Endothelial Toxicity of High Glucose and its by-Products in Diabetic Kidney Disease


Abstract
1. Introduction
2. Effect of High Glucose and by-Products on Renal Endothelial Cells Leading to DKD
2.1. Toxicity of High Glucose and Glucose by-Products
2.1.1. Glucose
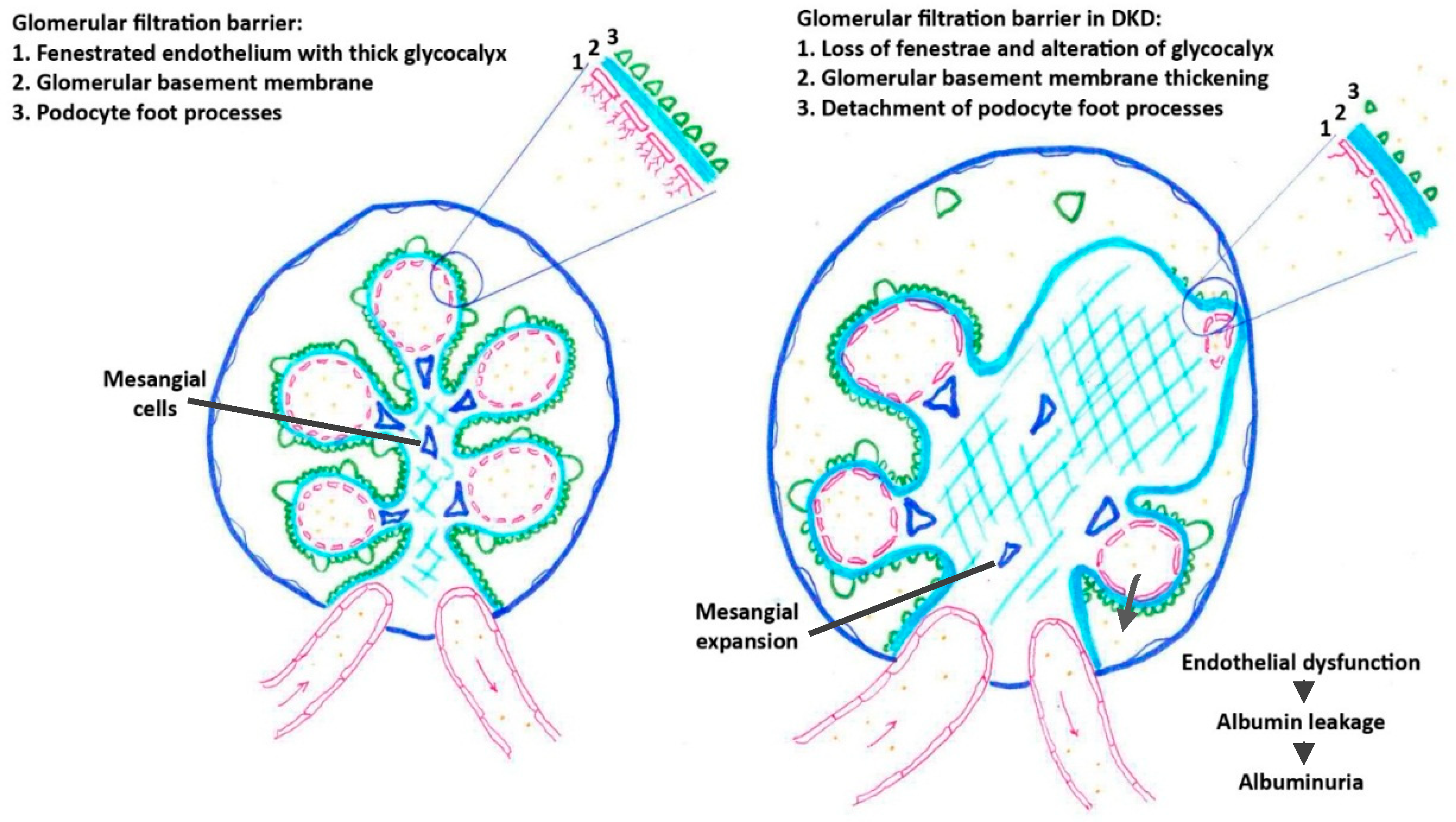
2.1.2. The Polyol Pathway
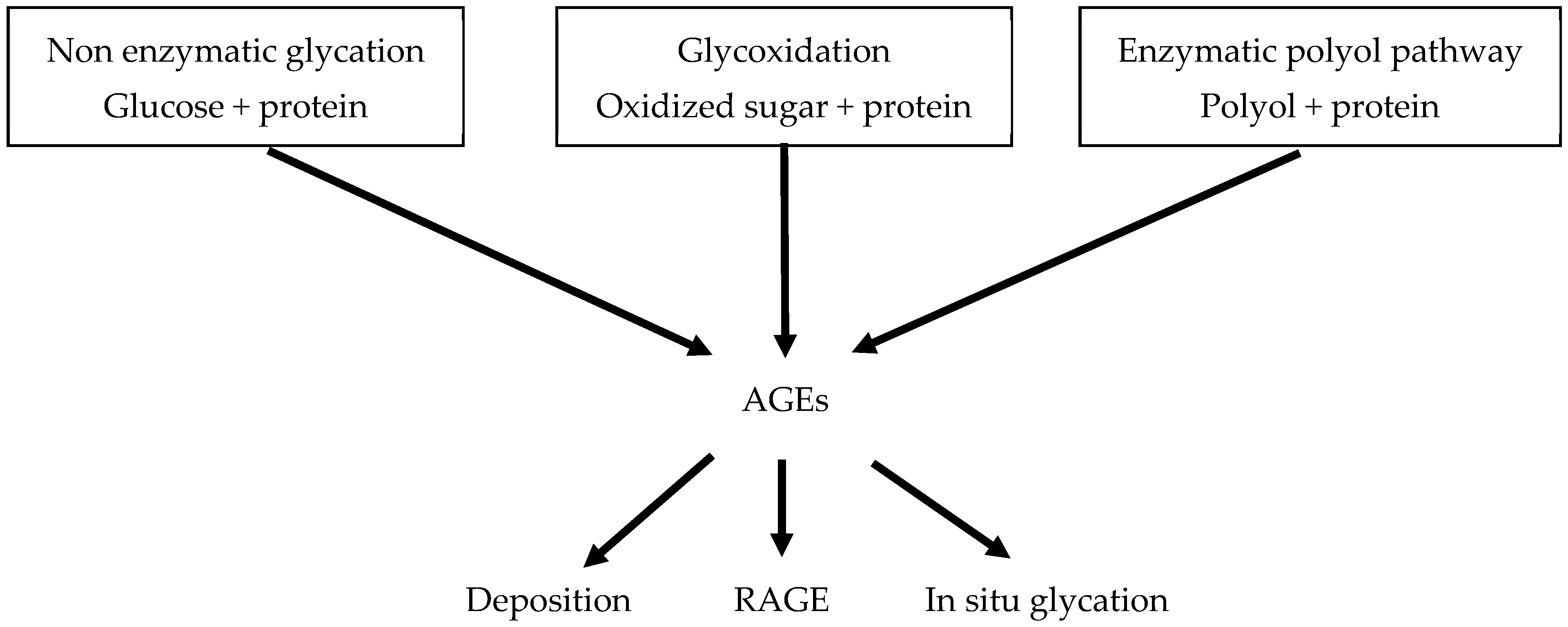
2.1.3. Advanced Glycation End Products (AGEs)
2.2. Mechanisms Inducing Endothelial Dysfunction Leading to DKD
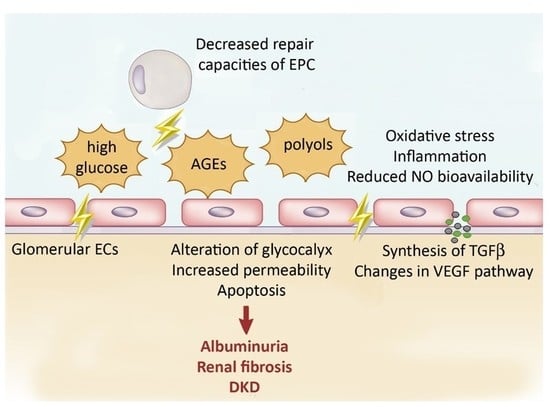
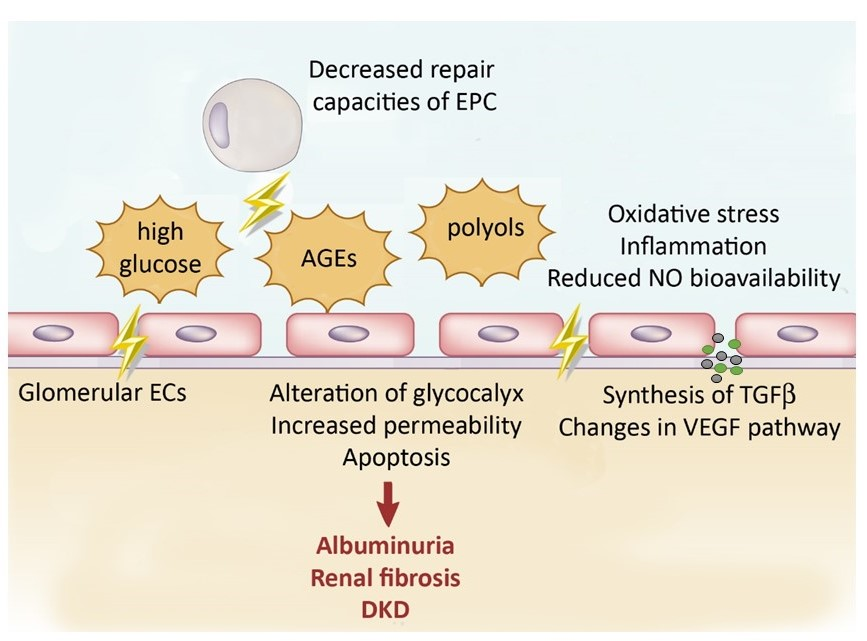
2.2.1. Effect of High Glucose and by-Products on Glomerular Filtration Barrier
Endothelial Permeability
Glycocalyx Alteration
Endothelial Apoptosis
2.2.2. Changes in VEGF Pathway
2.2.3. Synthesis of TGF-β
2.2.4. Oxidative Stress and Reduced NO Bioavailability
2.2.5. Proinflammatory Effects
2.2.6. Defect of endothelial progenitor cells
2.3. Therapies Targeting Endothelial Dysfunction in DKD
2.3.1. Glycemic Control and Diet Changes
2.3.2. Therapies Targeting Endothelial Injury as a Treatment for DKD?
2.3.3. New Antidiabetic Therapies
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADMA | Asymmetric Dimethylarginine |
| AGEs | advanced glycation end products |
| BH4 | tetrahydrobiopterin |
| COX-2 | Cyclooxygenase-2 |
| DKD | diabetic kidney disease |
| DPP-4 | Dipeptidyl peptidase-4 |
| EndMT | endothelial-mesenchymal transition |
| eNOS | endothelial nitric oxide synthase |
| EPC | endothelial progenitor cells |
| GECs | glomerular endothelial cells |
| GFR | glomerular filtration rate |
| GLP-1RA | glucagon-like peptide-1 receptor agonist |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| JNK | c-Jun NH2 terminal kinase |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NF-κB | nuclear factor-kappa B |
| NO | nitric oxide |
| PKC | protein kinase C |
| RAGE | receptor for advanced glycation endproducts |
| RAS | renin-angiotensin system |
| ROS | reactive oxygen species |
| SDF-1 | stromal cell-derived factor 1 |
| SGLT2 | Sodium glucose cotransporter 2 |
| TGF-β | transforming growth factor- β |
| VCAM-1 | vascular cell adhesion molecule 1 |
| VEGF | Vascular Endothelial Growth Factor |
| VEGFR2 | Vascular Endothelial Growth Factor Receptor 2 |
References
- Ritz, E.; Orth, S.R. Nephropathy in patients with type 2 diabetes mellitus. N. Engl. J. Med. 1999, 341, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Cooper, M.E.; Zimmet, P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Umanath, K.; Lewis, J.B. Update on Diabetic Nephropathy: Core Curriculum 2018. Am. J. Kidney Dis. 2018, 71, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Harris, R.C. Renal endothelial dysfunction in diabetic nephropathy. Cardiovasc. Hematol. Disord. Drug Targets 2014, 14, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Satchell, S.C.; Braet, F. Glomerular endothelial cell fenestrations: An integral component of the glomerular filtration barrier. Am. J. Physiol. Ren. Physiol. 2009, 296, F947–F956. [Google Scholar] [CrossRef] [PubMed]
- Satchell, S. The role of the glomerular endothelium in albumin handling. Nat. Rev. Nephrol. 2013, 9, 717–725. [Google Scholar] [CrossRef]
- Rabelink, T.J.; de Zeeuw, D. The glycocalyx—Linking albuminuria with renal and cardiovascular disease. Nat. Rev. Nephrol. 2015, 11, 667–676. [Google Scholar] [CrossRef]
- Gilbert, R.E. The endothelium in diabetic nephropathy. Curr. Atheroscler. Rep. 2014, 16, 410. [Google Scholar] [CrossRef] [PubMed]
- Weil, E.J.; Lemley, K.V.; Mason, C.C.; Yee, B.; Jones, L.I.; Blouch, K.; Lovato, T.; Richardson, M.; Myers, B.D.; Nelson, R.G. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 2012, 82, 1010–1017. [Google Scholar] [CrossRef]
- Sward, P.; Rippe, B. Acute and sustained actions of hyperglycaemia on endothelial and glomerular barrier permeability. Acta Physiol. 2012, 204, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Tervaert, T.W.; Mooyaart, A.L.; Amann, K.; Cohen, A.H.; Cook, H.T.; Drachenberg, C.B.; Ferrario, F.; Fogo, A.B.; Haas, M.; de Heer, E.; et al. Pathologic classification of diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Najafian, B.; Alpers, C.E.; Fogo, A.B. Pathology of human diabetic nephropathy. Contrib. Nephrol. 2011, 170, 36–47. [Google Scholar] [PubMed]
- Rajah, T.T.; Olson, A.L.; Grammas, P. Differential glucose uptake in retina- and brain-derived endothelial cells. Microvasc. Res. 2001, 62, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Alpert, E.; Gruzman, A.; Riahi, Y.; Blejter, R.; Aharoni, P.; Weisinger, G.; Eckel, J.; Kaiser, N.; Sasson, S. Delayed autoregulation of glucose transport in vascular endothelial cells. Diabetologia 2005, 48, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Daroux, M.; Prevost, G.; Maillard-Lefebvre, H.; Gaxatte, C.; D’Agati, V.D.; Schmidt, A.M.; Boulanger, E. Advanced glycation end-products: Implications for diabetic and non-diabetic nephropathies. Diabetes Metab. 2010, 36, 1–10. [Google Scholar] [CrossRef]
- Cheng, H.; Wang, H.; Fan, X.; Paueksakon, P.; Harris, R.C. Improvement of endothelial nitric oxide synthase activity retards the progression of diabetic nephropathy in db/db mice. Kidney Int. 2012, 82, 1176–1183. [Google Scholar] [CrossRef][Green Version]
- Kelly, D.J.; Zhang, Y.; Hepper, C.; Gow, R.M.; Jaworski, K.; Kemp, B.E.; Wilkinson-Berka, J.L.; Gilbert, R.E. Protein kinase C beta inhibition attenuates the progression of experimental diabetic nephropathy in the presence of continued hypertension. Diabetes 2003, 52, 512–518. [Google Scholar] [CrossRef]
- Cosentino, F.; Eto, M.; De Paolis, P.; van der Loo, B.; Bachschmid, M.; Ullrich, V.; Kouroedov, A.; Delli Gatti, C.; Joch, H.; Volpe, M.; et al. High glucose causes upregulation of cyclooxygenase-2 and alters prostanoid profile in human endothelial cells: Role of protein kinase C and reactive oxygen species. Circulation 2003, 107, 1017–1023. [Google Scholar] [CrossRef]
- Paeng, J.; Park, J.; Um, J.E.; Nam, B.Y.; Kang, H.Y.; Kim, S.; Oh, H.J.; Park, J.T.; Han, S.H.; Ryu, D.R.; et al. The locally activated renin-angiotensin system is involved in albumin permeability in glomerular endothelial cells under high glucose conditions. Nephrol. Dial. Transplant. 2017, 32, 61–72. [Google Scholar] [CrossRef]
- Ebefors, K.; Wiener, R.J.; Yu, L.; Azeloglu, E.U.; Yi, Z.; Jia, F.; Zhang, W.; Baron, M.H.; He, J.C.; Haraldsson, B.; et al. Endothelin receptor-A mediates degradation of the glomerular endothelial surface layer via pathologic crosstalk between activated podocytes and glomerular endothelial cells. Kidney Int. 2019, 96, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Friden, V.; Dasgupta, I.; Foster, R.R.; Welsh, G.I.; Tooke, J.E.; Haraldsson, B.; Mathieson, P.W.; Satchell, S.C. High glucose causes dysfunction of the human glomerular endothelial glycocalyx. Am. J. Physiol. Ren. Physiol. 2011, 300, F40–F48. [Google Scholar] [CrossRef] [PubMed]
- van den Born, J.; van Kraats, A.A.; Bakker, M.A.; Assmann, K.J.; Dijkman, H.B.; van der Laak, J.A.; Berden, J.H. Reduction of heparan sulphate-associated anionic sites in the glomerular basement membrane of rats with streptozotocin-induced diabetic nephropathy. Diabetologia 1995, 38, 1169–1175. [Google Scholar] [CrossRef] [PubMed][Green Version]
- van den Hoven, M.J.; Rops, A.L.; Bakker, M.A.; Aten, J.; Rutjes, N.; Roestenberg, P.; Goldschmeding, R.; Zcharia, E.; Vlodavsky, I.; van der Vlag, J.; et al. Increased expression of heparanase in overt diabetic nephropathy. Kidney Int. 2006, 70, 2100–2108. [Google Scholar] [CrossRef] [PubMed]
- van den Hoven, M.J.; Waanders, F.; Rops, A.L.; Kramer, A.B.; van Goor, H.; Berden, J.H.; Navis, G.; van der Vlag, J. Regulation of glomerular heparanase expression by aldosterone, angiotensin II and reactive oxygen species. Nephrol. Dial. Transplant. 2009, 24, 2637–2645. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Zhang, L.; Yao, Q.; Li, L.; Wang, B.; Zhang, J.; He, M. The receptor for advanced glycation endproducts mediates podocyte heparanase expression through NF-κB signaling pathway. Mol. Cell. Endocrinol. 2017, 470, 14–25. [Google Scholar] [CrossRef]
- Gil, N.; Goldberg, R.; Neuman, T.; Garsen, M.; Zcharia, E.; Rubinstein, A.M.; van Kuppevelt, T.; Meirovitz, A.; Pisano, C.; Li, J.P.; et al. Heparanase is essential for the development of diabetic nephropathy in mice. Diabetes 2012, 61, 208–216. [Google Scholar] [CrossRef]
- Packham, D.K.; Wolfe, R.; Reutens, A.T.; Berl, T.; Heerspink, H.L.; Rohde, R.; Ivory, S.; Lewis, J.; Raz, I.; Wiegmann, T.B.; et al. Sulodexide fails to demonstrate renoprotection in overt type 2 diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 123–130. [Google Scholar] [CrossRef]
- Lewis, E.J.; Lewis, J.B.; Greene, T.; Hunsicker, L.G.; Berl, T.; Pohl, M.A.; de Zeeuw, D.; Heerspink, H.L.; Rohde, R.D.; Atkins, R.C.; et al. Sulodexide for kidney protection in type 2 diabetes patients with microalbuminuria: A randomized controlled trial. Am. J. Kidney Dis. 2011, 58, 729–736. [Google Scholar] [CrossRef]
- Wang, Z.B.; Zhang, S.; Li, Y.; Wang, R.M.; Tong, L.C.; Wang, Y.; Liu, W.Y.; Su, D.F.; Tu, Y.; Zhang, L.C.; et al. LY333531, a PKCβ inhibitor, attenuates glomerular endothelial cell apoptosis in the early stage of mouse diabetic nephropathy via down-regulating swiprosin-1. Acta Pharmacol. Sin. 2017, 38, 1009–1023. [Google Scholar] [CrossRef]
- Peng, H.; Xing, Y.F.; Ye, Z.C.; Li, C.M.; Luo, P.L.; Li, M.; Lou, T.Q. High glucose induces activation of the local reninangiotensin system in glomerular endothelial cells. Mol. Med. Rep. 2014, 9, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wei, C.; Zhang, W.; Schlondorff, D.; Wu, J.; Cai, M.; He, W.; Baron, M.H.; Chuang, P.Y.; Liu, Z.; et al. Gene expression profiles of glomerular endothelial cells support their role in the glomerulopathy of diabetic mice. Kidney Int. 2018, 94, 326–345. [Google Scholar] [CrossRef] [PubMed]
- Ho, F.M.; Liu, S.H.; Liau, C.S.; Huang, P.J.; Lin-Shiau, S.Y. High glucose-induced apoptosis in human endothelial cells is mediated by sequential activations of c-Jun NH(2)-terminal kinase and caspase-3. Circulation 2000, 101, 2618–2624. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Advani, A. VEGF and the diabetic kidney: More than too much of a good thing. J. Diabetes Complicat. 2017, 31, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, B.; Hausknecht, B.; Boehmer, K.; Riess, R.; Brekken, R.A.; Hugo, C.P. Local VEGF activity but not VEGF expression is tightly regulated during diabetic nephropathy in man. Kidney Int. 2006, 69, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Li, X.; Xiao, W.; Fu, J.; Harris, R.C.; Lindenmeyer, M.; Cohen, C.D.; Guillot, N.; Baron, M.H.; Wang, N.; et al. BAMBI elimination enhances alternative TGF-β signaling and glomerular dysfunction in diabetic mice. Diabetes 2015, 64, 2220–2233. [Google Scholar] [CrossRef]
- Chen, L.; Yang, T.; Lu, D.W.; Zhao, H.; Feng, Y.L.; Chen, H.; Chen, D.Q.; Vaziri, N.D.; Zhao, Y.Y. Central role of dysregulation of TGF-β/Smad in CKD progression and potential targets of its treatment. Biomed. Pharmacother. 2018, 101, 670–681. [Google Scholar] [CrossRef]
- Sutariya, B.; Jhonsa, D.; Saraf, M.N. TGF-β: The connecting link between nephropathy and fibrosis. Immunopharmacol. Immunotoxicol. 2016, 38, 39–49. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 2008, 19, 2282–2287. [Google Scholar] [CrossRef]
- Li, J.; Qu, X.; Bertram, J.F. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 2009, 175, 1380–1388. [Google Scholar] [CrossRef]
- Huynh, P.; Chai, Z. Transforming growth factor beta (TGFβ) and related molecules in chronic kidney disease (CKD). Clin. Sci. 2019, 133, 287–313. [Google Scholar] [CrossRef] [PubMed]
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti-TGF-β1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Banal, C.; Chow, B.S.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, S.; Starnes, J.; Shi, S.; Lonis, B.; Tran, R. Diabetic nephropathy is associated with oxidative stress and decreased renal nitric oxide production. J. Am. Soc. Nephrol. 2007, 18, 2945–2952. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, E.A.; Hua, P.; Tian, R.X.; Raij, L. Human glomerular endothelium: Interplay among glucose, free fatty acids, angiotensin II, and oxidative stress. Am. J. Physiol. Ren. Physiol. 2010, 298, F125–F132. [Google Scholar] [CrossRef] [PubMed]
- Hanai, K.; Babazono, T.; Nyumura, I.; Toya, K.; Tanaka, N.; Tanaka, M.; Ishii, A.; Iwamoto, Y. Asymmetric dimethylarginine is closely associated with the development and progression of nephropathy in patients with type 2 diabetes. Nephrol. Dial. Transplant. 2009, 24, 1884–1888. [Google Scholar] [CrossRef] [PubMed]
- Komers, R.; Schutzer, W.E.; Reed, J.F.; Lindsley, J.N.; Oyama, T.T.; Buck, D.C.; Mader, S.L.; Anderson, S. Altered endothelial nitric oxide synthase targeting and conformation and caveolin-1 expression in the diabetic kidney. Diabetes 2006, 55, 1651–1659. [Google Scholar] [CrossRef]
- You, H.; Gao, T.; Cooper, T.K.; Morris, S.M., Jr.; Awad, A.S. Diabetic nephropathy is resistant to oral l-arginine or l-citrulline supplementation. Am. J. Physiol. Ren. Physiol. 2014, 307, F1292–F1301. [Google Scholar] [CrossRef]
- Schaffer, S.W.; Jong, C.J.; Mozaffari, M. Role of oxidative stress in diabetes-mediated vascular dysfunction: Unifying hypothesis of diabetes revisited. Vasc. Pharmacol. 2012, 57, 139–149. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Toth, E.; Racz, A.; Toth, J.; Kaminski, P.M.; Wolin, M.S.; Bagi, Z.; Koller, A. Contribution of polyol pathway to arteriolar dysfunction in hyperglycemia. Role of oxidative stress, reduced NO, and enhanced PGH(2)/TXA(2) mediation. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3096–H3104. [Google Scholar] [CrossRef] [PubMed]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, A.; Satoh, M.; Tomita, N.; Sasaki, T.; Kashihara, N. Deterioration of glomerular endothelial surface layer induced by oxidative stress is implicated in altered permeability of macromolecules in Zucker fatty rats. Diabetologia 2010, 53, 2056–2065. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Casalena, G.; Shi, S.; Yu, L.; Ebefors, K.; Sun, Y.; Zhang, W.; D’Agati, V.; Schlondorff, D.; Haraldsson, B.; et al. Glomerular Endothelial Mitochondrial Dysfunction Is Essential and Characteristic of Diabetic Kidney Disease Susceptibility. Diabetes 2017, 66, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Kiritoshi, S.; Nishikawa, T.; Sonoda, K.; Kukidome, D.; Senokuchi, T.; Matsuo, T.; Matsumura, T.; Tokunaga, H.; Brownlee, M.; Araki, E. Reactive oxygen species from mitochondria induce cyclooxygenase-2 gene expression in human mesangial cells: Potential role in diabetic nephropathy. Diabetes 2003, 52, 2570–2577. [Google Scholar] [CrossRef]
- Ha, H.; Lee, H.B. Reactive oxygen species amplify glucose signalling in renal cells cultured under high glucose and in diabetic kidney. Nephrology 2005, 10, S7–S10. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Dou, L.; Cerini, C.; Dignat-George, F.; Brunet, P. Vascular incompetence in dialysis patients—Protein-bound uremic toxins and endothelial dysfunction. Semin. Dial. 2011, 24, 327–337. [Google Scholar] [CrossRef]
- Barouch, F.C.; Miyamoto, K.; Allport, J.R.; Fujita, K.; Bursell, S.E.; Aiello, L.P.; Luscinskas, F.W.; Adamis, A.P. Integrin-mediated neutrophil adhesion and retinal leukostasis in diabetes. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1153–1158. [Google Scholar]
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial Progenitors: A Consensus Statement on Nomenclature. Stem Cells Transl. Med. 2017, 6, 1316–1320. [Google Scholar] [CrossRef]
- Wils, J.; Favre, J.; Bellien, J. Modulating putative endothelial progenitor cells for the treatment of endothelial dysfunction and cardiovascular complications in diabetes. Pharmacol. Ther. 2017, 170, 98–115. [Google Scholar] [CrossRef]
- Fadini, G.P.; Boscaro, E.; de Kreutzenberg, S.; Agostini, C.; Seeger, F.; Dimmeler, S.; Zeiher, A.; Tiengo, A.; Avogaro, A. Time course and mechanisms of circulating progenitor cell reduction in the natural history of type 2 diabetes. Diabetes Care 2010, 33, 1097–1102. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tepper, O.M.; Galiano, R.D.; Capla, J.M.; Kalka, C.; Gagne, P.J.; Jacobowitz, G.R.; Levine, J.P.; Gurtner, G.C. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 2002, 106, 2781–2786. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Ma, X.; Liu, J.; Fan, Y.; Deng, X. High glucose-induced endothelial progenitor cell dysfunction. Diabetes Vasc. Dis. Res. 2017, 14, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Jarajapu, Y.P.; Caballero, S.; Verma, A.; Nakagawa, T.; Lo, M.C.; Li, Q.; Grant, M.B. Blockade of NADPH oxidase restores vasoreparative function in diabetic CD34+ cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5093–5104. [Google Scholar] [CrossRef] [PubMed]
- Yuen, D.A.; Zhang, Y.; Thai, K.; Spring, C.; Chan, L.; Guo, X.; Advani, A.; Sivak, J.M.; Gilbert, R.E. Angiogenic dysfunction in bone marrow-derived early outgrowth cells from diabetic animals is attenuated by SIRT1 activation. Stem Cells Transl. Med. 2012, 1, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Yamagishi, S.; Matsui, T.; Noda, Y.; Jinnouchi, Y.; Sasaki, K.; Takeuchi, M.; Imaizumi, T. Serum levels of advanced glycation end products (AGEs) are inversely associated with the number and migratory activity of circulating endothelial progenitor cells in apparently healthy subjects. Cardiovasc. Ther. 2012, 30, 249–254. [Google Scholar] [CrossRef]
- Chen, Q.; Dong, L.; Wang, L.; Kang, L.; Xu, B. Advanced glycation end products impair function of late endothelial progenitor cells through effects on protein kinase Akt and cyclooxygenase-2. Biochem. Biophys. Res. Commun. 2009, 381, 192–197. [Google Scholar] [CrossRef]
- Shen, C.; Li, Q.; Zhang, Y.C.; Ma, G.; Feng, Y.; Zhu, Q.; Dai, Q.; Chen, Z.; Yao, Y.; Chen, L.; et al. Advanced glycation endproducts increase EPC apoptosis and decrease nitric oxide release via MAPK pathways. Biomed. Pharmacother. 2010, 64, 35–43. [Google Scholar] [CrossRef]
- Li, H.; Zhang, X.; Guan, X.; Cui, X.; Wang, Y.; Chu, H.; Cheng, M. Advanced glycation end products impair the migration, adhesion and secretion potentials of late endothelial progenitor cells. Cardiovasc. Diabetol. 2012, 11, 46. [Google Scholar] [CrossRef]
- Bhatwadekar, A.D.; Glenn, J.V.; Li, G.; Curtis, T.M.; Gardiner, T.A.; Stitt, A.W. Advanced glycation of fibronectin impairs vascular repair by endothelial progenitor cells: Implications for vasodegeneration in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1232–1241. [Google Scholar] [CrossRef]
- Effect of intensive therapy on the development and progression of diabetic nephropathy in the Diabetes Control and Complications Trial. The Diabetes Control and Complications (DCCT) Research Group. Kidney Int. 1995, 47, 1703–1720. [CrossRef] [PubMed]
- Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) Study Research Group. Intensive Diabetes Treatment and Cardiovascular Outcomes in Type 1 Diabetes: The DCCT/EDIC Study 30-Year Follow-up. Diabetes Care 2016, 39, 686–693. [CrossRef] [PubMed]
- Patel, A.; MacMahon, S.; Chalmers, J.; Neal, B.; Billot, L.; Woodward, M.; Marre, M.; Cooper, M.; Glasziou, P.; Grobbee, D.; et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008, 358, 2560–2572. [Google Scholar] [PubMed]
- Ravindran, S.; Kuruvilla, V.; Wilbur, K.; Munusamy, S. Nephroprotective Effects of Metformin in Diabetic Nephropathy. J. Cell. Physiol. 2017, 232, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Triggle, C.R.; Ding, H. Metformin is not just an antihyperglycaemic drug but also has protective effects on the vascular endothelium. Acta Physiol. 2017, 219, 138–151. [Google Scholar] [CrossRef]
- Uribarri, J.; Tuttle, K.R. Advanced glycation end products and nephrotoxicity of high-protein diets. Clin. J. Am. Soc. Nephrol. 2006, 1, 1293–1299. [Google Scholar] [CrossRef]
- Meek, R.L.; LeBoeuf, R.C.; Saha, S.A.; Alpers, C.E.; Hudkins, K.L.; Cooney, S.K.; Anderberg, R.J.; Tuttle, K.R. Glomerular cell death and inflammation with high-protein diet and diabetes. Nephrol. Dial. Transplant. 2013, 28, 1711–1720. [Google Scholar] [CrossRef]
- Bhattacharjee, N.; Barma, S.; Konwar, N.; Dewanjee, S.; Manna, P. Mechanistic insight of diabetic nephropathy and its pharmacotherapeutic targets: An update. Eur. J. Pharmacol. 2016, 791, 8–24. [Google Scholar] [CrossRef]
- Schneider, M.P.; Schneider, A.; Jumar, A.; Kistner, I.; Ott, C.; Schmieder, R.E. Effects of folic acid on renal endothelial function in patients with diabetic nephropathy: Results from a randomized trial. Clin. Sci. 2014, 127, 499–505. [Google Scholar] [CrossRef]
- Bolignano, D.; Cernaro, V.; Gembillo, G.; Baggetta, R.; Buemi, M.; D’Arrigo, G. Antioxidant agents for delaying diabetic kidney disease progression: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0178699. [Google Scholar] [CrossRef]
- Tsimihodimos, V.; Filippatos, T.D.; Elisaf, M.S. SGLT2 inhibitors and the kidney: Effects and mechanisms. Diabetes Metab. Syndr. 2018, 12, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, A.; Bhansali, A. SGLT2 Inhibitors Through the Windows of EMPA-REG and CANVAS Trials: A Review. Diabetes Ther. 2017, 8, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Mahaffey, K.W.; Jardine, M.J.; Bompoint, S.; Cannon, C.P.; Neal, B.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; et al. Canagliflozin and Cardiovascular and Renal Outcomes in Type 2 Diabetes Mellitus and Chronic Kidney Disease in Primary and Secondary Cardiovascular Prevention Groups. Circulation 2019, 140, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Khat, D.Z.; Husain, M. Molecular Mechanisms Underlying the Cardiovascular Benefits of SGLT2i and GLP-1RA. Curr. Diabetes Rep. 2018, 18, 45. [Google Scholar] [CrossRef] [PubMed]
- Shigiyama, F.; Kumashiro, N.; Miyagi, M.; Ikehara, K.; Kanda, E.; Uchino, H.; Hirose, T. Effectiveness of dapagliflozin on vascular endothelial function and glycemic control in patients with early-stage type 2 diabetes mellitus: DEFENCE study. Cardiovasc. Diabetol. 2017, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Oelze, M.; Hanf, A.; Kroller-Schon, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Godtel-Armbrust, U.; Xia, N.; et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Kokubo, H.; Bumdelger, B.; Yoshizumi, M.; Yamamotoya, T.; Matsunaga, Y.; Ueda, K.; Inoue, Y.; Inoue, M.K.; Fujishiro, M.; et al. The SGLT2 Inhibitor Luseogliflozin Rapidly Normalizes Aortic mRNA Levels of Inflammation-Related but Not Lipid-Metabolism-Related Genes and Suppresses Atherosclerosis in Diabetic ApoE KO Mice. Int. J. Mol. Sci. 2017, 18, 1704. [Google Scholar] [CrossRef]
- Khemais-Benkhiat, S.; Belcastro, E.; Idris-Khodja, N.; Park, S.H.; Amoura, L.; Abbas, M.; Auger, C.; Kessler, L.; Mayoux, E.; Toti, F.; et al. Angiotensin II-induced redox-sensitive SGLT1 and 2 expression promotes high glucose-induced endothelial cell senescence. J. Cell. Mol. Med. 2019. [CrossRef]
- Gaspari, T.; Spizzo, I.; Liu, H.; Hu, Y.; Simpson, R.W.; Widdop, R.E.; Dear, A.E. Dapagliflozin attenuates human vascular endothelial cell activation and induces vasorelaxation: A potential mechanism for inhibition of atherogenesis. Diabetes Vasc. Dis. Res. 2018, 15, 64–73. [Google Scholar] [CrossRef]
- Pollock, C.; Stefansson, B.; Reyner, D.; Rossing, P.; Sjostrom, C.D.; Wheeler, D.C.; Langkilde, A.M.; Heerspink, H.J.L. Albuminuria-lowering effect of dapagliflozin alone and in combination with saxagliptin and effect of dapagliflozin and saxagliptin on glycaemic control in patients with type 2 diabetes and chronic kidney disease (DELIGHT): A randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2019, 7, 429–441. [Google Scholar]
- Shi, S.; Srivastava, S.P.; Kanasaki, M.; He, J.; Kitada, M.; Nagai, T.; Nitta, K.; Takagi, S.; Kanasaki, K.; Koya, D. Interactions of DPP-4 and integrin β1 influences endothelial-to-mesenchymal transition. Kidney Int. 2015, 88, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Krasner, N.M.; Ido, Y.; Ruderman, N.B.; Cacicedo, J.M. Glucagon-like peptide-1 (GLP-1) analog liraglutide inhibits endothelial cell inflammation through a calcium and AMPK dependent mechanism. PLoS ONE 2014, 9, e97554. [Google Scholar] [CrossRef] [PubMed]
- Oeseburg, H.; de Boer, R.A.; Buikema, H.; van der Harst, P.; van Gilst, W.H.; Sillje, H.H. Glucagon-like peptide 1 prevents reactive oxygen species-induced endothelial cell senescence through the activation of protein kinase A. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1407–1414. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanisms | Evidence | References |
|---|---|---|
| GEC damage | Increased GEC fenestration [20]. Increased GECs permeability [17] via PKC [11] and RAS activation [20]. Induction of GEC apoptosis [30]. | [11,17,20,30] |
| Glycocalyx alterations | Reduced biosynthesis of glycosaminoglycans in GECs [22]. Increased synthesis of heparanase by GECs and podocytes [24]. | [22,24] |
| Changes in VEGF pathways | Alteration of VEGF synthesis by podocytes [9,34,35]. Decreased VEGF expression in GEC [34]. | [9,34,35] |
| Fibrosis | Stimulation of TGF-β secretion [4,38]. Induction of endothelial-mesenchymal transition (EndMT) [39,40]. | [4,38,39,40] |
| Oxidative stress | Increased ROS production by NAD(P)H oxidase activation and eNOS uncoupling [45,49,51]. Increased mitochondrial ROS production by ECs [50] via endothelin-1 pathway [54]. Formation of AGEs further increasing NADPH-oxidase-dependent ROS synthesis [16,52]. | [16,45,49,50,51,52,54]. |
| Reduced NO bioavailability | Defect in eNOS expression [16], dimerization [17], and phosphorylation [17,47], leading to eNOS uncoupling. Decreased NO bioavailability due to increased oxidative stress and AGEs [4,16]. Depletion of BH4 co-factor in endothelial cells [4]. Elevation of ADMA [46]. | [4,16,17,46,47] |
| Inflammation | Upregulation of endothelial adhesion molecule expression [4,57]. Increased expression of leukocyte counter-receptors [58]. Increased leukocyte adhesion to endothelial cells [58]. | [4,57,58] |
| Decreased EPC repair capacities | Reduction of EPC angiogenic properties: impaired proliferation, migration, and incorporation in vascular structures [9,61,62,63,67]. Increased EPC senescence [63] and apoptosis [67]. Modification of vascular basement membrane leading to reduced EPC attachment and spreading [70] | [9,61,62,63,67,70] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dou, L.; Jourde-Chiche, N. Endothelial Toxicity of High Glucose and its by-Products in Diabetic Kidney Disease. Toxins 2019, 11, 578. https://doi.org/10.3390/toxins11100578
Dou L, Jourde-Chiche N. Endothelial Toxicity of High Glucose and its by-Products in Diabetic Kidney Disease. Toxins. 2019; 11(10):578. https://doi.org/10.3390/toxins11100578
Chicago/Turabian StyleDou, Laetitia, and Noémie Jourde-Chiche. 2019. "Endothelial Toxicity of High Glucose and its by-Products in Diabetic Kidney Disease" Toxins 11, no. 10: 578. https://doi.org/10.3390/toxins11100578
APA StyleDou, L., & Jourde-Chiche, N. (2019). Endothelial Toxicity of High Glucose and its by-Products in Diabetic Kidney Disease. Toxins, 11(10), 578. https://doi.org/10.3390/toxins11100578