The Sequence and a Three-Dimensional Structural Analysis Reveal Substrate Specificity among Snake Venom Phosphodiesterases


Abstract
:1. Introduction
2. Results and Discussion
2.1. Sequence Alignment Analysis
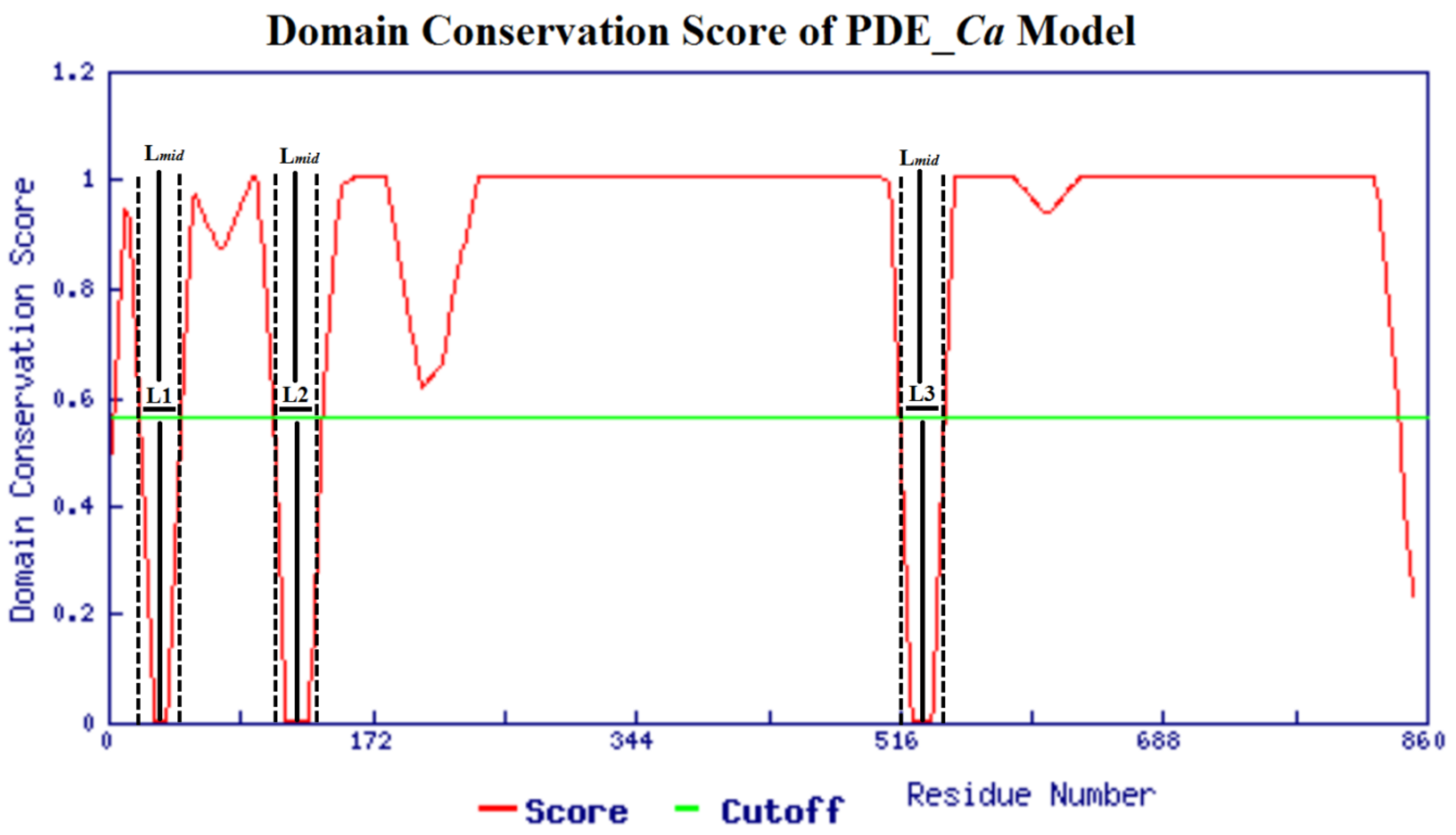
2.2. Domain Analysis
2.3. Glycosylation Sites
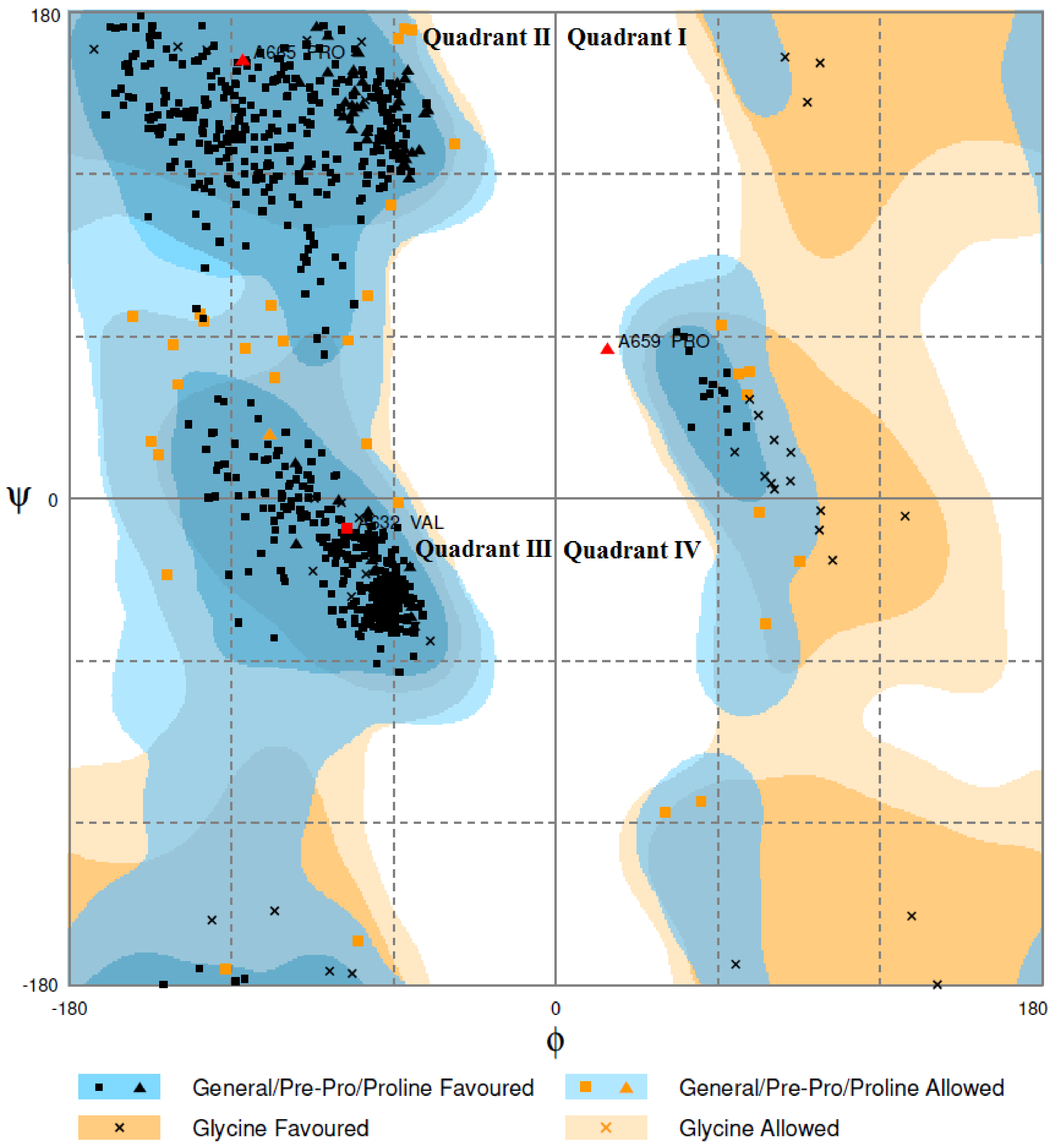
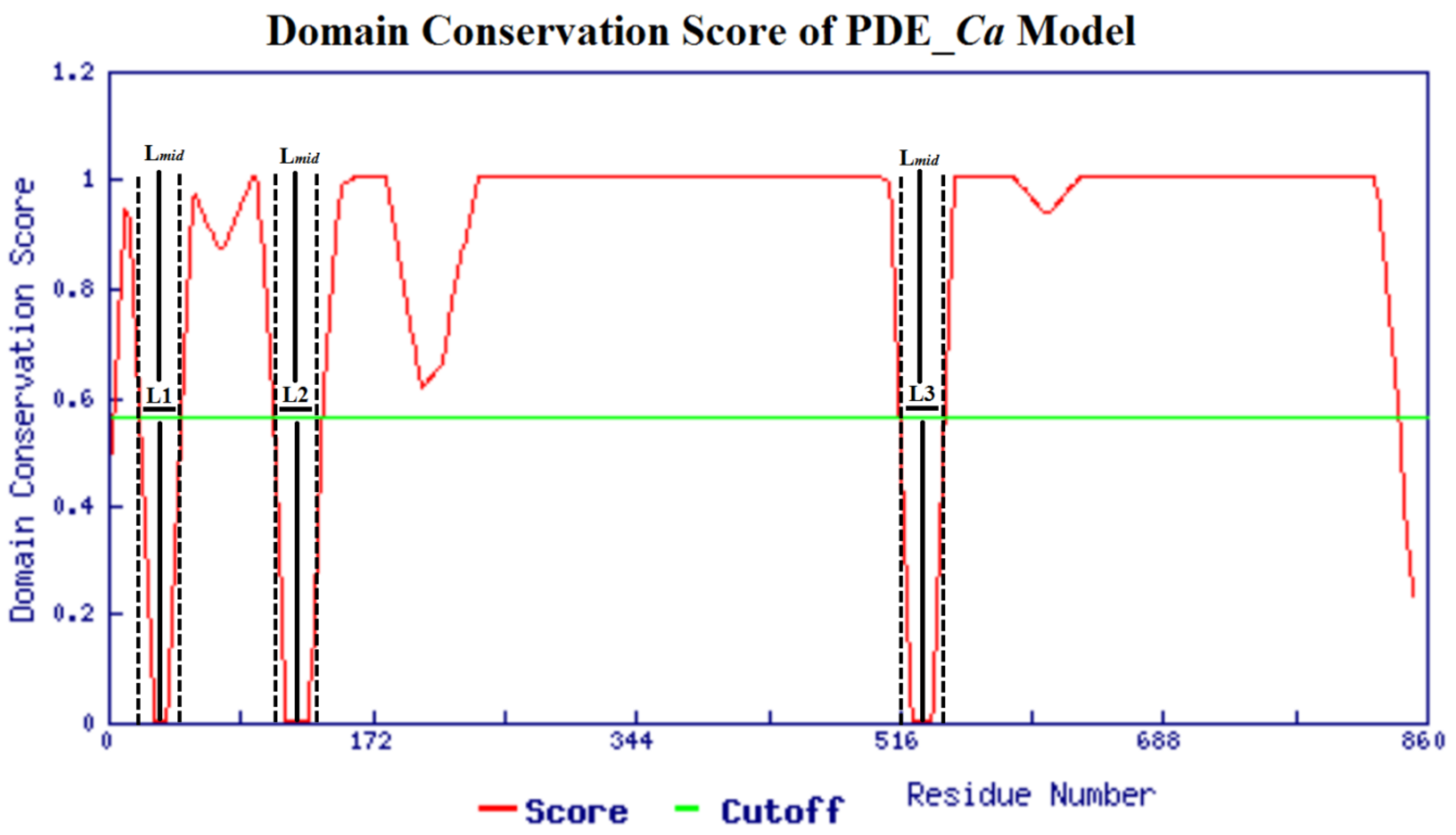
2.4. Homology Modeling and Model Evaluation
2.5. Molecular Dynamics Simulation
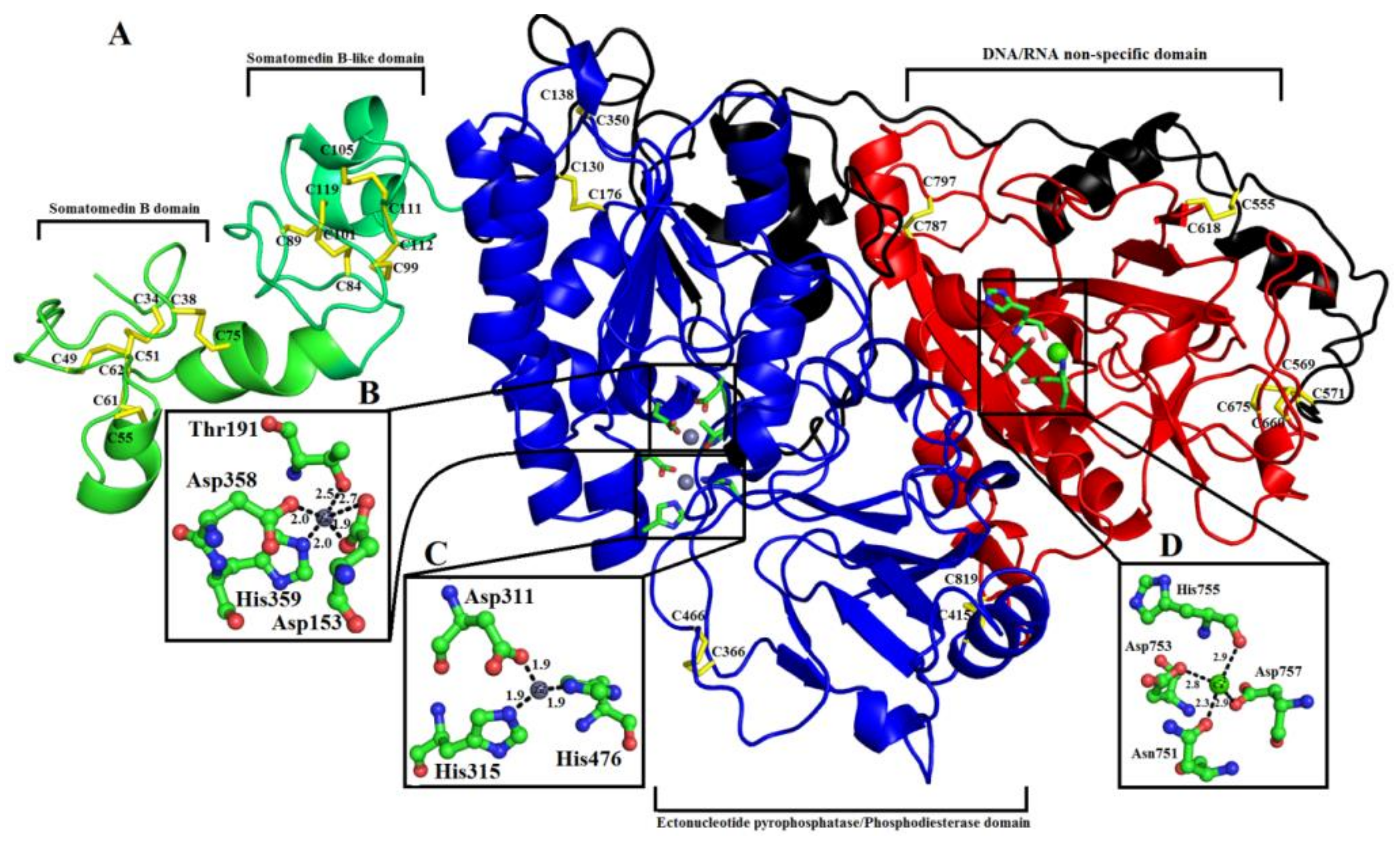
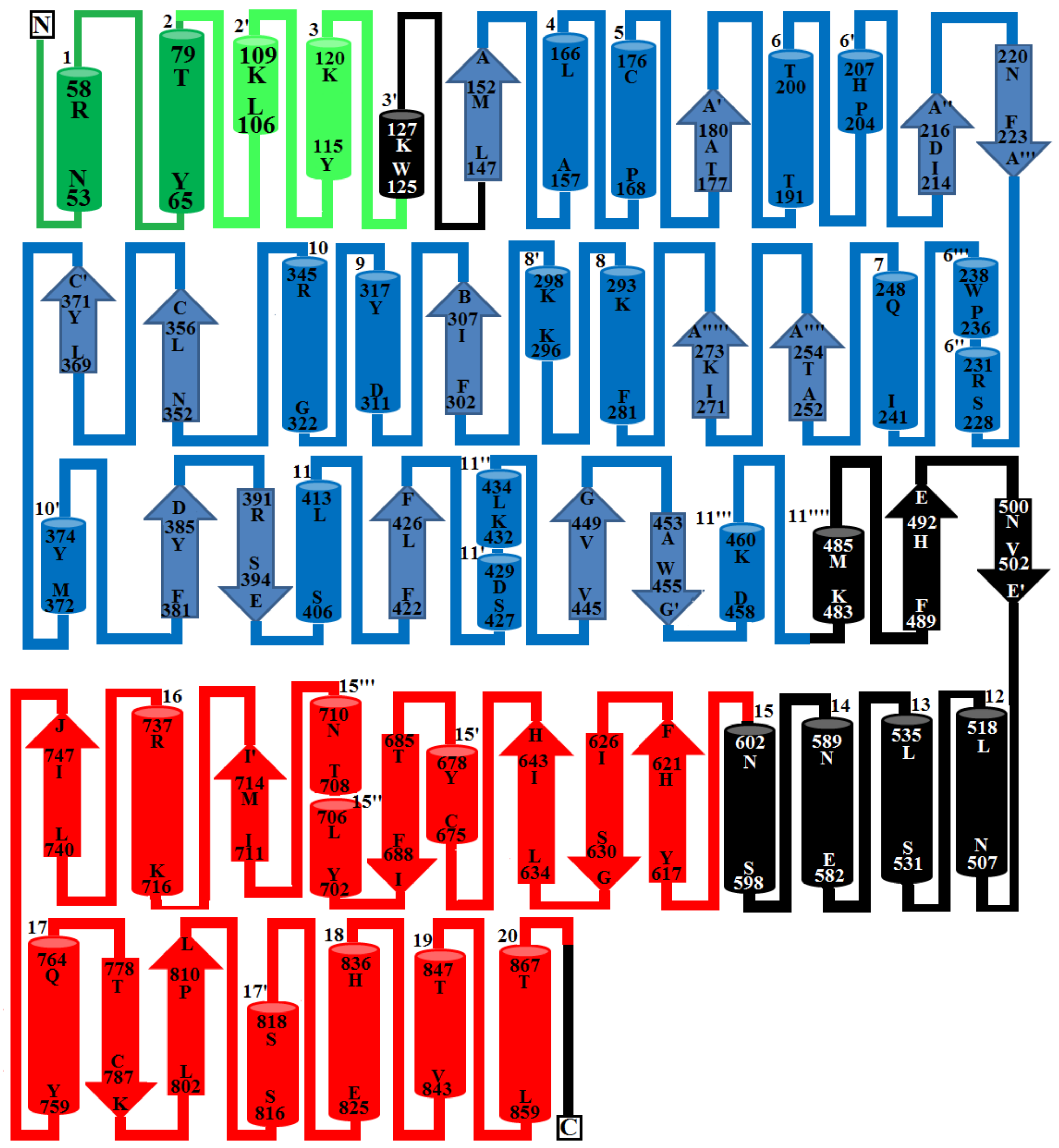
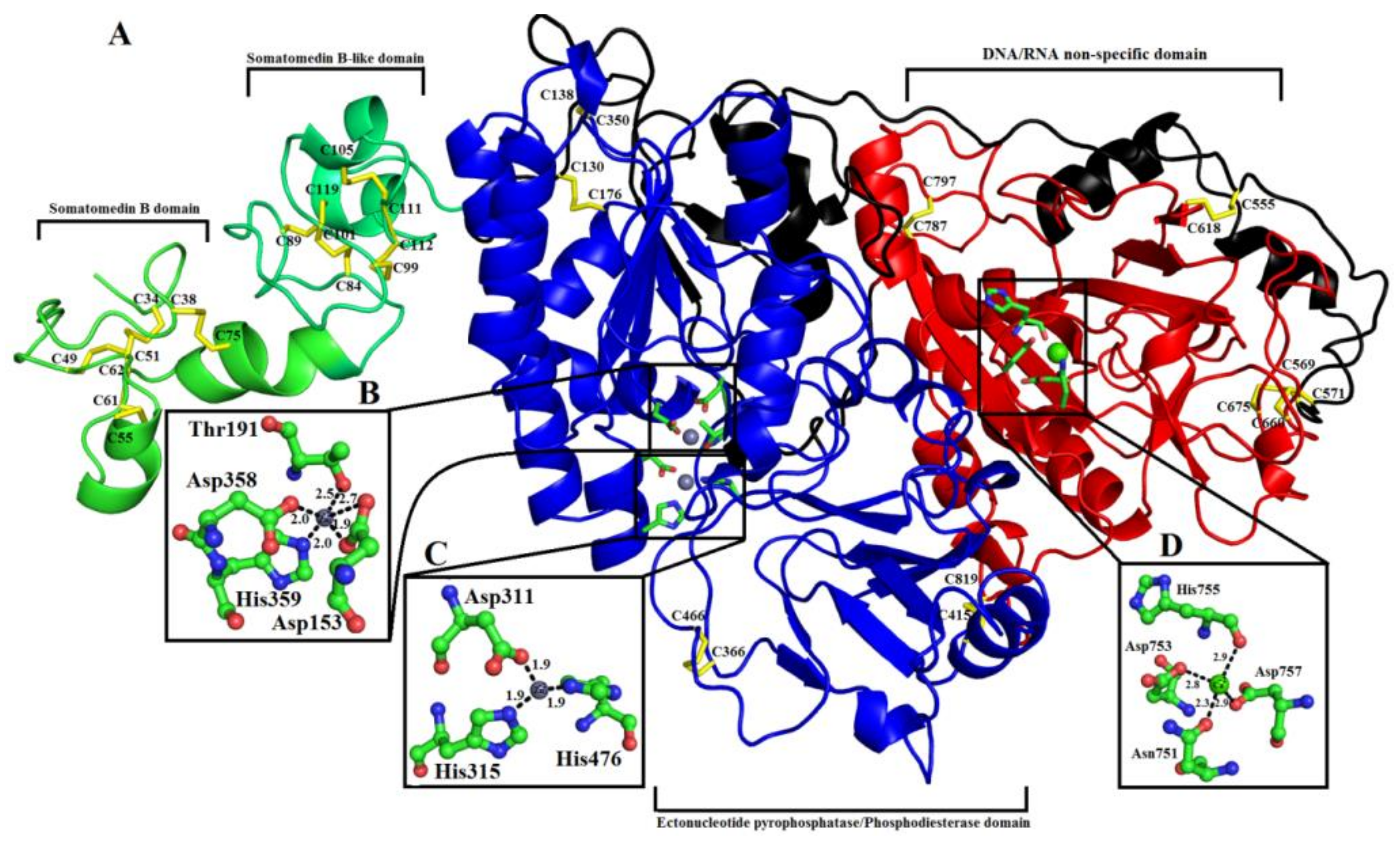
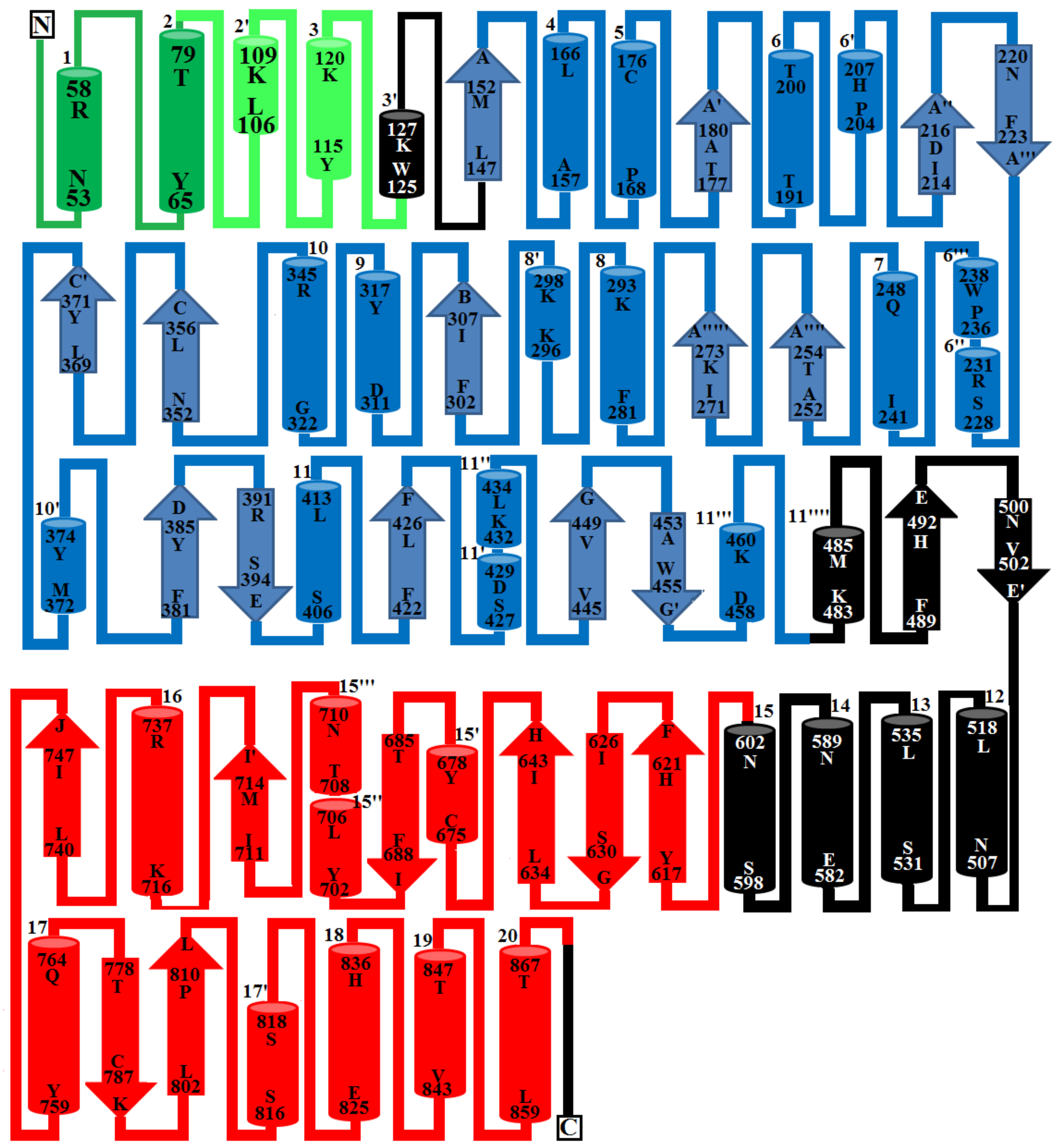
2.6. Overall Structure of PDE_Ca
2.6.1. Somatomedin B Domain
2.6.2. Somatomedin B-like Domain
2.6.3. Ectonucleotide Pyrophosphatase/Phosphodiesterase Domain
2.6.4. DNA/RNA Non-Specific Domain
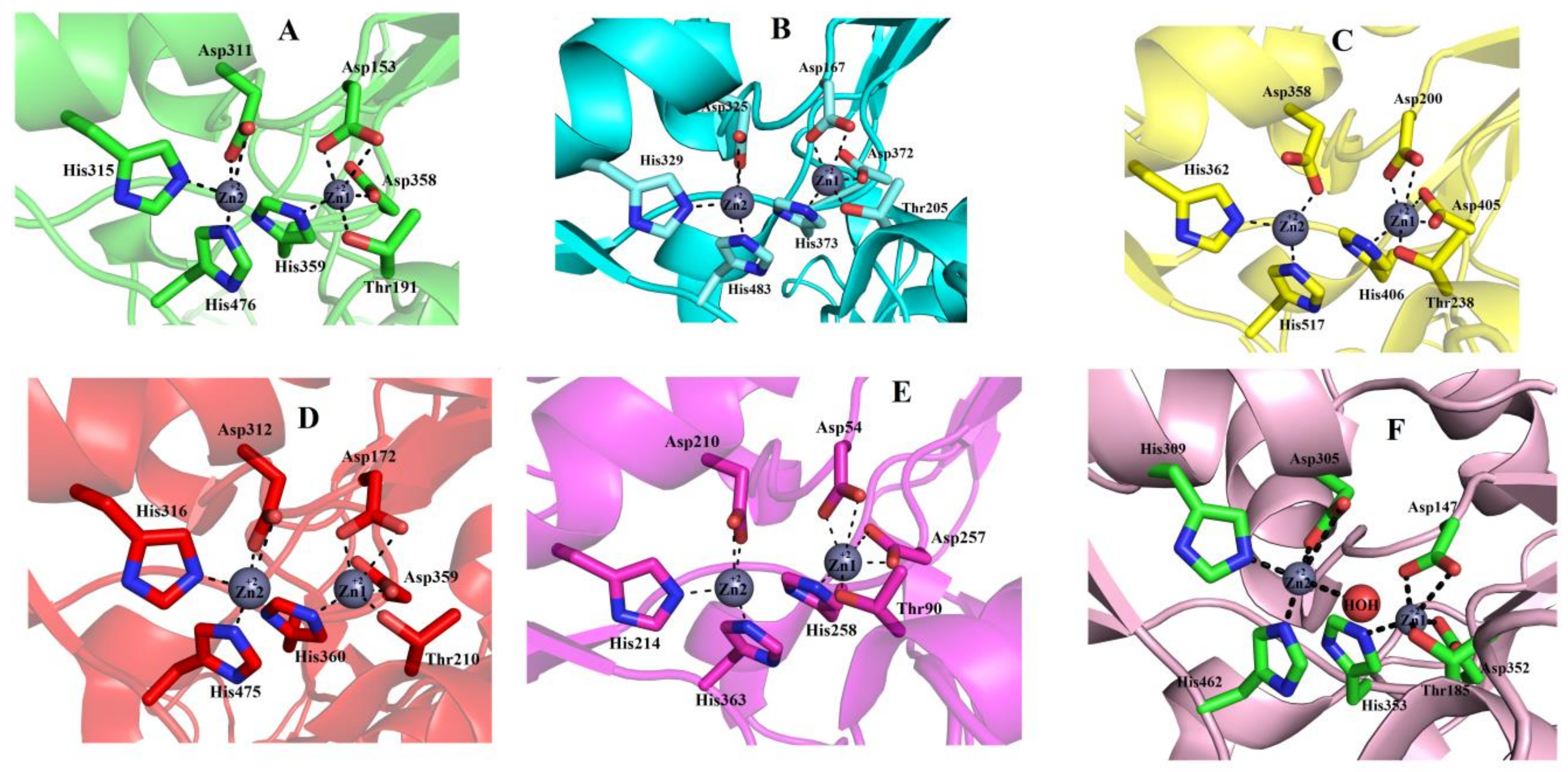
2.6.5. Metal Ion-Binding Sites
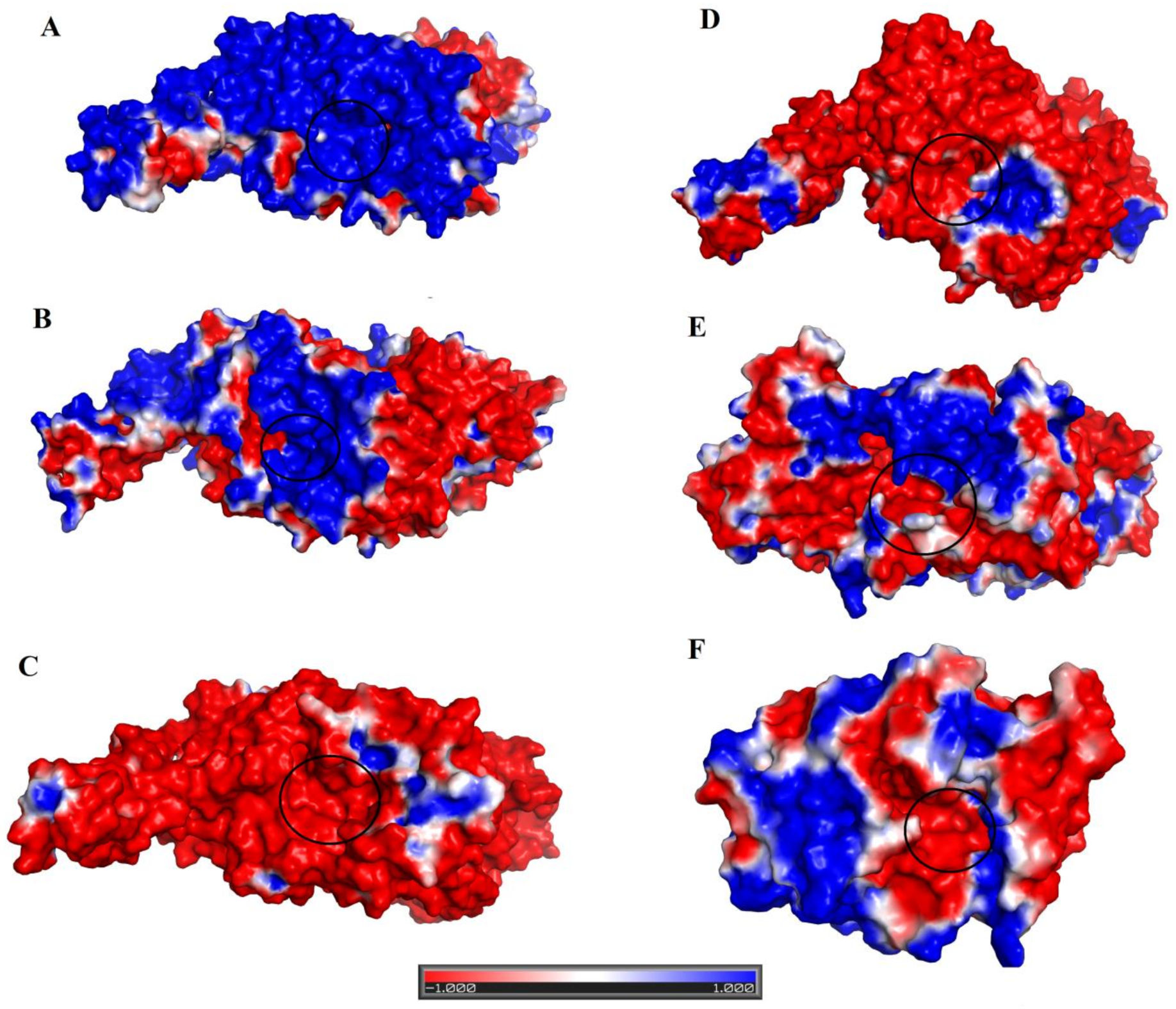
2.7. Structural Basis for Substrate Specificity of Snake Venom Phosphodiesterases
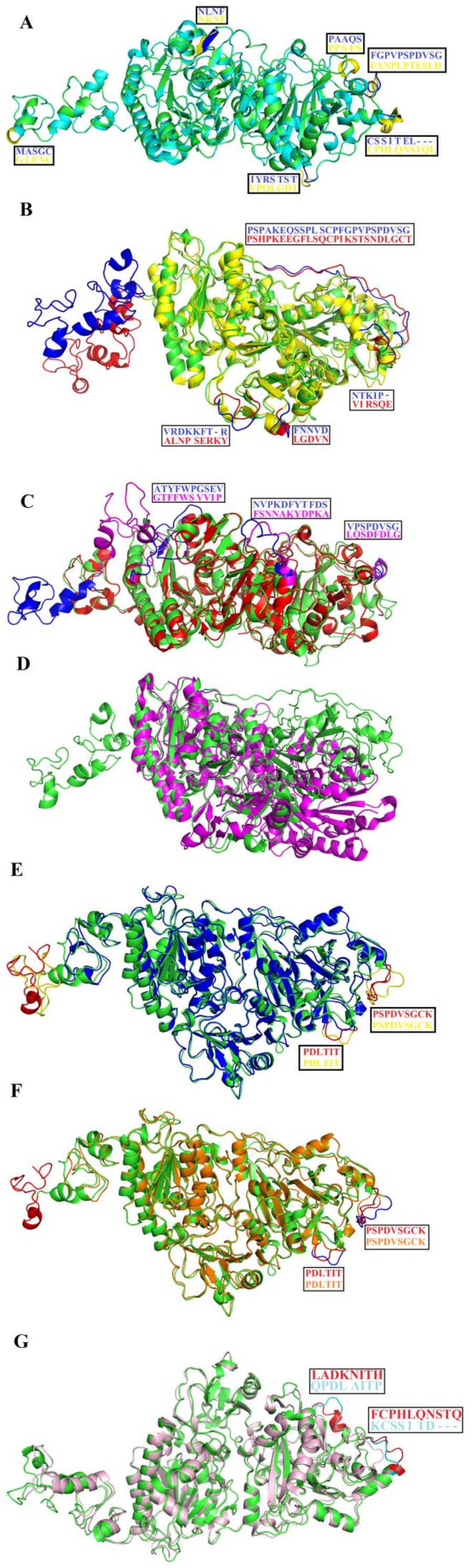
2.8. Structural Alignment between PDE_Ca, Human ENPP3, Mouse NPP1, Human Autotaxin, Xa NPP1, PDE_Vl, PDE_Ba and Naja atra atra PDE.
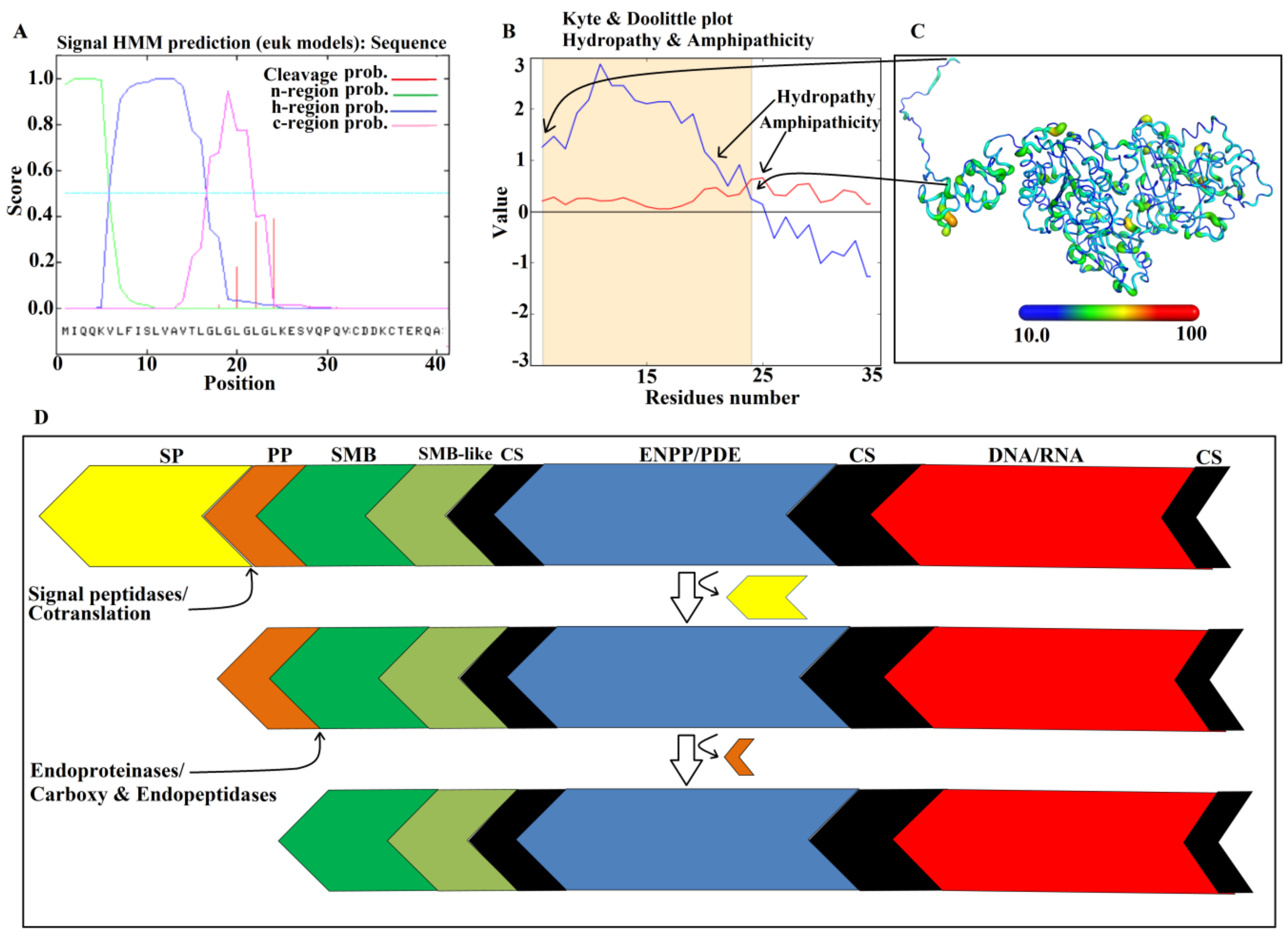
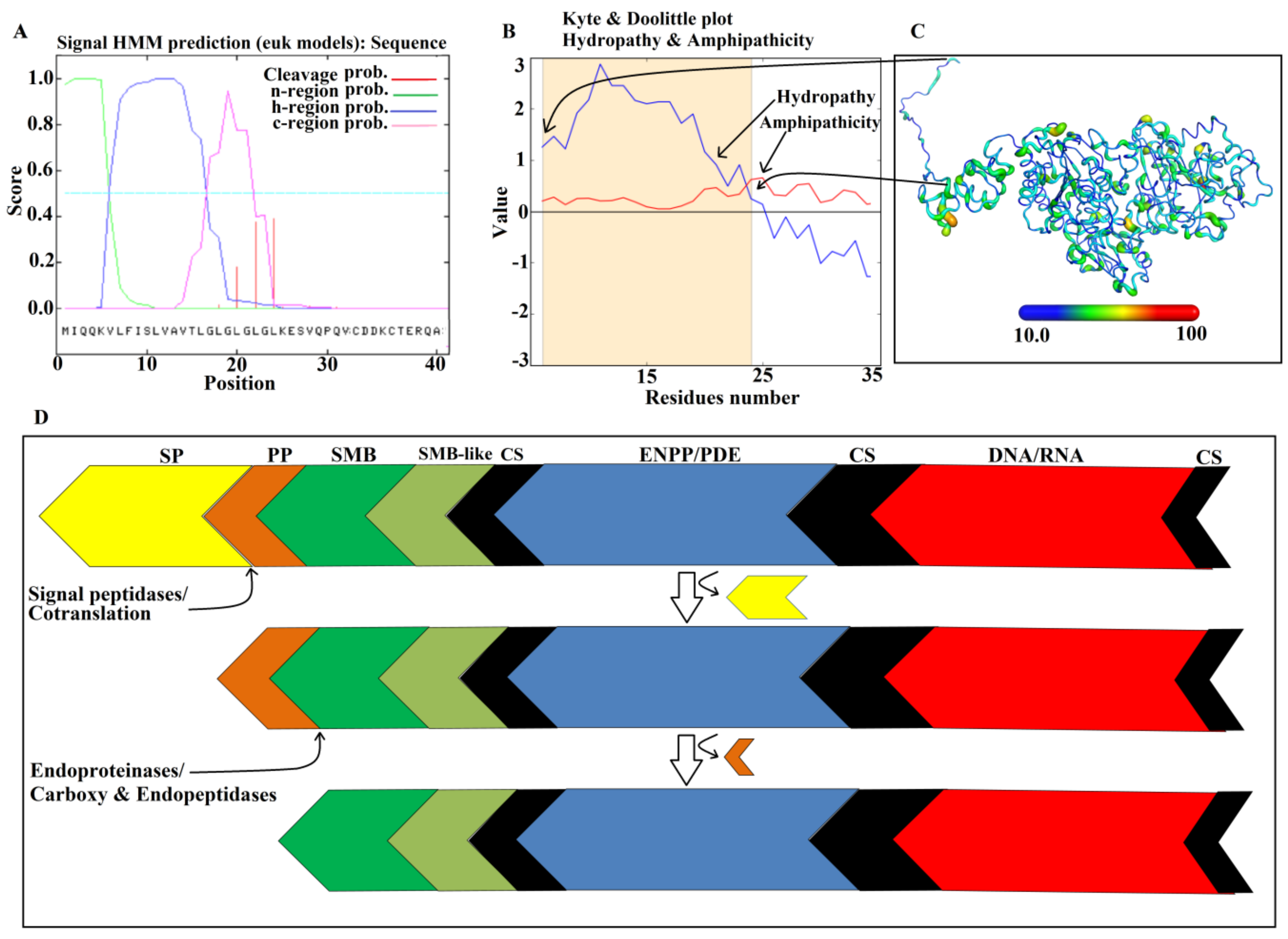
2.9. Maturation Mechanism for SVPDEs
3. Materials and Methods
3.1. Sequence Retrieval and Multiple Sequence Alignment
3.2. Sequence Logo Generated from Multiple Sequence Alignment
3.3. In Silico Analysis of the Domain and Biochemical Properties of the PDE_Ca
3.4. Prediction of Ligand Binding and Glycosylation Sites
3.5. Disulfide Bond Prediction
3.6. Homology Model Building of PDE_Ca
3.7. Molecular Dynamics Simulation
3.8. Model Validation
3.9. Structure Superimposition
3.10. Surface Charge Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kang, T.S.; Georgieva, D.; Genov, N.; Murakami, M.T.; Sinha, M.; Kumar, R.P.; Kaur, P.; Kumar, S.; Dey, S.; Sharma, S.; et al. Enzymatic toxins from snake venom: Structural characterization and mechanism of catalysis. FEBS J. 2011, 278, 4544–4576. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Masood, R.; Ali, I.; Ullah, K.; Ali, H.; Akbar, H.; Betzel, C. Thrombin-like enzymes from snake venom: Structural characterization and mechanism of action. Int. J. Biol. Macromol. 2018, 114, 788–811. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Chijiwa, T.; Oda-Ueda, N.; Ohno, M. Molecular diversity and accelerated evolution of C-type lectin-like proteins from snake venom. Toxicon 2005, 45, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kini, R.M.; Doley, R. Structure, function and evolution of three finger toxins: Mini proteins with multiple targets. Toxicon 2010, 56, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.W. A brief review of the scientific history of several lesser-known snake venom proteins: L-amino acid oxidases, hyaluronidases and phosphodiesterases. Toxicon 2013, 62, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Masood, R.; Spencer, P.J.; Murakami, M.T.; Arni, R.K. Crystallization and preliminary X-ray diffraction studies of an L-amino-acid oxidase from Lachesis muta venom. Acta Crystallogr. Sect. F Struct. Boil. Commun. 2014, 70, 1556–1559. [Google Scholar] [CrossRef]
- Ullah, A.; Magalhães, G.S.; Masood, R.; Mariutti, R.B.; Coronado, M.A.; Murakami, M.T.; Barbaro, K.C.; Arni, R.K. Crystallization and preliminary X-ray diffraction analysis of a novel sphingomyelinase D from Loxosceles gaucho venom. Acta Crystallogr. Sect. F Struct. Boil. Commun. 2014, 70, 1418–1420. [Google Scholar] [CrossRef]
- De Oliveira, L.M.F.; Ullah, A.; Masood, R.; Zelanis, A.; Spencer, P.J.; Serrano, S.M.; Arni, R.K. Rapid purification of serine proteinases from Bothrops alternatus and Bothrops moojeni venoms. Toxicon 2013, 76, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Ullah, A.; Coronado, M.; Murakami, M.T.; Betzel, C.; Arni, R.K. Crystallization and preliminary X-ray diffraction analysis of an L-amino-acid oxidase from Bothrops jararacussu venom. Acta Crystallogr. Sect. F Struct. Boil. Cryst. Commun. 2012, 68, 211–213. [Google Scholar] [CrossRef]
- Ullah, A.; Souza, T.D.A.C.B.D.; Masood, R.; Murakami, M.T.; Arni, R.K. Purification, crystallization and preliminary X-ray diffraction analysis of a class P-III metalloproteinase (BmMP-III) from the venom of Bothrops moojeni. Acta Crystallogr. Sect. F Struct. Boil. Cryst. Commun. 2012, 68, 1222–1225. [Google Scholar] [CrossRef]
- Ullah, A.; Souza, T.A.; Betzel, C.; Murakami, M.T.; Arni, R.K. Crystallographic portrayal of different conformational states of a Lys49 phospholipase A2 homologue: Insights into structural determinants for myotoxicity and dimeric configuration. Int. J. Biol. Macromol. 2012, 51, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Souza, T.A.; Abrego, J.R.; Betzel, C.; Murakami, M.T.; Arni, R.K. Structural insights into selectivity and cofactor binding in snake venom L-amino acid oxidases. Biochem. Biophys. Res. Commun. 2012, 421, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Nikai, T.; Sugihara, H. Phosphodiesterase from the venom of crotalus ruber ruber. Int. J. Biochem. 1987, 19, 115–119. [Google Scholar] [CrossRef]
- Stoynov, S.S.; Bakalova, A.T.; Dimov, S.I.; Mitkova, A.V.; Dolapchiev, L.B. Single-strand-specific DNase activity is an inherent property of the 140-kDa protein of the snake venom exonuclease. FEBS Lett. 1997, 409, 151–154. [Google Scholar] [CrossRef]
- da Silva, N.J., Jr.; Aird, S.D. Prey specificity, comparative lethality and compositional differences of coral snake venoms. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2001, 128, 425–456. [Google Scholar] [CrossRef]
- Sales, R.B.; Santoro, M.L. Nucleotidase and DNase activities in Brazilian snake venoms. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2008, 147, 85–95. [Google Scholar] [CrossRef]
- Dhananjaya, B.L.; D’Souza, C.J.M. An overview on nucleases (DNase, RNase, and phosphodiesterase) in snake venoms. Biochemistry 2010, 75, 1–6. [Google Scholar]
- Philipps, G.R. Purification and Characterization of Phosphodiesterase from Crotalus Venom. Hoppe Seylers Z. Physiol. Chem. 1975, 356, 1085–1096. [Google Scholar] [CrossRef]
- Sugihara, H.; Nikai, T.; Komori, Y.; Katada, H.; Mori, N. Purification and characterization of the phosphodiesterase from the venom of Akistrodon acutus (China). Jpn. J. Trop. Med. Hyg. 1984, 12, 247–254. [Google Scholar] [CrossRef]
- Björk, W. Purification of Phosphodiesterase from Bothrops atrox venoms, with special consideration of the elimination of monophosphatases. J. Biol. Chem. 1963, 238, 2487–2490. [Google Scholar]
- Valério, A.A.; Corradini, A.C.; Panunto, P.C.; Mello, S.M.; Hyslop, S. Purification and Characterization of a Phosphodiesterase from Bothrops alternatus Snake Venom. J. Protein Chem. 2002, 21, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Saad, S.M.; Khan, S.; Ashraf, M. Purification of phosphodiesterase I from Cerastes Vipera venom, biochemical and biological properties of the purified enzyme. Proc. Pak. Acad. Sci. 2009, 46, 1–12. [Google Scholar]
- Oka, J.; Ueda, K.; Hayaishi, O. Snake venom phosphodiesterase. Simple purification with blue sepharose and its application to poly (ADP-ribose) study. Biochem. Biophys. Res. Commun. 1978, 80, 841–848. [Google Scholar] [CrossRef]
- Tatsuki, T.; Iwanaga, S.; Suzuki, T. A simple method for preparation of snake venom phosphodiesterase almost free from 5’-nucleotidase. J. Biochem. 1975, 77, 831–836. [Google Scholar] [CrossRef]
- Laskowski, M., Sr. Purification and properties of venom phosphodiesterase. Methods Enzymol. 1980, 65, 276–284. [Google Scholar]
- Kini, R.M.; Gowda, T.V. Rapid method for separation and purification of four isoenzymes of Phosphodiesrerase from Trimeresurus flavorviridis (Habu snake) venom. J. Chromatogr. 1984, 291, 299–305. [Google Scholar] [CrossRef]
- Ballario, P.; Bergami, M.; Pedone, F. A simple method for the purification of phosphodiesterase from Vipera aspis venom. Anal. Biochem. 1977, 80, 646–651. [Google Scholar] [CrossRef]
- Sannaningaiah, D.; Subbaiah, G.K.; Kempaiah, K. Pharmacology of spider venom toxins. Toxin Rev. 2014, 33, 206–220. [Google Scholar] [CrossRef]
- Uzawa, S. Uber die phosphomonoesterase und die phosphodiesterase. J. Biochem. 1932, 15, 19–28. [Google Scholar] [CrossRef]
- Aird, S.D. Ophidian envenomation strategies and the role of purines. Toxicon 2002, 40, 335–393. [Google Scholar] [CrossRef]
- Aird, S.D. Taxonomic distribution and quantitative analysis of free purine and pyrimidine nucleosides in snake venoms. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2005, 140, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, M.B.; Stoggall, S.M.; Collis, M.G. Evidence that the adenosine receptor mediating relaxation in dog lateral saphenous vein and guinea-pig aorta is of the A2b subtype. Br. J. Pharmacol. 1991, 102, 198. [Google Scholar]
- Sobrevia, L.; Yudilevich, D.L.; Mann, G.E. Activation of A2-purinoceptors by adenosine stimulates L-arginine transport (system y+) and nitric oxide synthesis in human fetal endothelial cells. J. Physiol. 1997, 499, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Uzair, B.; Khan, B.A.; Sharif, N.; Shabbir, F.; Menaa, F. Phosphodiesterases (PDEs) from Snake Venoms: Therapeutic Applications. Protein Pept. Lett. 2018, 25, 612–618. [Google Scholar] [CrossRef]
- Iwanaga, S.; Suzuki, T. Enzymes in snake venom. In Snake Venoms; Lec, C.Y., Ed.; Springer: New York, NY, USA, 1979; Volume 19, pp. 61–158. [Google Scholar]
- Pollack, S.E.; Tetsuo, U.; David, S.A. Snake venom phosphodiesterase: A zinc metalloenzyme. J. Protein 1983, 2, 1–12. [Google Scholar] [CrossRef]
- Levy, Z.; Bdolah, A. Multiple molecular forms of snake venom phosphodiesterase from Vipera palastinae. Toxicon 1976, 14, 389–391. [Google Scholar] [CrossRef]
- Ferrè, F.; Clote, P. DiANNA 1.1: An extension of the DiANNA web server for ternary cysteine classification. Nucleic Acids Res. 2006, 34 (Suppl. 2), W182–W185. [Google Scholar] [CrossRef]
- Perron, S.; Mackessy, S.P.; Hyslop, R.M. Purification and characterization of exonuclease from rattlesnake venom. Acad. Sci. 1993, 25, 21–22. [Google Scholar]
- Mackessy, S.P. Venoms as Trophic Adaptations: An Ultrastructural Investigation of the Venom Apparatus and Biochemical Characterization of the Proteolytic Enzymes of the Northern Pacific Rattlesnake Crotalus Viridis Oreganus. Ph.D. Thesis, Washington State University, Pullman, WA, USA, 1989. [Google Scholar]
- Xue, Z.; Xu, D.; Wang, Y.; Zhang, Y. ThreaDom: Extracting protein domain boundary information from multiple threading alignments. Bioinformatics 2013, 29, i247–i256. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Trummal, K.; Aaspõllu, A.; Tõnismägi, K.; Samel, M.; Subbi, J.; Siigur, J.; Siigur, E. Phosphodiesterase from Vipera lebetina venom—Structure and characterization. Biochimie 2014, 106, 48–55. [Google Scholar] [CrossRef]
- Mitra, J.; Bhattacharyya, D. Phosphodiesterase from Daboia russelli russelli venom: Purification, partial characterization and inhibition of platelet aggregation. Toxicon 2014, 88, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Razzell, W.E.; Khorana, H.G. Studies on polynucleotides. III. Enzymic degradation; substrate specificity and properties of snake venom phosphodiesterase. J. Biol. Chem. 1959, 234, 2105–2113. [Google Scholar] [PubMed]
- Peng, L.; Xu, X.; Shen, D.; Zhang, Y.; Song, J.; Yan, X.; Guo, M. Purification and partial characterization of a novel phosphodiesterase from the venom of Trimeresurus stejnegeri: Inhibition of platelet aggregation. Biochimie 2011, 93, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.L.; Vaquero, T.S.; Paes Leme, A.F.; Serrano, S.M. NPP-BJ, a nucleotide pyrophosphatase/phosphodiesterase from Bothrops jararaca snake venom, inhibits platelet aggregation. Toxicon 2009, 54, 499–512. [Google Scholar] [CrossRef]
- Gupta, R.; Jung, E.; Brunak, S. Prediction of N-glycosylation Sites in Human Proteins. 2004. Available online: http://www.cbs.dtu.dk/services/NetNGlyc/ (accessed on 12 December 2018).
- Sulkowski, E.; Bjork, W.; Laskowski, M. A specific and nonspecific alkaline monophosphatase in the venom of Bothrops atrox and their occurrence in the purified venom phosphodiesterase. J. Biol. Chem. 1963, 238, 2477–2486. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Thornton, J.M. PROCHECK: Validation of protein structure coordinates. In International Tables of Crystallography: Volume F: Crystallography of Biological Macromolecules; Rossmann, M.G., Arnold, E., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001; pp. 722–725. [Google Scholar]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Protein Sci. 2016, 86, 3. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Gorelik, A.; Randriamihaja, A.; Illes, K.; Nagar, B. Structural basis for nucleotide recognition by the ectoenzyme CD203c. FEBS J. 2018, 285, 2481–2494. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Bowie, J.; Luthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Hospital, A.; Andrio, P.; Fenollosa, C.; Cicin-Sain, D.; Orozco, M.; Gelpí, J.L. MDWeb and MDMoby: An integrated web-based platform for molecular dynamics simulations. Bioinformatics 2012, 28, 1278–1279. [Google Scholar] [CrossRef]
- Jansen, S.; Perrakis, A.; Ulens, C.; Winkler, C.; Andries, M.; Joosten, R.P.; Van Acker, M.; Luyten, F.P.; Moolenaar, W.H.; Bollen, M. Structure of NPP1, an Ectonucleotide pyrophosphatase/phosphodiesterase involved in tissue calcification. Structure 2012, 20, 1948–1959. [Google Scholar] [CrossRef]
- Zalatan, J.G.; Fenn, T.D.; Brunger, A.T.; Herschlag, D. Structural and functional comparisons of nucleotide pyrophosphatase/phosphodiesterase and alkaline phosphatase: Implications for mechanism and evolution. Biochemistry 2006, 45, 9788–9803. [Google Scholar] [CrossRef]
- Stein, A.J.; Bain, G.; Prodanovich, P.; Santini, A.M.; Darlington, J.; Stelzer, N.M.P.; Sidhu, R.S.; Schaub, J.; Goulet, L.; Lonergan, D.; et al. Structural Basis for Inhibition of Human Autotaxin by Four Potent Compounds with Distinct Modes of Binding. Mol. Pharmacol. 2015, 88, 982–992. [Google Scholar] [CrossRef]
- Rokyta, D.R.; Margres, M.J.; Calvin, K. Post-transcriptional Mechanisms Contribute Little to Phenotypic Variation in Snake Venoms. G3 Genes Genomes Genet. 2015, 5, 2375–2382. [Google Scholar] [CrossRef] [Green Version]
- Hausmann, J.; Kamtekar, S.; Christodoulou, E.; Day, J.E.; Wu, T.; Fulkerson, Z.; Albers, H.M.; Van Meeteren, L.A.; Houben, A.J.S.; Van Zeijl, L.; et al. Structural basis for substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Boil. 2011, 18, 198–204. [Google Scholar] [CrossRef]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef] [PubMed]
- Philipps, G.R. Purification and characterization of phosphodiesterase I from Bothrops atrox. Biochim. Biophys. Acta 1976, 432, 237–244. [Google Scholar]
- Boman, H.G. On the specificity of the snake venom phosphodiesterase. Ann. N. Y. Acad. Sci. 1959, 81, 800–803. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Wang, H.; Ye, M. Structural Insight into the Substrate Specificity of Phosphodiesterases. Pharmacol. Itch 2011, 204, 121–134. [Google Scholar]
- Bendtsen, J.D.; Nielsen, H.; von Heijne, G.; Brunak, S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004, 340, 783–795. [Google Scholar] [CrossRef]
- Von Heijne, G. On the hydrophobic nature of signal sequences. Eur. J. Biochem. 1981, 116, 419–422. [Google Scholar] [CrossRef]
- Lively, M.O.; Walsh, K.A. Hen oviduct signal peptidase is an integral membrane protein. J. Biol. Chem. 1983, 258, 9488–9495. [Google Scholar]
- Ullah, A.; Mariutti, R.B.; Masood, R.; Caruso, I.P.; Costa, G.H.; de Freita, C.M.; Santos, C.R.; Zanphorlin, L.M.; Mutton, M.J.R.; Murakami, M.T.; et al. Crystal structure of mature 2S albumin from Moringa oleifera seeds. Biochem. Biophys. Res. Commun. 2015, 468, 365–371. [Google Scholar] [CrossRef]
- Khan, A.R.; James, M.N. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 1998, 7, 815–836. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Hofmann, K.; Baron, M.D. Institute for Animal Health Ash Road Pirbright, Surrey GU24 0, 2017 (NF U.K.).
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.D.; Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, W327–W331. [Google Scholar] [CrossRef] [PubMed]
- Wass, M.N.; Kelley, L.A.; Sternberg, M.J. 3DLigandSite: Predicting ligand-binding sites using similar structures. Nucleic Acids Res. 2010, 38, W469–W473. [Google Scholar] [CrossRef] [PubMed]
- De Castro, E.; Sigrist, C.J.A.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, N.; Hulo, A. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef]
- Yaseen, A.; Li, Y. Dinosolve: A protein disulfide bonding prediction server using context-based features to enhance prediction accuracy. BMC Bioinform. 2013, 14, S9. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theor. Comput. 2015, 8, 3696–3713. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulation. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Masood, R.; Hayat, Z.; Hafeez, A. Determining the Structures of the Snake and Spider Toxins by X-Rays. Methods Mol. Biol. 2020, 2068, 163–172. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | PDE_Ca | PDE_Pm | PDE_Ml | PDE_Pe | PDE_Oo | 5GZ4 | 6CO1 | 4B56 | 5IJQ |
|---|---|---|---|---|---|---|---|---|---|
| PDE_Ca | ---- | 85% | 93% | 92% | 96% | 85% | 64% | 50% | 47% |
| PDE_Pm | 85% | ----- | 92% | 94% | 95% | 84% | 63% | 50% | 48% |
| PDE_Ml | 93% | 92% | ----- | 87% | 90% | 86% | 63% | 50% | 46% |
| PDE_Pe | 92% | 94% | 87% | ------ | 95% | 85% | 64% | 50% | 49% |
| PDE_Oo | 96% | 95% | 90% | 95% | ------ | 85% | 64% | 50% | 48% |
| 5GZ4 | 85% | 84% | 86% | 85% | 85% | ----- | 64% | 50% | 47% |
| 6CO1 | 64% | 63% | 63% | 64% | 64% | 64% | ----- | 53% | 46% |
| 4B56 | 50% | 50% | 50% | 50% | 50% | 50% | 53% | ----- | 44% |
| 5IJQ | 47% | 48% | 46% | 49% | 48% | 47% | 46% | 44% | ----- |
| 1st Cysteine | 2nd Cysteine |
|---|---|
| 34 | 51 |
| 38 | 75 |
| 49 | 62 |
| 55 | 61 |
| 84 | 101 |
| 89 | 119 |
| 99 | 112 |
| 105 | 111 |
| 130 | 176 |
| 138 | 350 |
| 366 | 466 |
| 415 | 819 |
| 555 | 618 |
| 569 | 675 |
| 571 | 660 |
| 767 | 777 |
| Residue 1 | Residue 2 | Distance |
|---|---|---|
| NH1 ARG A 58 | OD1 ASP A 52 | 3.59 |
| NH1 ARG A 58 | OD2 ASP A 52 | 2.60 |
| NH2 ARG A 58 | OD1 ASP A 52 | 2.69 |
| NH2 ARG A 58 | OD2 ASP A 52 | 3.30 |
| NH2 ARG A 82 | OE1 GLU A 85 | 3.39 |
| NH1 ARG A 87 | OD1 ASP A 104 | 2.58 |
| NH1 ARG A 87 | OD2 ASP A 104 | 3.54 |
| NH2 ARG A 87 | OD1 ASP A 98 | 2.84 |
| NH2 ARG A 87 | OD2 ASP A 98 | 3.93 |
| NH2 ARG A 87 | OD1 ASP A 104 | 3.31 |
| NH2 ARG A 87 | OD2 ASP A 104 | 2.62 |
| NZ LYS A 102 | OD2 ASP A 98 | 2.69 |
| NZ LYS A 168 | OD2 ASP A 158 | 2.87 |
| ND1 HIS A 189 | OD2 ASP A 352 | 3.42 |
| NE2 HIS A 189 | OD2 ASP A 352 | 3.82 |
| NH1 ARG A 278 | OE1 GLU A 302 | 2.85 |
| NH2 ARG A 278 | OE1 GLU A 302 | 2.88 |
| NE2 HIS A 309 | OD1 ASP A 305 | 2.89 |
| NE2 HIS A 309 | OD2 ASP A 305 | 2.95 |
| NZ LYS A 337 | OD2 ASP A 122 | 3.84 |
| NH2 ARG A 339 | OD1 ASP A 287 | 3.88 |
| NH2 ARG A 339 | OD2 ASP A 287 | 2.58 |
| NE2 HIS A 353 | OD1 ASP A 147 | 3.04 |
| NE2 HIS A 353 | OD1 ASP A 352 | 3.46 |
| NE2 HIS A 353 | OD2 ASP A 352 | 2.96 |
| NH1 ARG A 384 | OD2 ASP A 205 | 2.62 |
| NH2 ARG A 384 | OD2 ASP A 205 | 3.69 |
| NH2 ARG A 384 | OD2 ASP A 436 | 3.73 |
| NZ LYS A 425 | OD2 ASP A 727 | 2.56 |
| NH1 ARG A 426 | OD1 ASP A 465 | 3.74 |
| NH1 ARG A 426 | OE1 GLU A 467 | 3.98 |
| NH2 ARG A 426 | OD1 ASP A 465 | 2.70 |
| NH2 ARG A 426 | OD2 ASP A 465 | 3.11 |
| NH2 ARG A 426 | OE1 GLU A 467 | 3.26 |
| NE2 HIS A 428 | OD1 ASP A 816 | 2.79 |
| NH2 ARG A 434 | OD2 ASP A 210 | 2.94 |
| NE2 HIS A 462 | OD1 ASP A 305 | 3.83 |
| NE2 HIS A 462 | OD2 ASP A 305 | 2.68 |
| NZ LYS A 469 | OE1 GLU A 155 | 3.97 |
| NE2 HIS A 515 | OD1 ASP A 502 | 2.80 |
| NE2 HIS A 515 | OD2 ASP A 502 | 2.97 |
| NH1 ARG A 588 | OE1 GLU A 534 | 3.19 |
| NH2 ARG A 588 | OE1 GLU A 534 | 2.60 |
| NH2 ARG A 645 | OD1 ASP A 643 | 3.26 |
| NH2 ARG A 645 | OD2 ASP A 643 | 2.59 |
| NZ LYS A 710 | OE1 GLU A 804 | 3.97 |
| NH1 ARG A 786 | OE1 GLU A 791 | 3.76 |
| NH2 ARG A 786 | OD1 ASP A 788 | 3.97 |
| NE2 HIS A 810 | OE1 GLU A 791 | 3.37 |
| NH1 ARG A 813 | OE1 GLU A 492 | 3.24 |
| NH2 ARG A 813 | OD1 ASP A 816 | 2.69 |
| NH1 ARG A 815 | OE1 GLU A 818 | 2.75 |
| NZ LYS A 840 | OE1 GLU A 818 | 3.80 |
| Protein | Average Volume (Å3) | Average Depth (Å) |
|---|---|---|
| PD_Ca model | 6608.25 | 21.39 |
| PD_Vl model | 3985.03 | 12.54 |
| 6C01 | 14,690.95 | 19.13 |
| 4B56 | 3651.33 | 16.30 |
| 2GSN | 10,107.28 | 18.58 |
| 4ZG7 | 11,367.42 | 17.94 |
| Protein | RMSD Value |
|---|---|
| PDE_Ca aligned 6C01 | 0.21 |
| PDE_Ca aligned 4B56 | 0.72 |
| PDE_Ca aligned 4ZG7 | 0.73 |
| PDE_Ca aligned 2GSN | 0.92 |
| PDE_Ca aligned PDE_Vl | 0.57 |
| PDE_Ca aligned PDE_Ba | 0.56 |
| PDE_Ca aligned 5GZ4 | 0.60 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, A.; Ullah, K.; Ali, H.; Betzel, C.; ur Rehman, S. The Sequence and a Three-Dimensional Structural Analysis Reveal Substrate Specificity among Snake Venom Phosphodiesterases. Toxins 2019, 11, 625. https://doi.org/10.3390/toxins11110625
Ullah A, Ullah K, Ali H, Betzel C, ur Rehman S. The Sequence and a Three-Dimensional Structural Analysis Reveal Substrate Specificity among Snake Venom Phosphodiesterases. Toxins. 2019; 11(11):625. https://doi.org/10.3390/toxins11110625
Chicago/Turabian StyleUllah, Anwar, Kifayat Ullah, Hamid Ali, Christian Betzel, and Shafiq ur Rehman. 2019. "The Sequence and a Three-Dimensional Structural Analysis Reveal Substrate Specificity among Snake Venom Phosphodiesterases" Toxins 11, no. 11: 625. https://doi.org/10.3390/toxins11110625