Gut Microbiota and Cardiovascular Uremic Toxicities

{kind=link}
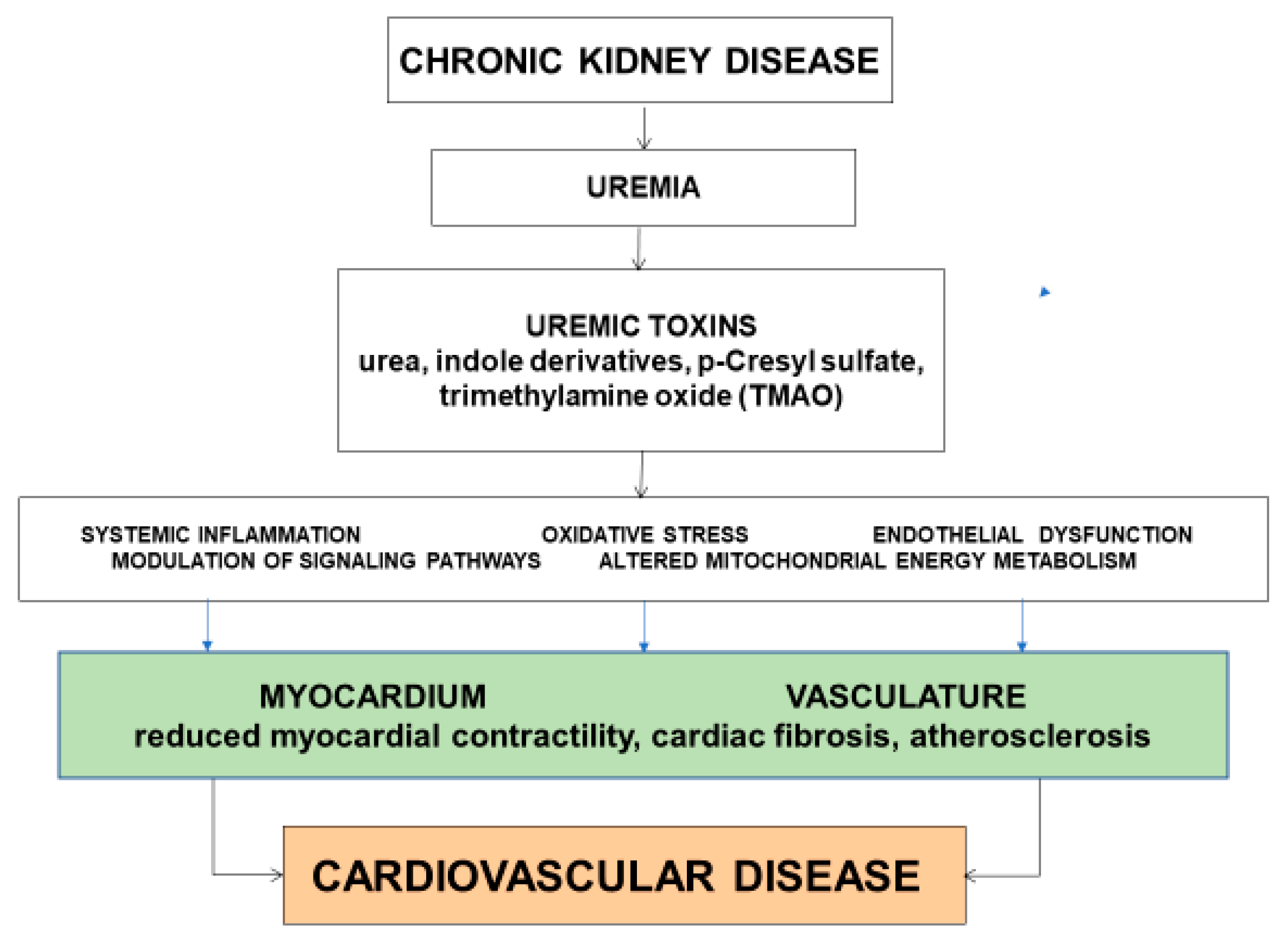
Abstract
1. Introduction
- (1)
- Small water-soluble molecules (molecular weight [MW] ≤ 500 Da), such as urea and phosphorus. Most toxins in this category are dialyzable with conventional dialysis. Some of them, such as urea and creatinine, have been used in the evaluation of renal excretory function and monitoring removal efficiency of dialysis treatment;
- (2)
- Middle molecules (MW ≥ 500 Da). β2-microglobulin is a prototype of this group. Removal of middle molecules appears to be more effective with peritoneal dialysis than with conventional hemodialysis (HD). This is probably because of the larger pore sizes and the longer dialysis duration of peritoneal dialysis. Hemodialysis techniques that increased the permeability membranes (high-flux HD) or convection (hemofiltration/hemodiafiltration) provide superior clearance of these solutes, such as parathyroid hormone (PTH), β2-microglobulin, and advanced glycosylation end products.
- (3)
- Protein-bound compounds, for example, 3-carboxy-4-methyl-5-propyl-2-furanpropionic acid (CMPF), indoxyl sulfate (IS), p-cresol sulfate (PCS), and homocysteine. Twenty-five of the 90 listed toxins (27.8%) are protein-bound, and 23 of them have an MW ≤ 500 D. Among these uremic toxins, organic anions, such as IS, CMPF, PCS, indole-3-acetic acid (IAA), and hippuric acid (HA) are low-molecular-weight compounds. Nevertheless, they should be classified as high-molecular-weight compounds in general circulation as they are firmly bound to plasma proteins, primarily albumin (molecular weight: 66 kDa). Therefore, this set of toxins is difficult to be dialyzed with conventional hemodialysis despite having molecular sizes small enough to pass through the dialysis membrane.
2. Origin and Metabolism of Uremic Toxins
2.1. Urea
2.2. Indole and Indole Components
2.3. p-Cresyl Sulfate
2.4. Trimethylamine N-Oxide (TMAO)
3. Altered Gut Microbiota in Chronic Kidney Disease
4. Altered Gut Microbiota in Cardiovascular Disease
5. Gut microbiota and Uremic Toxins
6. Uremic Toxins and Cardiovascular Disease
7. Effects of Uremic Toxins on the Myocardium
8. Effects of Uremic Toxins on the Vasculature
9. Therapeutic Interventions
9.1. Prebiotics
9.2. Probiotics
9.3. Synbiotics
9.4. Adsorbant AST-120
9.5. Genetically Engineered Bacteria
9.6. Dialysis Modalities
9.7. Peritoneal Dialysis
10. Summary and Conclusions
Funding
Conflicts of Interest
References
- Sarnak, M.; Levey, A. Cardiovascular disease and chronic renal disease: A new paradigm. Am. J. Kidney Dis. 2000, 35, S117–S131. [Google Scholar] [CrossRef]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney disease as a risk factor for development of cardiovascular disease: A statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 2003, 108, 2154–2169. [Google Scholar] [CrossRef] [PubMed]
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Gross, M.L.; Ritz, E. Hypertrophy and fibrosis in the cardiomyopathy of uremia—Beyond coronary heart disease. Semin. Dial. 2008, 21, 308–318. [Google Scholar]
- Weiner, D.E.; Tighiouart, H.; Elsayed, E.F.; Griffith, J.L.; Salem, D.N.; Levey, A.S.; Sarnak, M.J. The relationship between non-traditional risk factors and outcomes in individuals with stage 3-4 CKD. Am. J. Kidney Dis. 2008, 51, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.W.; Hostetter, T.H. Uremia. N. Engl. J. Med. 2007, 357, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. European Uremic Toxin Work Group (EUTox): Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Baurmeister, U.; Brunet, P.; Cohen, G.; Glorieux, G.; Jankowski, J. European Uremic Toxin Work G: A bench to bedside view of uremic toxins. J. Am. Soc. Nephrol. 2008, 19, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. on behalf of the European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Eloot, S.; Schepers, E.; Barreto, D.V.; Barreto, F.C.; Liabeuf, S.; Van Biesen, W.; Verbeke, F.; Glorieux, G.; Choukroun, G.; Massy, Z.; et al. Estimated glomerular filtration rate is a poor predictor of concentration for a broad range of uremic toxins. Clin. J. Am. Soc. Nephrol. 2011, 6, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Schepers, E.; Glorieux, G.; Vanholder, R. The gut: The forgotten organ in uremia? Blood Purif. 2010, 29, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.P.; Thadhani, R. New insights into uremia-induced alterations in metabolic pathways. Curr. Opin. Nephrol. Hypertens. 2011, 20, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.S.; Davies, S.S. Microbial metabolism of dietary components to bioactive metabolites: Opportunities for new therapeutic interventions. Genome Med. 2016, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Prado, R.; Esteras, R.; Perez-Gomez, M.V.; Gracia-Iguacel, C.; Gonzalez-Parra, E.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Nutrients turned into toxins: Microbiota modulation of nutrient properties in chronic kidney disease. Nutrients 2017, 9, 489. [Google Scholar] [CrossRef] [PubMed]
- Walser, M.; Bodenloss, L.J. Urea metabolism in man. J. Clin Investig. 1959, 38, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Smith, C.P. Urea nitrogen salvage mechanisms and their relevance to ruminants, non-ruminants and man. Nutr. Res. Rev. 2005, 18, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Aronov, P.A.; Luo, F.J.; Plummer, N.S.; Quan, Z.; Holmes, S.; Hostetter, T.H.; Meyer, T.W. Colonic contribution to uremic solutes. J. Am. Soc. Nephrol. 2011, 22, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.W.; Hostetter, T.H. Uremic solutes from colon microbes. Kidney Int. 2012, 81, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Koga, J.; Syono, K.; Ichikawa, T.; Adachi, T. Involvement of l-tryptophan aminotransferase in indole-3-acetic acid biosynthesis in Enterobacter cloacae. Biochim. Biophys. Acta 1994, 1209, 241–247. [Google Scholar] [CrossRef]
- Zhu, W.; Stevens, A.P.; Dettmer, K.; Gottfried, E.; Hoves, S.; Kreutz, M.; Holler, E.; Canelas, A.B.; Kema, I.; Oefner, P.J. Quantitative profiling of tryptophan metabolites in serum, urine, and cell culture supernatants by liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2011, 401, 3249–3261. [Google Scholar] [CrossRef] [PubMed]
- Sallée, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Griet Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Pan, C.F.; Chuang, C.K.; Sun, F.J.; Wang, D.J.; Chen, H.H.; Liu, H.L.; Wu, C.J. p-cresyl sulfate is a valuable predictor of clinical outcomes in pre-ESRD patients. Biomed. Res. Int. 2014, 526932. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Chuang, C.K.; Jayakumar, T.; Liu, H.L.; Pan, C.F.; Wang, T.J.; Chen, H.H.; Wu, C.J. Serum p-cresyl sulfate predicts cardiovascular disease and mortality in elderly hemodialysis patients. Arch. Med. Sci. 2013, 9, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Bammens, B.; Evenepoel, P.; Keuleers, H.; Verbeke, K.; Vanrenterghem, Y. Free serum concentrations of the protein-bound retention solute p-cresol predict mortality in hemodialysis patients. Kidney Int. 2006, 69, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Hsu, C.Y.; Tain, Y.L.; Ng, H.Y.; Cheng, B.C.; Yang, C.C.; Wu, C.H.; Chiou, T.T.Y.; Lee, Y.T.; Liao, S.C. Effects of AST-120 on blood concentrations of protein-bound uremic toxins and biomarkers of cardiovascular risk in chronic dialysis patients. Blood Purif. 2014, 37, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Koppe, L.; Pillon, N.J.; Vella, R.E.; Croze, M.L.; Pelletier, C.C.; Chambert, S.; Massy, Z.; Glorieux, G.; Vanholder, R.; Dugenet, Y.; et al. p-Cresyl sulfate promotes insulin resistance associated with CKD. J. Am. Soc. Nephrol. 2013, 24, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.P.; Luo, F.J.; Plummer, N.S.; Hostetter, T.H.; Meyer, T.W. The production of p-cresol sulfate and indoxyl sulfate in vegetarians versus omnivores. Clin. J. Am. Soc. Nephrol. 2012, 7, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.J.; de Aguiar Vallim, T.Q.; Wang, Z.; Shih, D.M.; Meng, Y.; Gregory, J.; Allayee, H.; Lee, R.; Graham, M.; Crooke, R.; Edwards, P.A.; Hazen, S.L.; Lusis, A.J. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013, 17, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.; Blaser, M.J. The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 2012, 13, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Bonder, M.J.; Cenit, M.C.; Tigchelaar, E.F.; Maatman, A.; Dekens, J.A.; Brandsma, E.; Marczynska, J.; Imhann, F.; Weersma, R.K.; et al. The gut microbiome contributes to a substantial proportion of the variation in blood lipids. Circ. Res. 2015, 117, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Lepage, P.; Leclerc, M.C.; Joossens, M.; Mondot, S.; Blottiere, H.M.; Raes, J.; Ehrlich, D.; Dore, J. A metagenomic insight into our gut’s microbiome. Gut 2013, 62, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Relman, D.A. The human microbiome: Ecosystem resilience and health. Nutr. Rev. 2012, 70 (Suppl. 1), S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Sommer, F.; Anderson, J.M.; Bharti, R.; Raes, J.; Rosenstiel, P. The resilience of the intestinal microbiota influences health and disease. Nat. Rev. Microbiol. 2017, 15, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; Desantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; Desantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.J.; Chang, C.S.; Cheng, C.H.; Chen, C.H.; Lee, W.C.; Hsu, Y.H.; Shu, K.H.; Tang, M.J. Colonic transit time in long-term dialysis patients. Am. J. Kidney Dis. 2004, 44, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Evidence for impaired assimilation of protein in chronic renal failure. Kidney Int. 2003, 64, 2196–2203. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, V.M.; Wei, G.; Baird, B.C.; Murtaugh, M.; Chonchol, M.B.; Raphael, K.L.; Greene, T.; Beddhu, S. High dietary fiber intake is associated with decreased inflammation and all-cause mortality in patients with chronic kidney disease. Kidney Int. 2012, 81, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Wandersman, C.; Delepelaire, P. Bacterial iron sources: From siderophores to hemophores. Annu. Rev. Microbiol. 2004, 58, 611–647. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, C.; Lofmark, S.; Edlund, C.; Jansson, J.K. Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology 2010, 156 Pt 11, 3216–3223. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4554–4561. [Google Scholar] [CrossRef] [PubMed]
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Fak, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed]
- Mitch, W.E. Effects of intestinal flora on nitrogen metabolism in patients with chronic renal failure. Am. J. Clin. Nutr. 1978, 31, 1594–1600. [Google Scholar] [CrossRef] [PubMed]
- Barrios, C.; Beaumont, M.; Pallister, T.; Villar, J.; Goodrich, J.K.; Clark, A.; Pascual, J.; Ley, R.E.; Spector, T.D.; Bell, J.T.; et al. Gut-microbiota-metabolite axis in early renal function decline. PLoS ONE 2015, 10, e0134311. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Fukuda, S.; Mukawa, C.; Yuri, A.; Kanemitsu, Y.; Matsumoto, Y.; Akiyama, Y.; Fukuda, N.N.; Tsukamoto, H.; Asaji, K.; et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. 2017, 92, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, M.; Ueno, M.; Itoh, Y.; Suda, W.; Hattori, M. Uremic toxin-producing gut microbiota in rats with chronic kidney disease. Nephron 2017, 135, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Sato, B.; Yoshikawa, D.; Ishii, H.; Suzuki, S.; Inoue, Y.; Takeshita, K.; Tanaka, M.; Kumagai, S.; Matsumoto, M.; Okumura, S.; et al. Relation of plasma indoxyl sulfate levels and estimated glomerular filtration rate to left ventricular diastolic dysfunction. Am. J. Cardiol. 2013, 111, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, S.; Hirashiki, A.; Okumura, T.; Yamada, T.; Okamoto, R.; Shinoda, N.; Takeshita, K.; Kondo, T.; Niwa, T.; Murohara, T. Association between indoxyl sulfate and cardiac dysfunction and prognosis in patients with dilated cardiomyopathy. Circ. J. 2013, 77, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Lu, Y.C.; Chiu, C.A.; Yu, T.H.; Hung, W.C.; Wang, C.P.; Lu, L.F.; Chung, F.M.; Lee, Y.J.; Tsai, I.T. Levels of indoxyl sulfate are associated with severity of coronary atherosclerosis. Clin. Investig. Med. 2013, 36, E42–E49. [Google Scholar] [CrossRef]
- Tsai, M.L.; Hsieh, I.C.; Hung, C.C.; Chen, C.C. Serum free indoxyl sulfate associated with in-stent restenosis after coronary artery stentings. Cardiovasc. Toxicol. 2015, 15, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Liu, H.L.; Pan, C.F.; Chuang, C.K.; Jayakumar, T.; Wang, T.J.; Chen, H.H.; Wu, C.J. Indoxyl sulfate predicts cardiovascular disease and renal function deterioration in advanced chronic kidney disease. Arch. Med. Res. 2012, 43, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Wu, V.; Wu, P.C.; Wu, C.J. Meta-analysis of the associations of p-cresyl sulfate (PCS) and indoxyl sulfate (IS) with cardiovascular events and all-cause mortality in patients with chronic renal failure. PLoS ONE 2015, 10, e0132589. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.S.; Chen, J.; Zou, J.Z.; Zhong, Y.H.; Teng, J.; Ji, J.; Chen, Z.W.; Liu, Z.H.; Shen, B.; Nie, Y.X.; et al. Association of indoxyl sulfate with heart failure among patients on hemodialysis. Clin. J. Am. Soc. Nephrol. 2015, 10, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Melamed, M.L.; Plantinga, L.; Shafi, T.; Parekh, R.; Meyer, T.W.; Hostetter, T.H.; Coresh, J.; Powe, N.R. Retained organic solutes, patient characteristics and all-cause and cardiovascular mortality in hemodialysis: Results from the retained organic solutes and clinical outcomes (ROSCO) investigators. BMC Nephrol. 2013, 14, 134. [Google Scholar] [CrossRef] [PubMed]
- Shafi, T.; Meyer, T.W.; Hostetter, T.H.; Melamed, M.L.; Parekh, R.S.; Hwang, S.; Banerjee, T.; Coresh, J.; Powe, N.R. Free levels of selected organic solutes and cardiovascular morbidity and mortality in hemodialysis patients: Results from the retained organic solutes and clinical outcomes (ROSCO) investigators. PLoS ONE 2015, 10, e0126048. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Wu, C.J.; Pan, C.F.; Chen, Y.C.; Sun, F.J.; Chen, H.H. Serum protein-bound uraemic toxins and clinical outcomes in haemodialysis patients. Nephrol. Dial. Transplant. 2010, 25, 3693–3700. [Google Scholar] [CrossRef] [PubMed]
- Shafi, T.; Sirich, T.L.; Meyer, T.W.; Hwang, S.; Melamed, M.L.; Banerjee, T.; Coresh, J.; Hostetter, T.H.; Powe, N.R. p-Cresol sulfate, indoxyl sulfate, hippurate and phenylacetylglutamine and cardiovascular outcomes in hemodialysis patients. In Proceedings of the American Society Nephrology Conference Abstract, Chicago, IL, USA, 15–20 November 2016. [Google Scholar]
- Meijers, B.K.; Claes, K.; Bammens, B.; de Loor, H.; Viaene, L.; Verbeke, K.; Kuypers, D.; Vanrenterghem, Y.; Evenepoel, P. p-Cresol and cardiovascular risk in mild-to-moderate kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.W.; Hsu, K.H.; Hsu, H.J.; Lee, C.C.; Sun, C.Y.; Tsai, C.J.; Wu, M.S. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients—A prospective cohort study. Nephrol. Dial. Transplant. 2012, 27, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. European Uremic Toxin Work Group (EUTox). Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Campbell, K.L.; Johnson, D.W.; Stanton, T.; Vesey, D.A.; Coombes, J.S.; Weston, K.S.; Hawley, C.M.; McWhinney, B.C.; Ungerer, J.P.; et al. Protein-bound uremic toxins, inflammation and oxidative stress: A cross-sectional study in stage 3-4 chronic kidney disease. Arch. Med. Res. 2014, 45, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Senthong, V.; Wang, Z.; Fan, Y.; Wu, Y.; Hazen, S.L.; Tang, W.H. Trimethylamine N-oxide and mortality risk in patients with peripheral artery disease. J. Am. Heart Assoc. 2016, 5, E004237. [Google Scholar] [CrossRef] [PubMed]
- Randrianarisoa, E.; Lehn-Stefan, A.; Wang, X.; Hoene, M.; Peter, A.; Heinzmann, S.S.; Zhao, X.; Königsrainer, I.; Königsrainer, A.; Balletshofer, B.; et al. Relationship of serum trimethylamine N-oxide (TMAO) Levels with early atherosclerosis in humans. Sci. Rep. 2016, 6, 26745. [Google Scholar] [CrossRef] [PubMed]
- Weisensee, D.; Schnaars, Y.; Schoeppe, W.; Bereiter-Hahn, J.; Löw-Friedrich, I. Potential uremic toxins modulate energy metabolism of cardiac myocytes in vitro. Exp. Nephrol. 1997, 5, 194–200. [Google Scholar] [PubMed]
- Peng, Y.S.; Ding, H.C.; Lin, Y.T.; Syu, J.P.; Chen, Y.; Wang, S.M. Uremic toxin p-cresol induces disassembly of gap junctions of cardiomyocytes. Toxicology 2012, 302, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Yisireyili, M.; Shimizu, H.; Saito, S.; Enomoto, A.; Nishijima, F.; Niwa, T. Indoxyl sulfate promotes cardiac fibrosis with enhanced oxidative stress in hypertensive rats. Life Sci. 2013, 92, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Xu, X.; Nie, L.; Xiao, T.; Guan, X.; He, T.; Yu, Y.; Liu, L.; Huang, Y.; Zhang, J.; et al. Indoxyl sulfate induces oxidative stress and hypertrophy in cardiomyocytes by inhibiting of the AMPK/UCP2 signaling pathway. Toxicol. Lett. 2015, 234, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Lekawanvijit, S.; Kompa, A.R.; Manabe, M.; Wang, B.H.; Langham, R.G.; Nishijima F Kelly, D.J.; Krum, H. Chronic kidney disease-induced cardiac fibrosis is ameliorated by reducing circulating levels of a non-dialysable uremic toxin, indoxyl sulfate. PLoS ONE 2012, 7, e41281. [Google Scholar] [CrossRef] [PubMed]
- Makrecka-Kuka, M.; Volska, K.; Antone, U.; Vilskersts, R.; Grinberga, S.; Bandere, D.; Liepinsh, E.; Dambrova, M. Trimethylamine N-oxide impairs pyruvate and fatty acid oxidation in cardiac mitochondria. Toxicol. Lett. 2017, 267, 32–38. [Google Scholar] [CrossRef] [PubMed]
- D’Apolito, M.; Du, X.; Pisanelli, D.; Pettoello-Mantovani, M.; Campanozzi, A.; Giacco, F.; Maffione, A.B.; Colia, A.L.; Brownlee, M.; Giardino, I. Urea-induced ROS cause endothelial dysfunction in chronic renal failure. Atherosclerosis 2015, 239, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Kraus, L.M.; Kraus, A.P., Jr. Carbamoylation of amino acids and proteins in uremia. Kidney Int. 2001, 78, S102–S107. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Nicholls, S.J.; Rodriguez, E.R.; Kummu, O.; Hörkkö, S.; Barnard, J.; Reynolds, W.F.; Topol, E.J.; DiDonato, J.A.; Hazen, S.L. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 2007, 13, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, F.H.; Tang, W.H.; Hazen, S.L. Protein carbamylation and cardiovascular disease. Kidney Int. 2015, 88, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Kalantar-Zadeh, K.; Wang, Z.; Fu, X.; Tang, W.H.; Hazen, S.L. Protein carbamylation predicts mortality in ESRD. J. Am. Soc. Nephrol. 2013, 24, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Tsuruoka, S.; Ioka, T.; Ando, H.; Ito, C.; Akimoto, T.; Fujimura, A.; Asano, Y.; Kusano, E. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006, 69, 1780–1785. [Google Scholar] [CrossRef] [PubMed]
- Pletinck, A.; Glorieux, G.; Schepers, E.; Cohen, G.; Gondouin, B.; Van Landschoot, M.; Eloot, S.; Rops, A.; Van de Voorde, J.; De Vriese, A.; et al. Protein-bound uremic toxins stimulate crosstalk between leukocytes and vessel wall. J. Am. Soc. Nephrol. 2013, 24, 1981–1994. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via. Biochem. Biophys. Res. Commun. 2016, 481, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Mitch, W.E.; Remuzzi, G. Diets for patients with chronic kidney disease, still worth prescribing. J. Am. Soc. Nephrol. 2004, 15, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Franch, H.A.; Mih, W.E. Navigating between the Scylla and Charybdis of prescribing dietary protein for chronic kidney diseases. Annu. Rev. Nutr. 2009, 29, 341–364. [Google Scholar] [CrossRef] [PubMed]
- Roberfroid, M.; Gibson, G.R.; Hoyles, L.; McCartney, A.L.; Rastall, R.; Rowland, I.; Wolvers, D.; Watzl, B.; Szajewska, H.; Stahl, B.; et al. Prebiotic effects: Metabolic and health benefits. Br. J. Nutr. 2010, 104 (Suppl. 2), S1–S63. [Google Scholar] [CrossRef] [PubMed]
- Adamberg, K.; Kolk, K.; Jaagura, M.; Vilu, R.; Adamberg, S. The composition and metabolism of faecal microbiota is specifically modulated by different dietary polysaccharides and mucin: An isothermal microcalorimetry study. Benef. Microbes 2017, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Liu, S.M.; Lau, W.L.; Khazaeli, M.; Nazertehrani, S.; Farzaneh, S.H.; Kieffer, D.A.; Adams, S.H.; Martin, R.J. High amylose resistant starch diet ameliorates oxidative stress, inflammation, and progression of chronic kidney disease. PLoS ONE 2014, 9, e114881. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, D.A.; Piccolo, B.D.; Vaziri, N.D.; Liu, S.; Lau, W.L.; Khazaeli, M.; Nazertehrani, S.; Moore, M.E.; Marco, M.L.; Martin, R.J.; et al. Resistant starch alters gut microbiome and metabolomic profiles concurrent with amelioration of chronic kidney disease in rats. Am. J. Physiol. Ren. Physiol. 2016, 310, F857–F871. [Google Scholar] [CrossRef] [PubMed]
- Salmean, Y.A.; Segal, M.S.; Langkamp-Henken, B.; Canales, M.T.; Zello, G.A.; Dahl, W.J. Foods with added fiber lower serum creatinine levels in patients with chronic kidney disease. J. Ren. Nutr. 2013, 23, e29–e32. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; De Preter, V.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. p-Cresyl sulfate serum concentrations in haemodialysis patients are reduced by the prebiotic oligofructose-enriched inulin. Nephrol. Dial. Transplant. 2010, 25, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Sirich, T.L.; Plummer, N.S.; Gardner, C.D.; Hostetter, T.H.; Meyer, T.W. Effect of increasing dietary fiber on plasma levels of colon-derived solutes in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2014, 9, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, R.; Pechenyak, B.; Vyas, U.; Ranganathan, P.; Weinberg, A.; Liang, P.; Mallappallil, M.C.; Norin, A.J.; Friedman, E.A.; Saggi, S.J. Randomized controlled trial of strain-specific probiotic formulation (Renadyl) in dialysis patients. Biomed. Res. Int. 2014, 2014, 568571. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.K.; Wu, Y.Y.; Yang, Y.F.; Ting, I.W.; Lin, C.C.; Yen, T.H.; Chen, J.H.; Wang, C.H.; Huang, C.C.; Lin, H.C. The effect of probiotics on serum levels of cytokine and endotoxin in peritoneal dialysis patients: A randomised, double-blind, placebo-controlled trial. Benef. Microbes 2015, 6, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Simenhoff, M.L.; Dunn, S.R.; Zollner, G.P.; Fitzpatrick, M.E.; Emery, S.M.; Sandine, W.E.; Ayres, J.W. Biomodulation of the toxic and nutritional effects of small bowel bacterial overgrowth in end-stage kidney disease using freeze-dried Lactobacillus acidophilus. Miner. Electrolyte Metab. 1996, 22, 92–96. [Google Scholar] [PubMed]
- Takayama, F.; Taki, K.; Niwa, T. Bifidobacterium in gastro-resistant seamless capsule reduces serum levels of indoxyl sulfate in patients on hemodialysis. Am. J. Kidney Dis. 2003, 41, S142–S145. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, N.; Patel, B.; Ranganathan, P.; Marczely, J.; Dheer, R.; Chordia, T.; Dunn, S.R.; Friedman, E.A. Probiotic amelioration of azotemia in 5/6th nephrectomized Sprague-Dawley rats. Sci. World J. 2005, 5, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Guida, B.; Germanò, R.; Trio, R.; Russo, D.; Memoli, B.; Grumetto, L.; Barbato, F.; Cataldi, M. Effect of short-term synbiotic treatment on plasma p-cresol levels in patients with chronic renal failure: A randomized clinical trial. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Johnson, D.W.; Morrison, M.; Pascoe, E.M.; Coombes, J.D.S.; Forbes, J.M.; Szeto, C.-C.; McWhinney, B.C.; Ungerer, J.P.J.; Campbell, K.L. Synbiotics easing renal failure by improving gut microbiology (SYNERGY): A randomized trial. Clin. J. Am. Soc. Nephrol. 2016, 11, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Shibahara, N.; Takagi, S.; Inoue, T.; Katsuoka, Y. AST-120, an oral adsorbent, delays the initiation of dialysis in patients with chronic kidney diseases. Ther. Apheresis Dial. 2007, 11, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Kawagoe, Y.; Matsuda Ueda, Y.; Shimada, N.; Ebihara, I.; Koide, H. Oral ADSORBENT AST-120 decreases carotid intima-media thickness and arterial stiffness in patients with chronic renal failure. Kidney Blood Press. Res. 2004, 27, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, H.; Shibahara, N. Cardiorenal protective effect of the oral uremic toxin ABSORBENT AST-120 in chronic heart disease patients with moderate CKD. J. Nephrol. 2010, 23, 535–540. [Google Scholar] [PubMed]
- Owada, A.; Nakao, M.; Koike, J.; Ujiie, K.; Tomita, K.; Shiigai, T. Effects of oral adsorbent AST-120 on the progression of chronic renal failure: A randomized controlled study. Effects of oral adsorbent AST-120 on the progression of chronic renal failure: A randomized controlled study. Kidney Int. Suppl. 1997, 63, S188–S190. [Google Scholar] [PubMed]
- Prakash, S.; Chang, T.M. Microencapsulated genetically engineered live, E. coli DH5 cells administered orally to maintain normal plasma urea level in uremic rats. Nat. Med. 1996, 2, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Devlin, A.S.; Marcobal, A.; Dodd, D.; Nayfach, S.; Plummer, N.; Meyer, T.; Pollard, K.S.; Sonnenburg, J.L.; Fischbach, M.A. Modulation of a Circulating Uremic Solute via Rational Genetic Manipulation of the Gut Microbiota. Cell Host Microbe 2016, 20, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Maduell, F.; Moreso, F.; Pons, M.; Ramos, R.; Mora-Macià, J.; Carreras, J.; Soler, J.; Torres, F.; Campistol, J.M.; Martinez-Castelao, A.; ESHOL Study Group. High-efficiency postdilution online hemodiafiltration reduces all-cause mortality in hemodialysis patients. J. Am. Soc. Nephrol. 2013, 24, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.A.; Bots, M.L.; Canaud, B.; Davenport, A.; Grooteman, M.P.; Kircelli, F.; Locatelli, F.; Maduell, F.; Morena, M.; Nubé, M.J.; et al. HDF Pooling Project Investigators. Haemodiafiltration and mortality in end-stage kidney disease patients: A pooled individual participant data analysis from four randomized controlled trials. Nephrol. Dial. Transplant. 2016, 31, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Boschetti-de-Fierro, A.; Voigt, M.; Storr, M.; Krause, B. MCO Membranes: Enhanced Selectivity in High-Flux Class. Sci. Rep. 2015, 5, 18448. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, A.H.; Rosenkranz, A.R.; Lyko, R.; Krieter, D.H. Effects of Hemodialysis Therapy Using Dialyzers with Medium Cut-Off Membranes on Middle Molecules. Contrib. Nephrol. 2017, 191, 158–167. [Google Scholar] [PubMed]
- Zickler, D.; Schindler, R.; Willy, K.; Martus, P.; Pawlak, M.; Storr, M.; Hulko, M.; Boehler, T.; Glomb, M.A.; Liehr, K.; et al. Medium Cut-Off (MCO) Membranes Reduce Inflammation in Chronic Dialysis Patients-A Randomized Controlled Clinical Trial. PLoS ONE 2017, 12, e0169024. [Google Scholar] [CrossRef] [PubMed]
- Flessner, M.F. Peritoneal transport physiology: Insights from basic research. J. Am. Soc. Nephrol. 1991, 2, 122–135. [Google Scholar] [PubMed]
- Bammens, B.; Evenepoel, P.; Verbeke, K.; Vanrenterghem, Y. Removal of middle molecules and protein-bound solutes by peritoneal dialysis and relation with uremic symptoms. Kidney Int. 2003, 64, 2238–2243. [Google Scholar] [CrossRef] [PubMed]
- Lameire, N.; Vanholder, R.; De Smet, R. Uremic toxins and peritoneal dialysis. Kidney Int. Suppl. 2001, 78, S292–S297. [Google Scholar] [CrossRef] [PubMed]
- Leypoldt, J.K. Solute Transport across the Peritoneal Membrane. J. Am. Soc. Nephrol. 2002, 13 (Suppl. 1), S84–S91. [Google Scholar] [PubMed]
- Eloot, S.; Vanholder, R.; Dequidt, C.; Van Biesen, W. Removal of Different Classes of Uremic Toxins in APD vs. CAPD: A Randomized Cross-Over Study. Perit. Dial. Int. 2015, 35, 436–442. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velasquez, M.T.; Centron, P.; Barrows, I.; Dwivedi, R.; Raj, D.S. Gut Microbiota and Cardiovascular Uremic Toxicities. Toxins 2018, 10, 287. https://doi.org/10.3390/toxins10070287
Velasquez MT, Centron P, Barrows I, Dwivedi R, Raj DS. Gut Microbiota and Cardiovascular Uremic Toxicities. Toxins. 2018; 10(7):287. https://doi.org/10.3390/toxins10070287
Chicago/Turabian StyleVelasquez, Manuel T., Patricia Centron, Ian Barrows, Rama Dwivedi, and Dominic S. Raj. 2018. "Gut Microbiota and Cardiovascular Uremic Toxicities" Toxins 10, no. 7: 287. https://doi.org/10.3390/toxins10070287
APA StyleVelasquez, M. T., Centron, P., Barrows, I., Dwivedi, R., & Raj, D. S. (2018). Gut Microbiota and Cardiovascular Uremic Toxicities. Toxins, 10(7), 287. https://doi.org/10.3390/toxins10070287