Tytgat, J. Scorpion toxins specific for Na+-channels. FEBS J. 1999, 264, 287–300. [Google Scholar] [CrossRef]
- Sunagar, K.; Undheim, E.; Chan, A.; Koludarov, I.; Muñoz-Gómez, S.; Antunes, A.; Fry, B. Evolution Stings: The Origin and Diversification of Scorpion Toxin Peptide Scaffolds. Toxins 2013, 5, 2456–2487. [Google Scholar] [CrossRef]
- Santibáñez-López, C.E.; Possani, L.D. Overview of the Knottin scorpion toxin-like peptides in scorpion venoms: Insights on their classification and evolution. Toxicon 2015, 107, 317–326. [Google Scholar] [CrossRef]
- Juárez, P.; Comas, I.; González-Candelas, F.; Calvete, J.J. Evolution of snake venom disintegrins by positive Darwinian selection. Mol. Biol. Evol. 2008, 25, 2391–2407. [Google Scholar] [CrossRef]
- Dowell, N.L.; Giorgianni, M.W.; Kassner, V.A.; Selegue, J.E.; Sanchez, E.E.; Carroll, S.B. The deep origin and recent loss of venom toxin genes in rattlesnakes. Curr. Bio. 2016, 26, 2434–2445. [Google Scholar] [CrossRef]
- Weinberger, H.; Moran, Y.; Gordon, D.; Turkov, M.; Kahn, R.; Gurevitz, M. Positions under positive selection—Key for selectivity and potency of scorpion alpha toxins. Mol. Biol. Evol. 2010, 27, 1025–1034. [Google Scholar] [CrossRef]
- Haney, R.A.; Clarke, T.H.; Gadgil, R.; Fitzpatrick, R.; Hayashi, C.Y.; Ayoub, N.A.; Garb, J.E. Effects of gene duplication, positive selection, and shifts in gene expression on the evolution of the venom gland transcriptome in widow spiders. Genome Biol. Evol. 2016, 8, 228–242. [Google Scholar] [CrossRef]
- Santibáñez-López, C.E.; Cid-Uribe, J.I.; Zamudio, F.Z.; Batista, C.V.F.; Ortiz, E.; Possani, L.D. Venom gland transcriptomic and venom proteomic analyses of the scorpion Megacormus gertschi Díaz-Najera, 1966 (Scorpiones: Euscorpiidae: Megacorminae). Toxicon 2017, 133, 95–109. [Google Scholar] [CrossRef]
- Santibáñez-López, C.; Cid-Uribe, J.; Batista, C.; Ortiz, E.; Possani, L. Venom Gland Transcriptomic and Proteomic Analyses of the Enigmatic Scorpion Superstitionia donensis (Scorpiones: Superstitioniidae), with Insights on the Evolution of Its Venom Components. Toxins 2016, 8, 367. [Google Scholar] [CrossRef]
- Romero-Gutiérrez, M.; Santibáñez-López, C.; Jiménez-Vargas, J.; Batista, C.; Ortiz, E.; Possani, L. Transcriptomic and Proteomic Analyses Reveal the Diversity of Venom Components from the Vaejovid Scorpion Serradigitus gertschi. Toxins 2018, 10, 359. [Google Scholar] [CrossRef] [PubMed]
- Luna-Ramírez, K.; Quintero-Hernández, V.; Juárez-González, V.R.; Possani, L.D. Whole Transcriptome of the Venom Gland from Urodacus yaschenkoi Scorpion. PLoS ONE 2015, 10, e0127883. [Google Scholar] [CrossRef] [PubMed]
- Kazemi-Lomedasht, F.; Khalaj, V.; Bagheri, K.P.; Behdani, M.; Shahbazzadeh, D. The first report on transcriptome analysis of the venom gland of Iranian scorpion, Hemiscorpius lepturus. Toxicon 2017, 125, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Zeng, X.-C.; Zeng, X.; Nie, Y.; Zhang, L.; Wu, S.; Bao, A. Transcriptomic analysis of the venom glands from the scorpion Hadogenes troglodytes revealed unique and extremely high diversity of the venom peptides. J. Proteom. 2017, 150, 40–62. [Google Scholar] [CrossRef] [PubMed]
- Santibáñez-López, C.E.; Ojanguren-Affilastro, A.; Sharma, P.P. Another one bites the dust: Taxonomic sampling of a key genus in phylogenomic datasets reveals more non-monophyletic groups in traditional scorpion classification. Inv. Syst. 2019, in press. [Google Scholar] [CrossRef]
- Schwartz, E.F.; Capes, E.M.; Diego-Garcia, E.; Zamudio, F.Z.; Fuentes, O.; Possani, L.D.; Valdivia, H.H. Characterization of hadrucalcin, a peptide from Hadrurus gertschi scorpion venom with pharmacological activity on ryanodine receptors. Br. J. Pharmacol. 2009, 157, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.F.; Diego-Garcia, E.; Rodríguez de la Vega, R.C.; Possani, L.D. Transcriptome analysis of the venom gland of the Mexican scorpion Hadrurus gertschi (Arachnida: Scorpiones). BMC Genom. 2007, 8, 119. [Google Scholar] [CrossRef]
- Chen, Z.-Y.; Hu, Y.-T.; Yang, W.-S.; He, Y.-W.; Feng, J.; Wang, B.; Zhao, R.-M.; Ding, J.-P.; Cao, Z.-J.; Li, W.-X.; et al. Hg1, Novel Peptide Inhibitor Specific for Kv1.3 Channels from First Scorpion Kunitz-type Potassium Channel Toxin Family. J. Biol. Chem. 2012, 287, 13813–13821. [Google Scholar] [CrossRef]
- Rokyta, D.R.; Ward, M.J. Venom-gland transcriptomics and venom proteomics of the black- back scorpion (Hadrurus spadix) reveal detectability challenges and an unexplored realm of animal toxin diversity. Toxicon 2017, 128, 23–37. [Google Scholar] [CrossRef]
- Diego-García, E.; Schwartz, E.F.; D’Suze, G.; González, S.A.R.; Batista, C.V.F.; García, B.I.; de la Vega, R.C.R.; Possani, L.D. Wide phylogenetic distribution of Scorpine and long-chain β-KTx-like peptides in scorpion venoms: Identification of “orphan” components. Peptides 2007, 28, 31–37. [Google Scholar] [CrossRef]
- Diego-García, E.; Abdel-Mottaleb, Y.; Schwartz, E.F.; de la Vega, R.C.R.; Tytgat, J.; Possani, L.D. Cytolytic and K+ channel blocking activities of β-KTx and scorpine-like peptides purified from scorpion venoms. CMLS Cell. Mol. Life Sci. 2007, 65, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Tytgat, J. The scorpine family of defensins: Gene structure, alternative polyadenylation and fold recognition. CMLS Cell. Mol. Life Sci. 2004, 61, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Conde, R.; Zamudio, F.Z.; Rodríguez, M.H.; Possani, L.D. Scorpine, an anti-malaria and anti-bacterial agent purified from scorpion venom. FEBS Lett. 2000, 471, 165–168. [Google Scholar] [CrossRef]
- Santibáñez-López, C.E.; Kriebel, R.; Ballesteros, J.A.; Rush, N.; Witter, Z.; Williams, J.; Janies, D.A.; Sharma, P.P. Integration of phylogenomics and molecular modeling reveals lineage-specific diversification of toxins in scorpions. PeerJ 2018, 6, e5902. [Google Scholar] [CrossRef]
- Sharma, P.P.; Baker, C.M.; Cosgrove, J.G.; Johnson, J.E.; Oberski, J.T.; Raven, R.J.; Harvey, M.S.; Boyer, S.L.; Giribet, G. A revised dated phylogeny of scorpions—Phylogenomic support for ancient divergence of the temperate Gondwanan family Bothriuridae. Mol. Phylogen. Evol. 2018, 122, 37–45. [Google Scholar] [CrossRef]
- Fet, V.; Soleglad, M.E. Cladistic analysis of superfamily Iuroidea, with emphasis on subfamily Hadrurinae (Scorpiones: Iurida). Bol. Soc. Entomol. Aragonesa 2008, 43, 255–281. [Google Scholar]
- Soleglad, M.; Fet, V. Further observations on scorpion genera Hadrurus and Hoffmannihadrurus (Scorpiones, Caraboctonidae). Zookeys 2010, 59, 1–13. [Google Scholar] [CrossRef]
- Sharma, P.P.; Fernández, R.; Esposito, L.A.; González-Santillán, E.; Monod, L. Phylogenomic resolution of scorpions reveals multilevel discordance with morphological phylogenetic signal. Proc. Biol. Sci. 2015, 282, 20142953. [Google Scholar] [CrossRef]
- Bryson, R.W., Jr.; Murphy, R.W.; Lathrop, A.; Lazcano-Villareal, D. Evolutionary drivers of phylogeographical diversity in the highlands of Mexico: A case study of the Crotalus triseriatus species group of montane rattlesnakes. J. Biogeogr. 2010, 38, 697–710. [Google Scholar] [CrossRef]
- Becerra, J.X. Timing the origin and expansion of the Mexican tropical dry forest. Proc. Natl. Acad. Sci. USA 2005, 102, 10919–10923. [Google Scholar] [CrossRef]
- Ferrusquia-Villafranca, I.; González-Guzmán, L.I. Northern Mexico’s landscape, part II: The biotic setting across time. In Biodiversity, Ecosystems, and Conservation in Northern Mexico; Oxford University Press: Oxford, UK, 2005; pp. 39–51. [Google Scholar]
- Undheim, E.A.B.; Mobli, M.; King, G.F. Toxin structures as evolutionary tools: Using conserved 3D folds to study the evolution of rapidly evolving peptides. BioEssays 2016, 38, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Khabbazian, M.; Kriebel, R.; Rohe, K.; Ané, C. Fast and accurate detection of evolutionary shifts in Ornstein-Uhlenbeck models. Methods Ecol. Evol. 2016, 7, 811–824. [Google Scholar] [CrossRef]
- Cressler, C.E.; Butler, M.A.; King, A.A. Detecting Adaptive Evolution in Phylogenetic Comparative Analysis Using the Ornstein–Uhlenbeck Model. Syst. Biol. 2015, 64, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Kriebel, R.; Khabbazian, M.; Sytsma, K.J. A continuous morphological approach to study the evolution of pollen in a phylogenetic context: An example with the order Myrtales. PLoS ONE 2017, 12, e0187228. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M. Studying smaller and neglected organisms in modern evolutionary venomics implementing rnaseq (transcriptomics)—A critical guide. Toxins 2018, 10, 292. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, E.P.; Rautsaw, R.M.; Strickland, J.L.; Holding, M.L.; Hogan, M.P.; Mason, A.J.; Rokyta, D.R.; Parkinson, C.L. Comparative venom-gland transcriptomics and venom proteomics of four sidewinder rattlesnake (Crotalus cerastes) lineages reveal little differential expression despite individual variation. Sci. Rep. 2018, 8, 15534. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Cid-Uribe, J.I.; Santibáñez-López, C.E.; Meneses, E.P.; Batista, C.V.F.; Jiménez-Vargas, J.M.; Ortiz, E.; Possani, L.D. The diversity of venom components of the scorpion species Paravaejovis schwenkmeyeri (Scorpiones: Vaejovidae) revealed by transcriptome and proteome analyses. Toxicon 2018, 151, 47–62. [Google Scholar] [CrossRef]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.H.; Davis, F.G.; et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef]
- Santibáñez-López, C.E.; González-Santillán, E.; Monod, L.; Sharma, P.P. Phylogenomics facilitates stable scorpion systematics_ Reassessing the relationships of Vaejovidae and a new higher-level classification of Scorpiones (Arachnida). Mol. Phylogen. Evol. 2019, 135, 22–30. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Hormiga, G. A New Orthology Assessment Method for Phylogenomic Data: Unrooted Phylogenetic Orthology. Mol. Biol. Evol. 2016, 33, 2117–2134. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotech. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Dongen, S. Graph Clustering by Low Simulation. Ph.D. Thesis, University of Utrecht, Utrecht, The Netherlands, 2000. [Google Scholar]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Tange, O. Gnu parallel-the command-line power tool. USENIX Mag. 2011, 36, 42–47. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Mirarab, S.; Warnow, T. ASTRAL-II: Coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 2015, 31, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.J. Estimating absolute rates of molecular evolution and divergence times: A penalized likelihood approach. Mol. Biol. Evol. 2002, 19, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E. Molecular dating of phylogenies by likelihood methods: A comparison of models and a new information criterion. Mol. Phylogen. Evol. 2013, 67, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Santibáñez-López, C.E.; Kriebel, R.; Sharma, P.P. eadem figura manet: Measuring morphological convergence in diplocentrid scorpions (Arachnida: Scorpiones: Diplocentridae) under a multilocus phylogenetic framework. Inv. Syst. 2017, 31, 233–248. [Google Scholar] [CrossRef]
- Herzig, V.; Wood, D.L.A.; Newell, F.; Chaumeil, P.A.; Kaas, Q.; Binford, G.J.; Nicholson, G.M.; Gorse, D.; King, G.F. ArachnoServer 2.0, an updated online resource for spider toxin sequences and structures. Nucleic Acids Res. 2010, 39, D653–D657. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Frost, S.D.W.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Osorio, D.; Rondón-Villarrea, P.; Torres, R. Peptides: A package for data mining of antimicrobial peptides. R J. 2015, 7, 4–14. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santibáñez-López, C.E.; Graham, M.R.; Sharma, P.P.; Ortiz, E.; Possani, L.D. Hadrurid Scorpion Toxins: Evolutionary Conservation and Selective Pressures. Toxins 2019, 11, 637. https://doi.org/10.3390/toxins11110637
Santibáñez-López CE, Graham MR, Sharma PP, Ortiz E, Possani LD. Hadrurid Scorpion Toxins: Evolutionary Conservation and Selective Pressures. Toxins. 2019; 11(11):637. https://doi.org/10.3390/toxins11110637
Chicago/Turabian StyleSantibáñez-López, Carlos E., Matthew R. Graham, Prashant P. Sharma, Ernesto Ortiz, and Lourival D. Possani. 2019. "Hadrurid Scorpion Toxins: Evolutionary Conservation and Selective Pressures" Toxins 11, no. 11: 637. https://doi.org/10.3390/toxins11110637
APA StyleSantibáñez-López, C. E., Graham, M. R., Sharma, P. P., Ortiz, E., & Possani, L. D. (2019). Hadrurid Scorpion Toxins: Evolutionary Conservation and Selective Pressures. Toxins, 11(11), 637. https://doi.org/10.3390/toxins11110637