A Data-Independent Methodology for the Structural Characterization of Microcystins and Anabaenopeptins Leading to the Identification of Four New Congeners




Abstract
1. Introduction
2. Results and Discussion
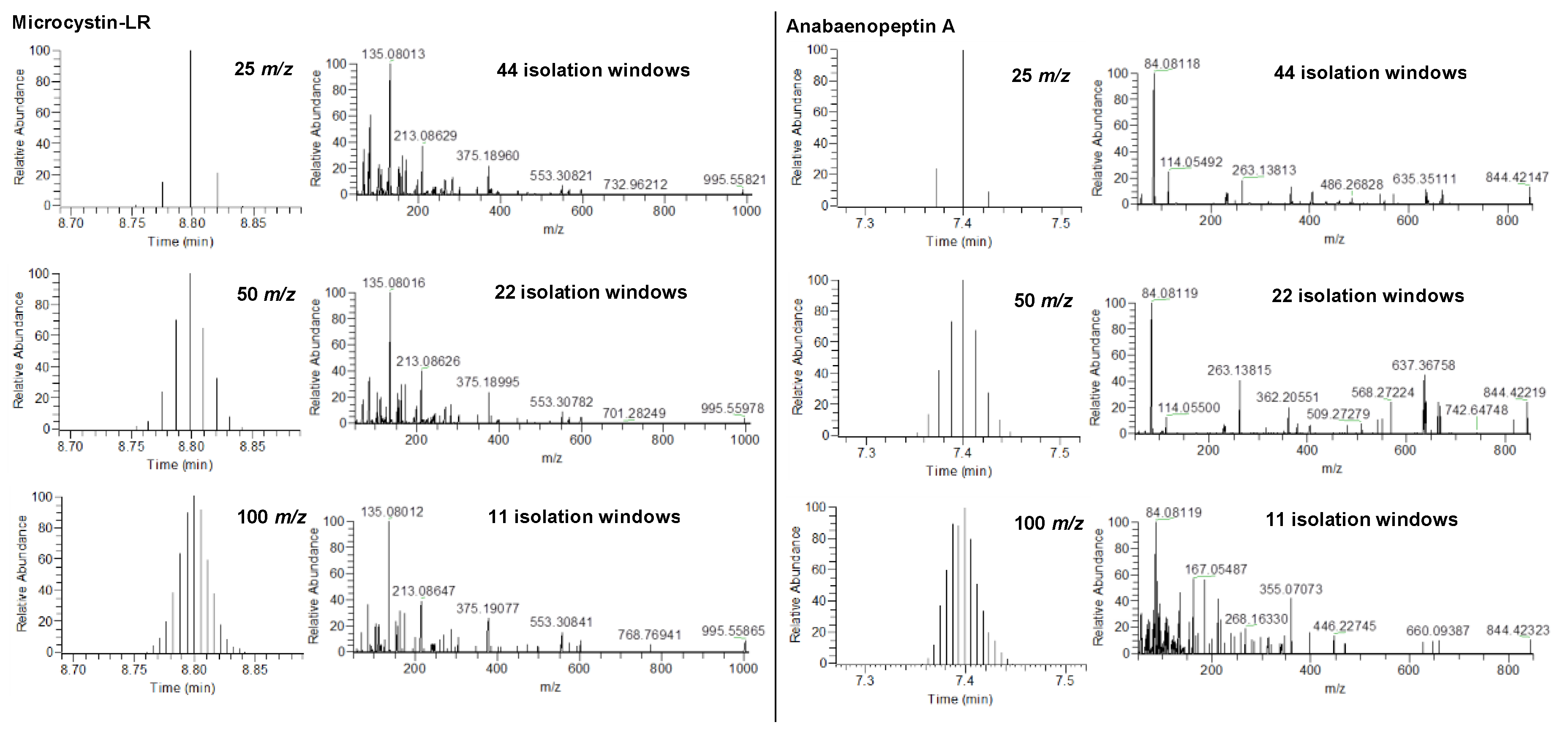
2.1. HRMS Parameters for Suspect Screening via DIA
2.2. Building in-House Databases
2.3. First Features Selection with Compound Discoverer
2.4. Confirmation of Suspects Using MS/MS Spectra
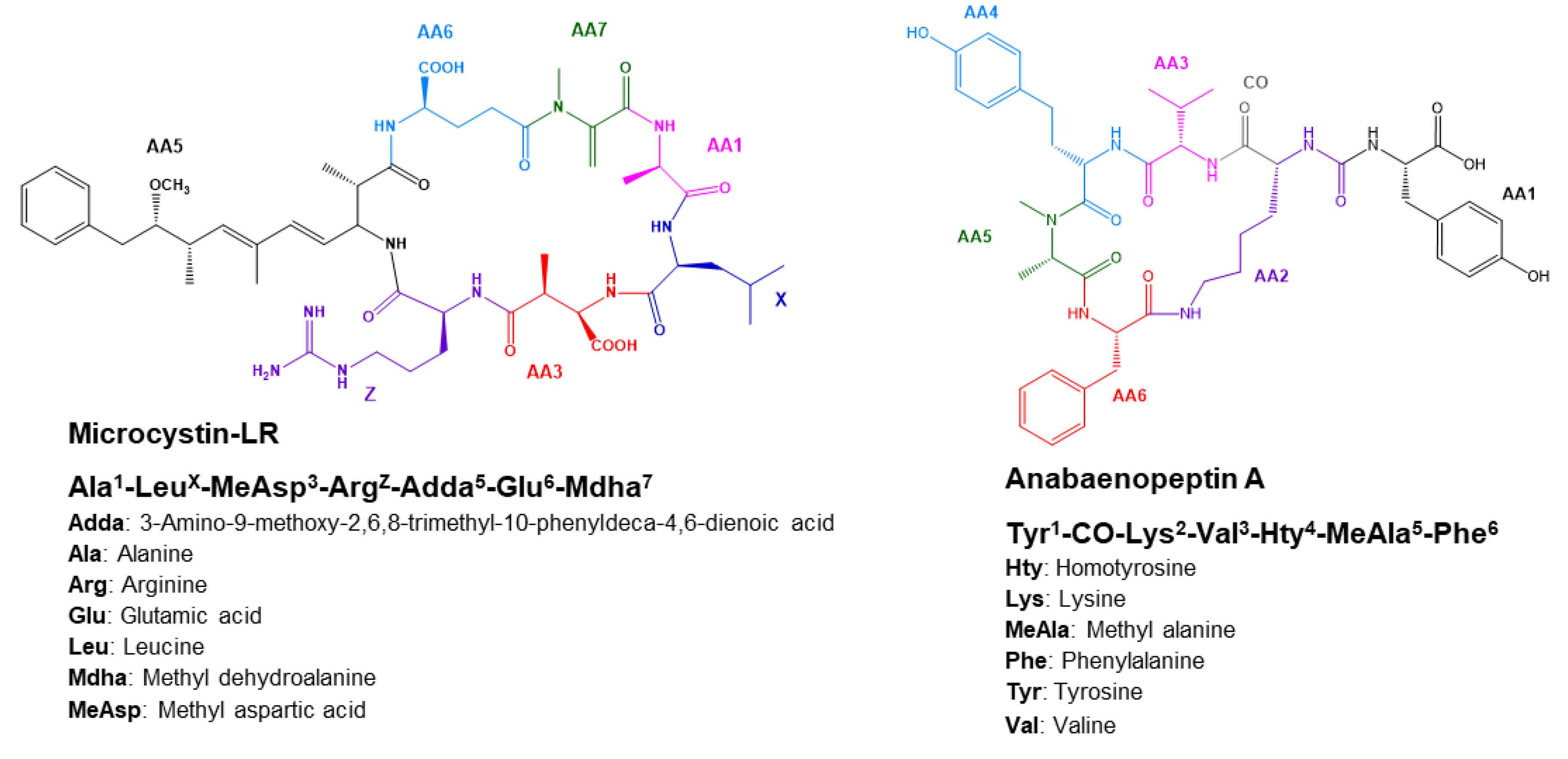
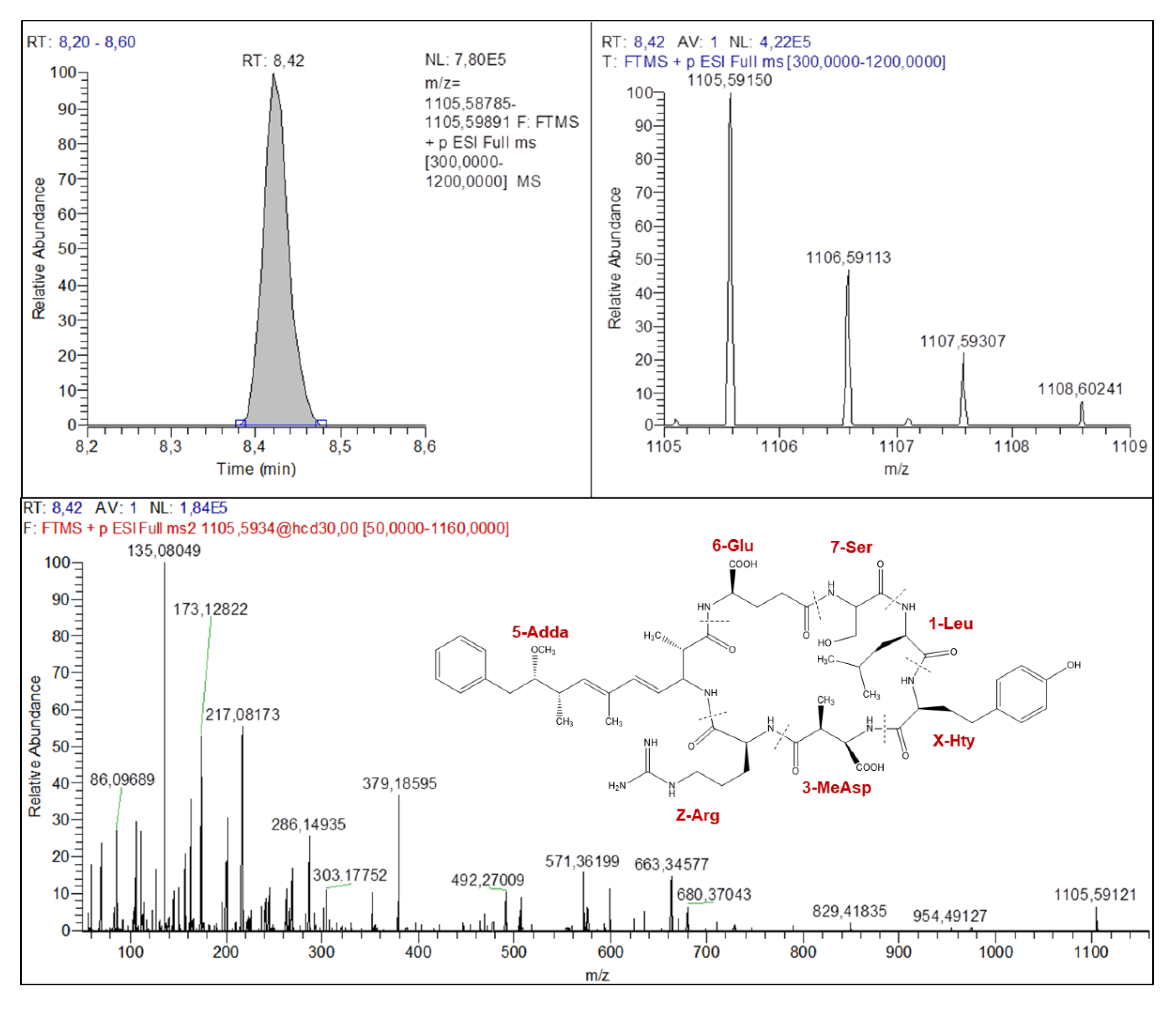
2.4.1. Microcystins Structures Elucidation
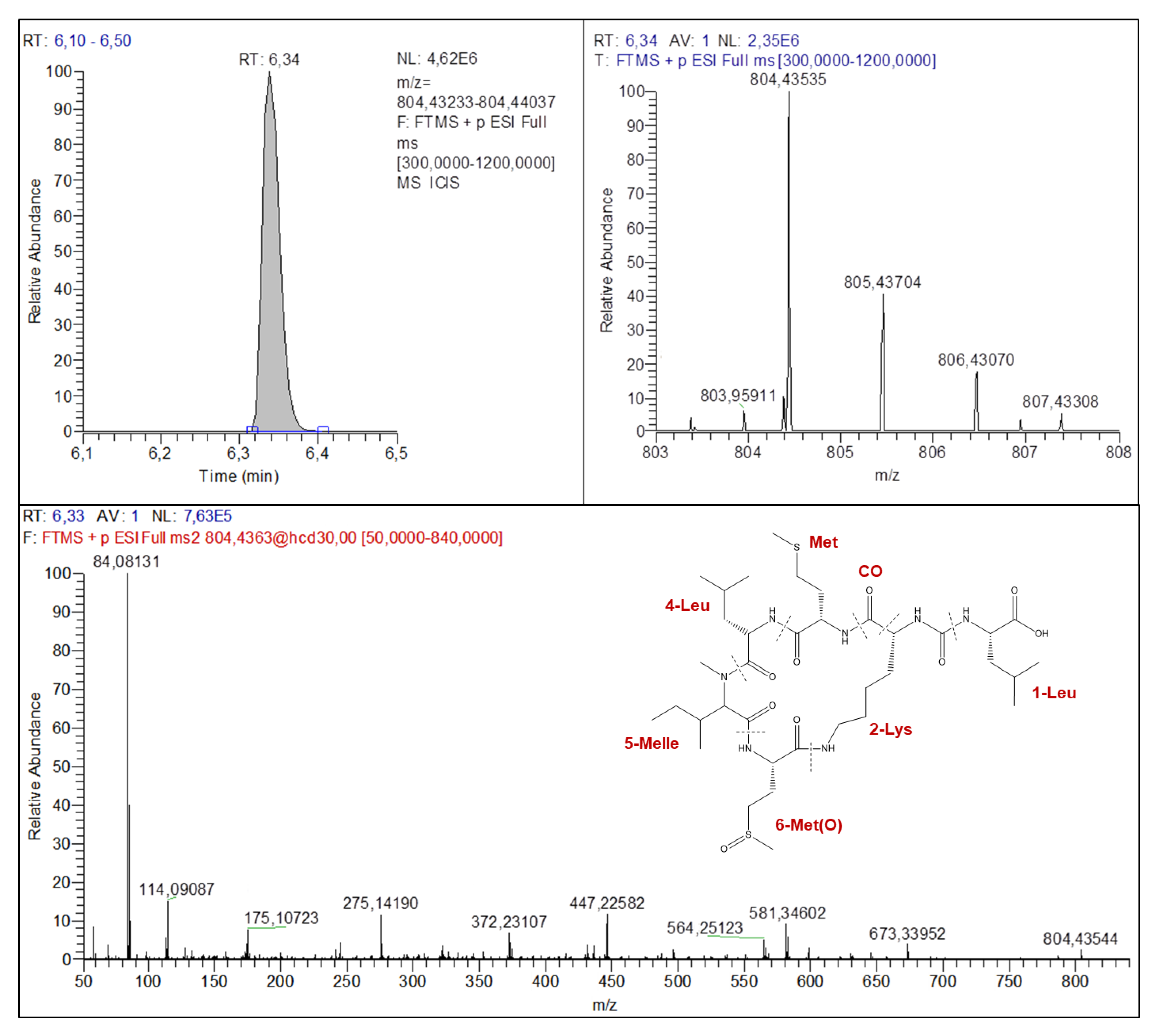
2.4.2. Anabaenopeptins Structures Elucidation
2.5. Quantification and Semi-Quantification
3. Conclusions
4. Materials and Methods
4.1. Chemicals, Reagents and Stock Solutions
4.2. Sample Collection, Preparation and Quantification
4.3. Instrumental Conditions
4.3.1. On-Line Solid Phase Extraction and Chromatographic Conditions
4.3.2. HRMS Conditions
4.4. Suspect Screening Using DIA Methodology
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bartram, J.; Chorus, I. Toxic Cyanobacteria in Water: A Guide to Their Public Health Consequences, Monitoring and Management; CRC Press: Boca Raton, FL, USA, 1999. [Google Scholar]
- Carmichael, W.W.; Azevedo, S.; An, J.S.; Molica, R.; Jochimsen, E.M.; Lau, S.; Rinehart, K.L.; Shaw, G.R.; Eaglesham, G.K. Human fatalities from cyanobacteria: Chemical and biological evidence for cyanotoxins. Environ. Health Perspect. 2001, 109, 663–668. [Google Scholar] [CrossRef] [PubMed]
- USEPA. Guidelines and Recommandations. Available online: https://www.epa.gov/nutrient-policy-data/guidelines-and-recommendations#what2 (accessed on 2 February 2019).
- World Health Organization. Guidelines for Drinking-Water Quality; World Health Organization: Geneva, Switzerland, 2017; p. 541. [Google Scholar]
- Janssen, E.M.-L. Cyanobacterial peptides beyond microcystins—A review on co-occurrence, toxicity, and challenges for risk assessment. Water Res. 2019, 151, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.; Vasconcelos, V. Molecular mechanisms of microcystin toxicity in animal cells. Int. J. Mol. Sci. 2010, 11, 268–287. [Google Scholar] [CrossRef] [PubMed]
- Spoof, L.; Błaszczyk, A.; Meriluoto, J.; Cegłowska, M.; Mazur-Marzec, H. Structures and activity of new anabaenopeptins produced by Baltic Sea cyanobacteria. Mar. Drugs 2016, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Sedmak, B.; Carmeli, S.; Eleršek, T. “Non-toxic” cyclic peptides induce lysis of cyanobacteria—An effective cell population density control mechanism in cyanobacterial blooms. Microb. Ecol. 2008, 56, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Welker, M.; Von Döhren, H. Cyanobacterial peptides—Nature’s own combinatorial biosynthesis. FEMS Microbiol. Rev. 2006, 30, 530–563. [Google Scholar] [CrossRef] [PubMed]
- Meriluoto, J.; Spoof, L.; Codd, G.A. Handbook of Cyanobacterial Monitoring and Cyanotoxin Analysis; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Ortiz, X.; Korenkova, E.; Jobst, K.J.; MacPherson, K.A.; Reiner, E.J. A high throughput targeted and non-targeted method for the analysis of microcystins and anatoxin—A using on-line solid phase extraction coupled to liquid chromatography–quadrupole time-of-flight high resolution mass spectrometry. Anal. Bioanal. Chem. 2017, 409, 4959–4969. [Google Scholar] [CrossRef]
- Bletsou, A.A.; Jeon, J.; Hollender, J.; Archontaki, E.; Thomaidis, N.S. Targeted and non-targeted liquid chromatography-mass spectrometric workflows for identification of transformation products of emerging pollutants in the aquatic environment. TrAC Trends Anal. Chem. 2015, 66, 32–44. [Google Scholar] [CrossRef]
- Crimmins, B.S.; Holsen, T.M. Non-targeted Screening in Environmental Monitoring Programs. In Advancements of Mass Spectrometry in Biomedical Research; Springer: Basel, Switzerland, 2019; pp. 731–741. [Google Scholar]
- Teta, R.; Della Sala, G.; Glukhov, E.; Gerwick, L.; Gerwick, W.H.; Mangoni, A.; Costantino, V. Combined LC–MS/MS and molecular networking approach reveals new cyanotoxins from the 2014 cyanobacterial bloom in green lake, seattle. Environ. Sci. Technol. 2015, 49, 14301–14310. [Google Scholar] [CrossRef]
- Bogialli, S.; Bortolini, C.; Di Gangi, I.M.; Di Gregorio, F.N.; Lucentini, L.; Favaro, G.; Pastore, P. Liquid chromatography-high resolution mass spectrometric methods for the surveillance monitoring of cyanotoxins in freshwaters. Talanta 2017, 170, 322–330. [Google Scholar] [CrossRef]
- Yilmaz, M.; Foss, A.J.; Miles, C.O.; Özen, M.; Demir, N.; Balcı, M.; Beach, D.G. Comprehensive multi-technique approach reveals the high diversity of microcystins in field collections and an associated isolate of Microcystis aeruginosa from a Turkish lake. Toxicon 2019, 167, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Schymanski, E.L.; Singer, H.P.; Slobodnik, J.; Ipolyi, I.M.; Oswald, P.; Krauss, M.; Schulze, T.; Haglund, P.; Letzel, T.; Grosse, S. Non-target screening with high-resolution mass spectrometry: Critical review using a collaborative trial on water analysis. Anal. Bioanal. Chem. 2015, 407, 6237–6255. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Ogawa, K.; Matsuura, K.; Murata, H.; Suzuki, M.; Watanabe, M.F.; Itezono, Y.; Nakayama, N. Structural determination of geometrical isomers of microcystins LR and RR from cyanobacteria by two-dimensional NMR spectroscopic techniques. Chem. Res. Toxicol. 1990, 3, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Shin, H.J.; Matsuda, H.; Ishida, K.; Yamaguchi, K. A cyclic peptide, anabaenopeptin B, from the cyanobacterium Oscillatoria agardhii. Phytochemistry 1997, 44, 449–452. [Google Scholar] [CrossRef]
- Grach-Pogrebinsky, O.; Sedmak, B.; Carmeli, S. Seco [d-Asp3] microcystin-RR and [d-Asp3, d-Glu (OMe)6] microcystin-RR, two new microcystins from a toxic water bloom of the cyanobacterium planktothrix rubescens. J. Nat. Prod. 2004, 67, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.D.; Strangman, W.K.; Barbera, A.E.; Mallin, M.A.; McIver, M.R.; Wright, J.L. Microcystins and two new micropeptin cyanopeptides produced by unprecedented Microcystis aeruginosa blooms in North Carolina’s Cape Fear River. Harmful Algae 2014, 31, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Welker, M.; Fastner, J.; Erhard, M.; von Döhren, H. Applications of MALDI-TOF MS analysis in cyanotoxin research. Environ. Toxicol. Int. J. 2002, 17, 367–374. [Google Scholar] [CrossRef]
- Czarnecki, O.; Henning, M.; Lippert, I.; Welker, M. Identification of peptide metabolites of Microcystis (Cyanobacteria) that inhibit trypsin—Like activity in planktonic herbivorous Daphnia (Cladocera). Environ. Microbiol. 2006, 8, 77–87. [Google Scholar] [CrossRef]
- Chorus, I. Cyanotoxins: Occurrence, Causes, Consequences; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Haruštiaková, D.; Welker, M. Chemotype diversity in P lanktothrix rubescens (cyanobacteria) populations is correlated to lake depth. Environ. Microbiol. Rep. 2017, 9, 158–168. [Google Scholar] [CrossRef]
- Bouhaddada, R.; Nélieu, S.; Nasri, H.; Delarue, G.; Bouaïcha, N. High diversity of microcystins in a Microcystis bloom from an Algerian lake. Environ. Pollut. 2016, 216, 836–844. [Google Scholar] [CrossRef]
- Di Gregorio, F.N.; Bogialli, S.; Ferretti, E.; Lucentini, L. First evidence of MC-HtyR associated to a Plankthothrix rubescens blooming in an Italian lake based on a LC-MS method for routinely analysis of twelve microcystins in freshwaters. Microchem. J. 2017, 130, 329–335. [Google Scholar] [CrossRef]
- Ballot, A.; Sandvik, M.; Rundberget, T.; Botha, C.J.; Miles, C.O. Diversity of cyanobacteria and cyanotoxins in Hartbeespoort Dam, South Africa. Mar. Freshw. Res. 2014, 65, 175–189. [Google Scholar] [CrossRef]
- Sanz, M.; Andreote, A.; Fiore, M.; Dörr, F.; Pinto, E. Structural characterization of new peptide variants produced by cyanobacteria from the Brazilian Atlantic coastal Forest using liquid chromatography coupled to quadrupole time-of-flight tandem mass spectrometry. Mar. Drugs 2015, 13, 3892–3919. [Google Scholar] [CrossRef] [PubMed]
- Diehnelt, C.W.; Dugan, N.R.; Peterman, S.M.; Budde, W.L. Identification of microcystin toxins from a strain of Microcystis aeruginosa by liquid chromatography introduction into a hybrid linear ion trap-fourier transform ion cyclotron resonance mass spectrometer. Anal. Chem. 2006, 78, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Benke, P.; Kumar, M.V.; Pan, D.; Swarup, S. A mass spectrometry-based unique fragment approach for the identification of microcystins. Analyst 2015, 140, 1198–1206. [Google Scholar] [CrossRef]
- Qi, Y.; Rosso, L.; Sedan, D.; Giannuzzi, L.; Andrinolo, D.; Volmer, D.A. Seven new microcystin variants discovered from a native Microcystis aeruginosa strain–unambiguous assignment of product ions by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2015, 29, 220–224. [Google Scholar] [CrossRef]
- Moschet, C.; Lew, B.M.; Hasenbein, S.; Anumol, T.; Young, T.M. LC-and GC-QTOF-MS as complementary tools for a comprehensive micropollutant analysis in aquatic systems. Environ. Sci. Technol. 2017, 51, 1553–1561. [Google Scholar] [CrossRef]
- Roy-Lachapelle, A.; Duy, S.V.; Munoz, G.; Dinh, Q.T.; Bahl, E.; Simon, D.F.; Sauvé, S. Analysis of multiclass cyanotoxins (microcystins, anabaenopeptins, cylindrospermopsin and anatoxins) in lake waters using on-line SPE liquid chromatography high-resolution Orbitrap mass spectrometry. Anal. Methods 2019, 11, 3126–3133. [Google Scholar] [CrossRef]
- Letzel, T.; Lucke, T.; Schulz, W.; Sengl, M.; Letzel, M. OMI (Organic Molecule Identification) in water using LC-MS (/MS): Steps from “unknown” to “identified”: A contribution to the discussion In a class of its own. Lab More Int. 2014, 4, 24–28. [Google Scholar]
- Egertson, J.D.; Kuehn, A.; Merrihew, G.E.; Bateman, N.W.; MacLean, B.X.; Ting, Y.S.; Canterbury, J.D.; Marsh, D.M.; Kellmann, M.; Zabrouskov, V. Multiplexed MS/MS for improved data-independent acquisition. Nat. Methods 2013, 10, 744. [Google Scholar] [CrossRef]
- Doerr, A. DIA mass spectrometry. Nat. Methods 2014, 12, 35. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.; Chen, X.; Zhong, L.; Yin, Y. Development of data-independent acquisition workflows for metabolomic analysis on a quadrupole-orbitrap platform. Talanta 2017, 164, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Ang, J.E.; Revell, V.; Mann, A.; Mäntele, S.; Otway, D.T.; Johnston, J.D.; Thumser, A.E.; Skene, D.J.; Raynaud, F. Identification of human plasma metabolites exhibiting time-of-day variation using an untargeted liquid chromatography–mass spectrometry metabolomic approach. Chronobiol. Int. 2012, 29, 868–881. [Google Scholar] [CrossRef] [PubMed]
- Nikolskiy, I.; Mahieu, N.G.; Chen, Y.J.; Tautenhahn, R.; Patti, G.J. An untargeted metabolomic workflow to improve structural characterization of metabolites. Anal. Chem. 2013, 85, 7713–7719. [Google Scholar] [CrossRef] [PubMed]
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted metabolomics strategies—Challenges and emerging directions. J. Am. Soc. Mass Spectrom. 2016, 27, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Flores, C.; Caixach, J. An integrated strategy for rapid and accurate determination of free and cell-bound microcystins and related peptides in natural blooms by liquid chromatography–electrospray-high resolution mass spectrometry and matrix-assisted laser desorption/ionization time-of-flight/time-of-flight mass spectrometry using both positive and negative ionization modes. J. Chromatogr. A 2015, 1407, 76–89. [Google Scholar] [PubMed]
- Tsuji, K.; Naito, S.; Kondo, F.; Ishikawa, N.; Watanabe, M.F.; Suzuki, M.; Harada, K.I. Stability of microcystins from cyanobacteria: Effect of light on decomposition and isomerization. Environ. Sci. Technol. 1994, 28, 173–177. [Google Scholar] [CrossRef]
- Sano, T.; Kaya, K. Oscillamide Y, a chymotrypsin inhibitor from toxic Oscillatoria agardhii. Tetrahedron Lett. 1995, 36, 5933–5936. [Google Scholar] [CrossRef]
- Gkelis, S.; Lanaras, T.; Sivonen, K. Cyanobacterial toxic and bioactive peptides in freshwater bodies of Greece: Concentrations, occurrence patterns, and implications for human health. Mar. Drugs 2015, 13, 6319–6335. [Google Scholar] [CrossRef]
- Beversdorf, L.; Weirich, C.; Bartlett, S.; Miller, T. Variable cyanobacterial toxin and metabolite profiles across six eutrophic lakes of differing physiochemical characteristics. Toxins 2017, 9, 62. [Google Scholar] [CrossRef]
- Kokociński, M.; Dziga, D.; Spoof, L.; Stefaniak, K.; Jurczak, T.; Mankiewicz-Boczek, J.; Meriluoto, J. First report of the cyanobacterial toxin cylindrospermopsin in the shallow, eutrophic lakes of western Poland. Chemosphere 2009, 74, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Pick, F.R. Blooming algae: A Canadian perspective on the rise of toxic cyanobacteria. Can. J. Fish. Aquat. Sci. 2016, 73, 1149–1158. [Google Scholar] [CrossRef]
- USEPA. EPA Drinking Water Health Advisories for Cyanotoxins. Available online: https://www.epa.gov/cyanohabs/epa-drinking-water-health-advisories-cyanotoxins (accessed on 26 September 2019).
- Biosynthesis. Amino Acid Masses Tables. Available online: https://www.biosyn.com/tew/amino-acid-masses-tables.aspx (accessed on 16 September 2019).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parent and Fragment Ions | Known MC (Certified Standard) | Known MC (No Certified Standard) | Unknown MC | |||
|---|---|---|---|---|---|---|
| MC-LR | [GluOMe6] MC-LR | [M(O)1] MC-LR | [M(O)1, GluOMe6] MC-LR | [Asp3]MC-RHar | [Leu1, Ser7] MC-HtyR | |
| M+H+ | 995.55927 | 1009.57104 | 1071.55340 | 1085.56928 | 1038.57291 | 1105.59150 |
| Isotope #1 | 996.55629 | 1010.56808 | 1072.55682 | 1086.5709 | 1039.57413 | 1106.59113 |
| Isotope #2 | 997.56067 | 1011.57490 | 1073.55194 | 1087.57513 | 1107.59307 | |
| Isotope #3 | 998.56488 | 1074.56341 | 1088.56762 | 1108.60241 | ||
| M+2H2+ | 536.27992 | 519.78065 | ||||
| Isotope #1 | 536.77931 | 520.29199 | ||||
| Isotope #2 | 537.28075 | 520.79363 | ||||
| Isotope #3 | 521.29156 | |||||
| M+H+-H2O | 977.56032 | 991.56022 | 1053.54316 | 1067.55874 | ||
| M+H+-CO | 967.54996 | 981.57442 | 1077.59665 | |||
| M+H+-CH2NHC(NH)NH2) (Arg) | 999.49677 | |||||
| M+H+-AA6 | 866.51198 | 976.54852 | ||||
| M+H+-134 (Adda) | 861.47956 | 875.49571 | 937.48531 | 951.49270 | 904.49915 | 971.51668 |
| M+H+-134 (Adda)-NH3 | 844.44971 | 858.46904 | 920.45409 | 934.46807 | 887.47064 | 954.49127 |
| Z+Adda+AA6+AA3+AA1-CO+H+ | 847.43655 | 861.45323 | ||||
| Z+Adda+AA6+AA3+H+ | 728.39793 | 742.41144 | 728.39593 | 742.41195 | 728.39601 | 728.39614 |
| AA3+Z+Adda+AA6-H2O+H+ | 710.38705 | 724.40291 | 710.38457 | 724.40284 | 710.38447 | |
| Z+Adda+AA6+CO+H+ | 625.33379 | 639.34874 | 625.33299 | 639.34839 | 625.33274 | |
| AA3+Z+Adda+H+ | 599.35556 | 599.35522 | 599.35471 | 599.35514 | 599.35420 | 599.36213 |
| Z+Adda+AA6+H+ | 599.35556 | 613.36953 | 599.35471 | 613.36946 | 613.37004 | 599.36213 |
| AA3+Z+Adda-CO+H+ | 571.35843 | 571.35829 | 571.35844 | 571.35823 | 571.35963 | 571.36199 |
| Z+Adda+AA6-CO+H+ | 571.35843 | 585.37421 | 571.35844 | 585.37418 | 585.37388 | 571.36199 |
| [AA7+AA1+X+AA3+Z+NH2+2H]+ | 570.33513 | 570.33402 | 646.33282 | 646.33296 | 613.35189 | 680.37043 |
| AA7+AA1+X+AA3+Z+H+ | 553.31097 | 553.30853 | 629.30526 | 629.30531 | 596.32510 | 663.34577 |
| AA7+AA1+X+AA3+Z-H2O+H+ | 535.29685 | 535.29715 | 611.29594 | 611.29603 | 578.31403 | |
| AA7+AA1+X+AA3+Z-CO+H+ | 525.31401 | 525.31395 | 601.31109 | 601.31177 | 568.33044 | 635.35001 |
| [AA1+X+AA3+Z+NH2+2H]+ | 487.29752 | 563.29411 | 416.26012 | 593.34189 | ||
| AA1+X+AA3+Z+H+ | 470.26987 | 470.26974 | 546.26878 | 546.27001 | 513.28930 | 576.31199 |
| AA1+X+AA3+Z-NH3+H+ | 453.23973 | 453.23985 | 529.24225 | 529.24229 | 496.26170 | 559.28760 |
| AA1+X+AA3+Z-H2O+H+ | 452.25983 | 452.25967 | 528.25943 | 528.25977 | 495.26546 | |
| AA1+X+AA3+Z-CO-NH3+H+ | 468.26542 | |||||
| Z+Adda-134+AA6-NH3+H+ | 448.25002 | 462.27024 | 448.25379 | 462.27020 | 462.27011 | |
| Adda-134+AA6+AA7+AA1-NH3+H+ | 446.22694 | 460.24297 | 522.22418 | 536.24198 | 492.27009 | |
| [X+AA3+Z+NH2+2H]+ | 416.26101 | 416.26113 | ||||
| X+AA3+Z+H+ | 399.23512 | 399.23409 | 399.23411 | 399.23417 | 442.25185 | 463.22949 |
| AA7+AA1+X+AA3+H+ | 397.20653 | 397.20649 | 426.20885 | 507.24592 | ||
| AA6+AA7+AA1+X+H+ | 397.20653 | 411.22257 | 473.20550 | 487.22124 | 440.22463 | 507.24592 |
| X+AA3+Z-NH3+H+ | 382.20854 | 382.20868 | 382.20836 | 382.20855 | 425.22561 | 446.20174 |
| Adda-134+AA6+AA7-NH3+H+ | 375.19117 | 389.20689 | 375.19028 | 389.20690 | 375.19269 | 379.18595 |
| Adda-134+AA6+AA7-NH3-CO+H+ | 347.19498 | 361.21108 | 361.21113 | 347.19155 | 351.19024 | |
| Adda-134+AA6-NH3+H+ | 292.15384 | 306.16894 | 292.15371 | 306.16887 | 292.15414 | |
| [AA3+Z+NH2+2H]+ | 303.17697 | 303.17668 | 303.17683 | 303.17739 | 303.17752 | |
| AA7+AA1+X-NH3+H+ | 294.15521 | |||||
| X+AA3+H+ | 272.13442 | 307.12806 | ||||
| AA3+Z-NH2+H+ | 286.14888 | 286.14981 | 286.14997 | 286.14989 | 286.14832 | 286.14935 |
| AA7+AA1+X+H+ | 268.16531 | 268.16581 | 344.16287 | 344.16366 | 311.18244 | 378.20111 |
| AA1+Z+H+ | 242.16093 | |||||
| AA6+AA7+CO+ | 253.08124 | 253.08129 | 239.06653 | 243.06043 | ||
| AA6+AA7+H+ | 213.08659 | 227.10269 | 213.08735 | 227.10254 | 213.08693 | 217.08173 |
| AA6+AA7-CO+H+ | 185.09586 | 189.08683 | ||||
| [Z+NH2+2H]+ | 174.13423 | 174.13431 | 174.13459 | |||
| Adda-134-NH3+H+ | 163.11149 | 163.11151 | 163.11156 | 163.11148 | 163.11138 | 163.11150 |
| AA7+AA1+H+ | 155.08136 | 155.08127 | 231.07989 | 231.07983 | 155.08138 | 201.12293 |
| AA7+AA1-CO+H+ | 127.08639 | 127.08636 | 203.08461 | 203.08484 | 127.08664 | 173.12822 |
| Adda frag (Ph-CH2-CH(O+Me) | 135.08040 | 135.08041 | 135.08073 | 135.08055 | 135.08049 | 135.08049 |
| X Immonium ion | 86.09695 | 86.09680 | 86.09682 | 129.11388 | 129.11358 | |
| Ser Immonium ion | 60.04481 | |||||
| Leu Immonium ion | 86.09677 | 86.09689 | ||||
| Parent and Fragment Ions | Known AP (Certified Standard) | Known AP (No Certified Standard) | Unknown AP | ||||
|---|---|---|---|---|---|---|---|
| AP-A | AP-C | AP-F | Ferintoic acid A | Oscillamide Y | AP731 | AP803 | |
| M+H+ | 844.42399 | 809.45396 | 851.47649 | 867.43760 | 858.43789 | 732.39224 | 804.43535 |
| Isotope #1 | 845.42487 | 810.45755 | 852.47948 | 868.44117 | 859.44270 | 733.39499 | 805.43704 |
| Isotope #2 | 846.42939 | 811.46099 | 853.48112 | 869.44461 | 860.44575 | 734.39684 | 806.43070 |
| Isotope #3 | 837.43126 | 861.44882 | 735.39821 | 807.43308 | |||
| M+H+-NH3 | 792.42755 | ||||||
| M+H+-H2O | 826.41253 | 791.44322 | 833.46495 | 849.42755 | 840.42805 | 714.38127 | 786.42485 |
| M+H+-H2O-CO | 821.43177 | 758.42793 | |||||
| M+H+-AA6residue | 603.34925 | 657.39861 | |||||
| M+H+-AA1 | 681.36103 | 695.37553 | |||||
| M+H+-AA1-H2O | 663.34863 | 663.34859 | 677.36401 | 663.34841 | 677.36411 | 567.31335 | 673.33952 |
| M+H+-CO-AA1-H2O | 635.35366 | 635.35363 | 635.35349 | 649.37013 | 539.31810 | 645.34572 | |
| M+H+-AA4-AA5 | 528.28961 | 547.32259 | 589.34485 | 605.30624 | 596.30578 | 548.26999 | 564.25123 |
| M+H+-AA3-AA4 | 591.29012 | ||||||
| M+H+-AA3-AA4-H2O | 550.26467 | 515.29601 | 573.28032 | 550.26427 | 502.22842 | 542.29930 | |
| M+H+-AA3-AA4-CO | 540.28143 | 563.29597 | 540.28113 | 532.31511 | |||
| M+H+-AA1-CO-AA6residue-H2O | 479.29701 | 581.34602 | |||||
| M+H+-AA1-CO-AA6resisue | 428.28596 | 516.32042 | |||||
| M+H+-AA1-AA4-AA6residue-H2O | 394.20841 | 496.25720 | |||||
| M+H+-AA1-AA3-AA4 | 405.21182 | 405.21189 | 405.21165 | 405.21192 | 405.21185 | 373.17124 | 447.22582 |
| Lys+AA3+AA5+AA6+H+ | 460.29013 | 474.30651 | 460.28997 | 474.30632 | 428.24910 | 534.27652 | |
| Lys+AA5+AA6+CO+H+ | 389.21756 | 389.21743 | 357.17623 | 431.23184 | |||
| AA6+Lys+CO+AA3+H+ | 403.23383 | 403.23379 | 385.20775 | 435.17195 | |||
| AA3+AA4+AA5+H+ | 362.20673 | 362.20676 | 376.22245 | 362.20651 | 376.22195 | 284.19651 | 372.23107 |
| AA5+AA6+H+ | 233.12808 | 233.12801 | 233.12811 | 233.12811 | 233.12809 | 201.08693 | 275.14190 |
| AA3+AA4+H+ | 277.15417 | 277.15409 | 277.15416 | 213.16084 | 245.13166 | ||
| AA4+AA5+H+ | 263.13861 | 263.13851 | 263.13865 | 263.13866 | 263.13866 | 185.12842 | 241.19079 |
| AA1+H+ | 175.11875 | ||||||
| AA1+CO+ | 201.09792 | ||||||
| [AA1+2H]+ | 130.11017 | ||||||
| Ph-CH2-OH | 107.04936 | 107.04913 | 107.04945 | 107.04961 | |||
| Lys Immonium ion | 84.08123 | 84.08122 | 84.08120 | 84.08119 | 84.08120 | 84.08134 | 84.08131 |
| AA1 Immonium Ion | 136.07545 | 129.11359 | 120.08070 | 86.09692 | |||
| Phe Immonium | 120.08100 | 120.08110 | 120.08110 | ||||
| Sample No. | CYN | [Asp3]MC-RR | MC-RR | MC-YR | MC-LR | [Asp3]MC-LR | MC-HiIR | MC-LA | MC-LY | AP-A | AP-B |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ND | ND | ND | ND | 90 ± 28 | ND | ND | 486 ± 105 | ND | ND | ND |
| 2 | ND | ND | ND | ND | ND | ND | ND | 364 ± 70 | ND | ND | 95 ± 17 |
| 3 | ND | ND | 491 ± 95 | 76 ± 6 | 1010 ± 21 | ND | ND | ND | ND | ND | ND |
| 4 | ND | ND | ND | ND | 106 ± 10 | ND | ND | 1165 ± 60 | ND | ND | ND |
| 5 | ND | ND | ND | ND | 47 ± 5* | ND | ND | ND | ND | 1290 ± 259 | 851 ± 116 |
| 6 | ND | ND | ND | ND | ND | ND | ND | ND | ND | 188 ± 66 | 348 ± 38 |
| 7 | ND | ND | ND | ND | 254 ± 29 | ND | ND | ND | 41 ± 13 * | ND | 124 ± 32 |
| 8 | 153 ± 66 | ND | ND | ND | 62 ± 5 | ND | ND | ND | ND | ND | ND |
| 9 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| 10 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| 11 | ND | 41,364 ± 3885 | 840 ± 87 | 259 ± 17 | 416 ± 39 | 1073 ± 116 | ND | ND | ND | 3178 ± 97 | 5836 ± 187 |
| 12 | ND | 123 ± 8 | 5691 ± 506 | 2692 ± 382 | 3263 ± 179 | ND | 321 ± 98 | ND | 39 ± 36 * | 137 ± 25 | 239 ± 46 |
| Sample No. | [GluOMe6] MC-LR | [M(O)1] MC-LR | [M(O)1, GluOMe6] MC-LR | [Asp3] MC-RHar | [Leu1, Ser7] MC-HtyR | AP-C | AP-F | Ferintoic acid A | Oscillamide Y | AP731 | AP803 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 596 ± 36 | 57 ± 11 | 197 ± 28 | ND | ND | ND | ND | ND | ND | ND | ND |
| 5 | ND | ND | ND | ND | ND | ND | 175 ± 34 | ND | 484 ± 55 | ND | 1035 ± 108 |
| 9 | ND | ND | ND | 201 ± 47 | ND | ND | ND | ND | ND | ND | ND |
| 11 | ND | ND | ND | ND | ND | 75 ± 9 | 221 ± 15 | ND | 88 ± 10 | 109 ± 7 | ND |
| 12 | ND | ND | ND | ND | 124 ± 23 | ND | ND | 60 ± 11 | ND | ND | ND |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy-Lachapelle, A.; Solliec, M.; Sauvé, S.; Gagnon, C. A Data-Independent Methodology for the Structural Characterization of Microcystins and Anabaenopeptins Leading to the Identification of Four New Congeners. Toxins 2019, 11, 619. https://doi.org/10.3390/toxins11110619
Roy-Lachapelle A, Solliec M, Sauvé S, Gagnon C. A Data-Independent Methodology for the Structural Characterization of Microcystins and Anabaenopeptins Leading to the Identification of Four New Congeners. Toxins. 2019; 11(11):619. https://doi.org/10.3390/toxins11110619
Chicago/Turabian StyleRoy-Lachapelle, Audrey, Morgan Solliec, Sébastien Sauvé, and Christian Gagnon. 2019. "A Data-Independent Methodology for the Structural Characterization of Microcystins and Anabaenopeptins Leading to the Identification of Four New Congeners" Toxins 11, no. 11: 619. https://doi.org/10.3390/toxins11110619
APA StyleRoy-Lachapelle, A., Solliec, M., Sauvé, S., & Gagnon, C. (2019). A Data-Independent Methodology for the Structural Characterization of Microcystins and Anabaenopeptins Leading to the Identification of Four New Congeners. Toxins, 11(11), 619. https://doi.org/10.3390/toxins11110619