Mechanisms of Action and Cell Death Associated with Clostridium perfringens Toxins



Abstract
1. Introduction
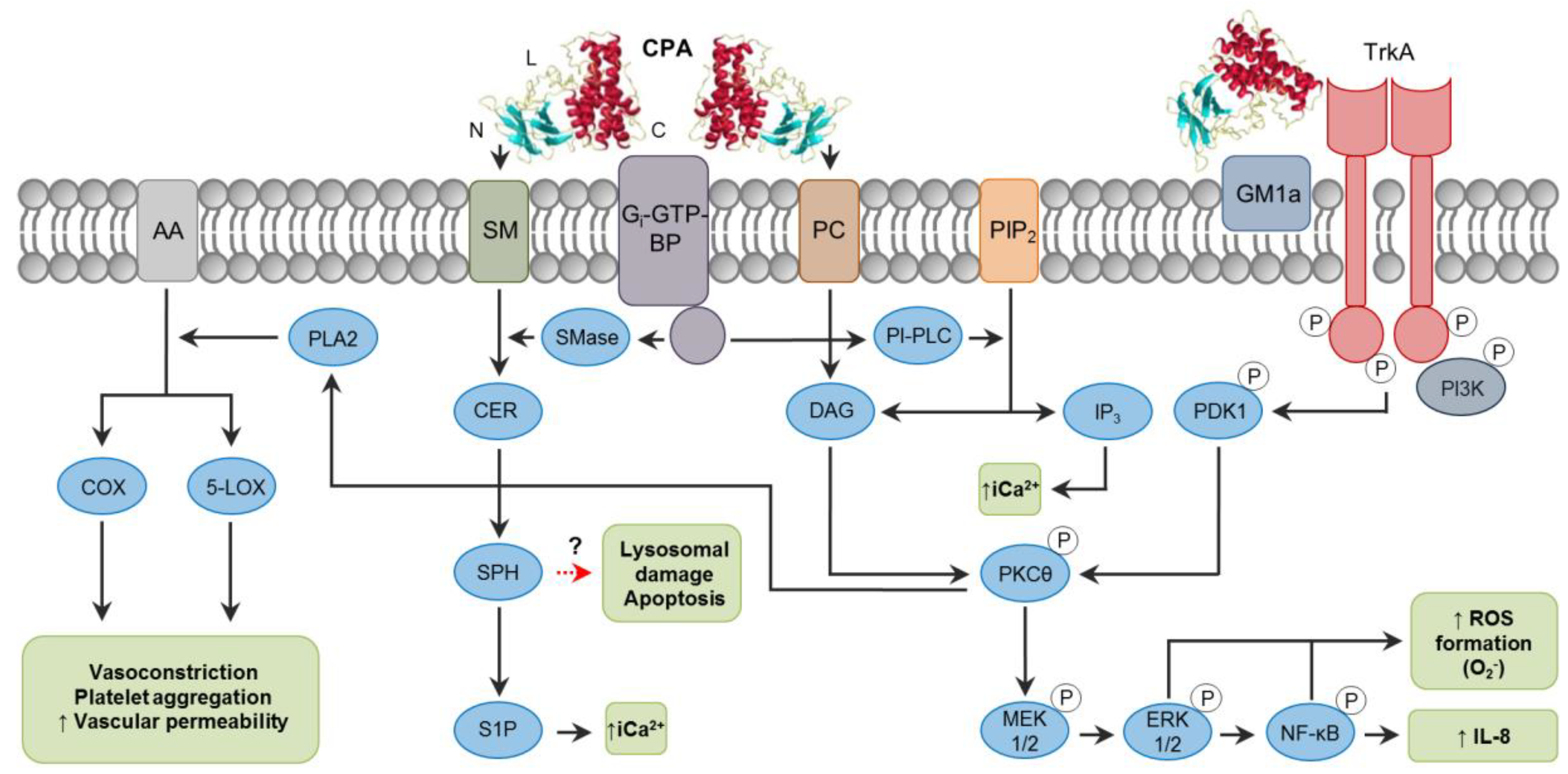
2. C. perfringens Alpha Toxin (CPA)
2.1. Toxin Genetics, Structure and Role in Disease
2.2. General Mechanism of Action
2.3. Mechanisms of Cell Death
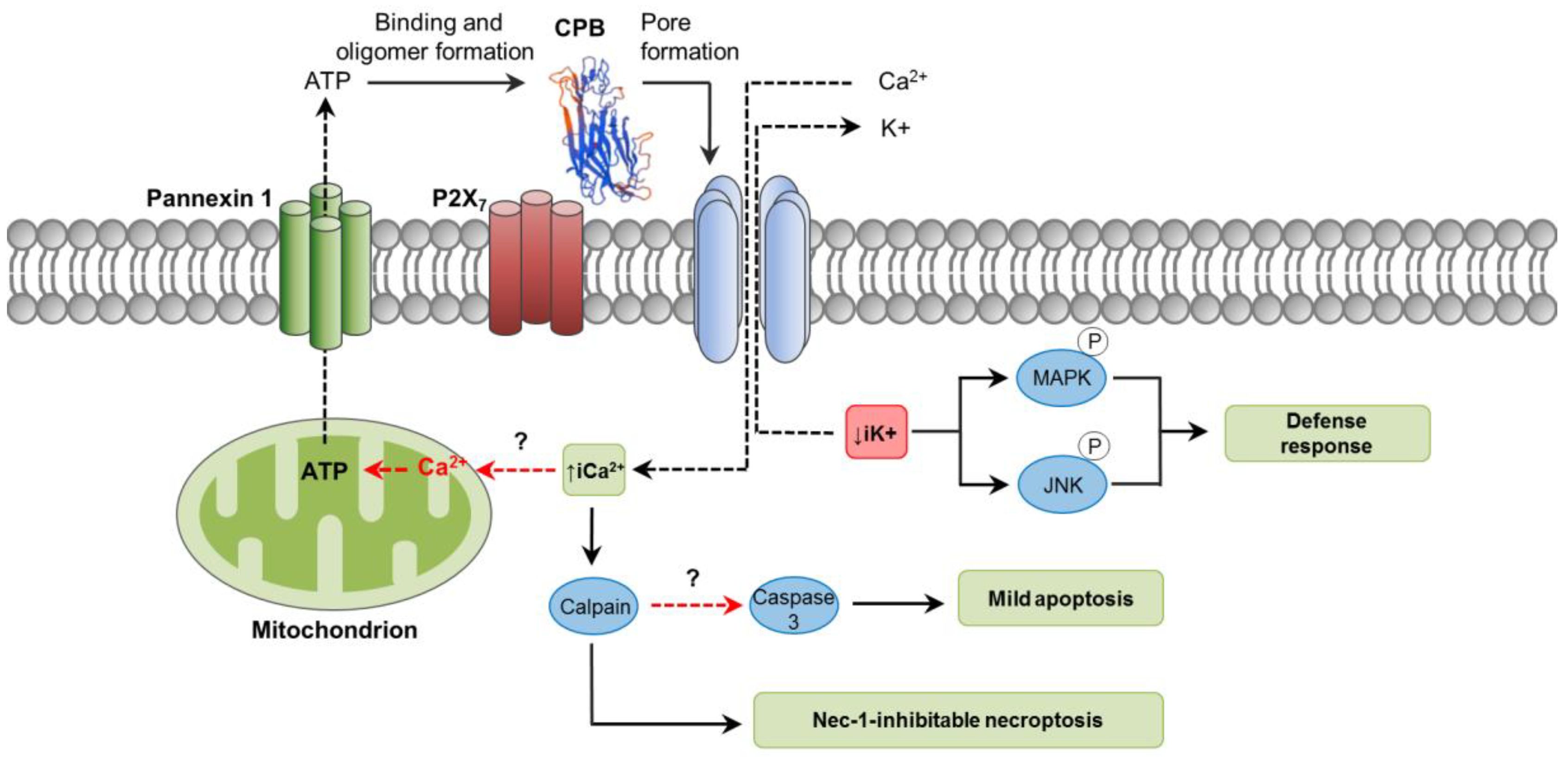
3. C. perfringens Beta Toxin (CPB)
3.1. Toxin Genetics, Structure and Role in Disease
3.2. General Mechanism of Action
3.3. Mechanisms of Cell Death
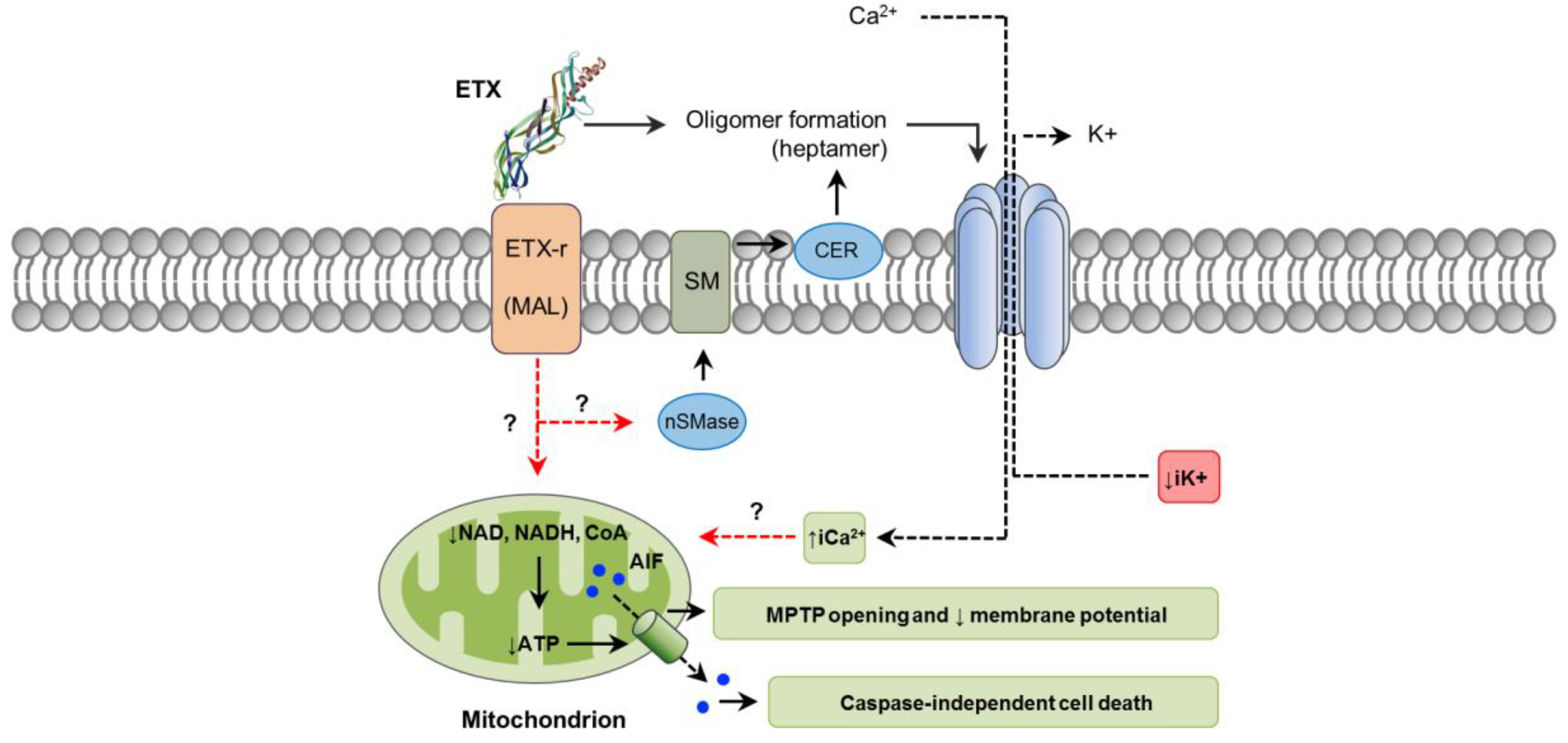
4. C. perfringens Epsilon Toxin (ETX)
4.1. Toxin Genetics, Structure and Role in Disease
4.2. General Mechanism of Action
4.3. Mechanisms of Cell Death
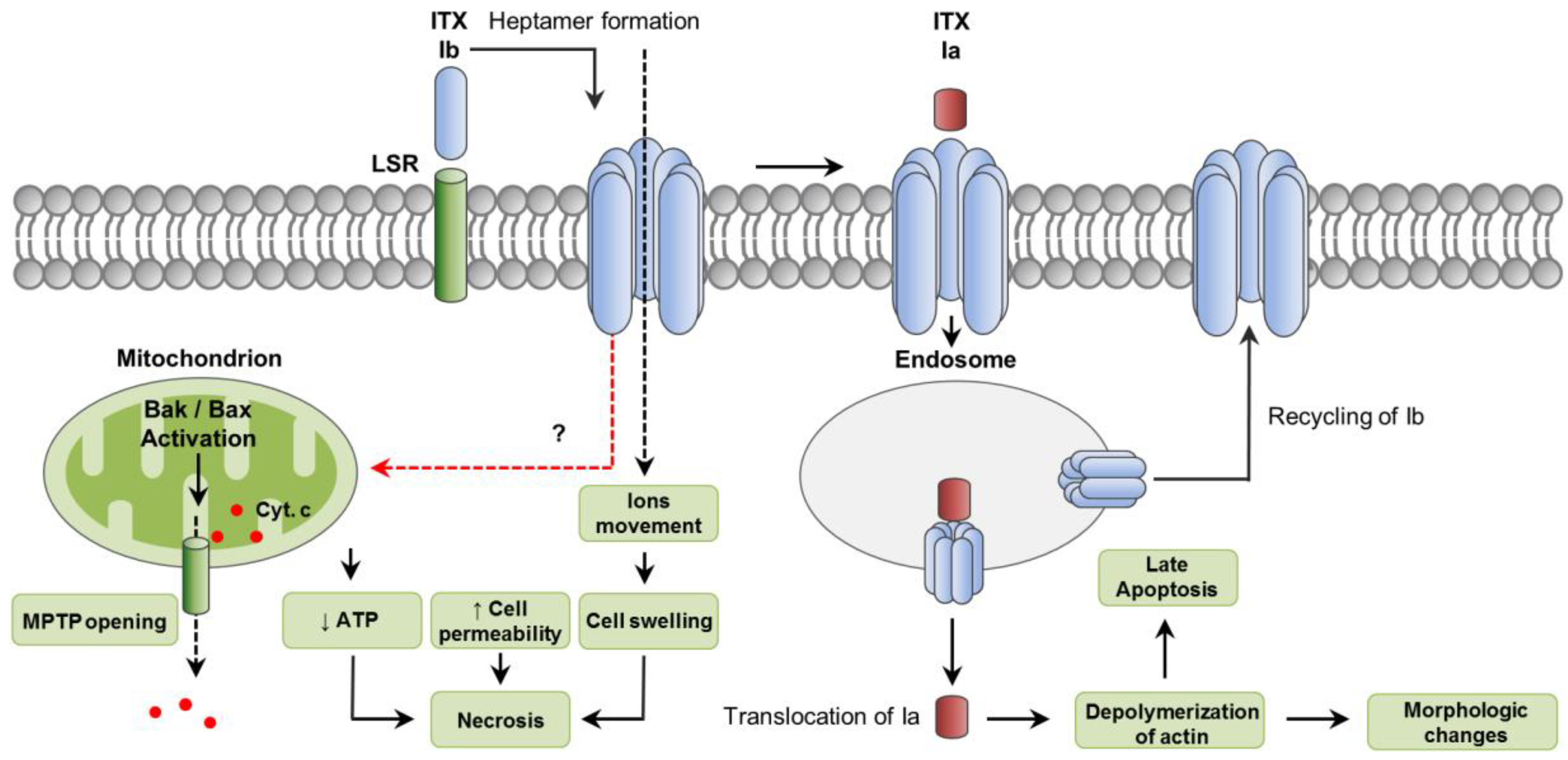
5. C. perfringens Iota Toxin (ITX)
5.1. Toxin Genetics, Structure and Role in Disease
5.2. General Mechanism of Action
5.3. Mechanisms of Cell Death
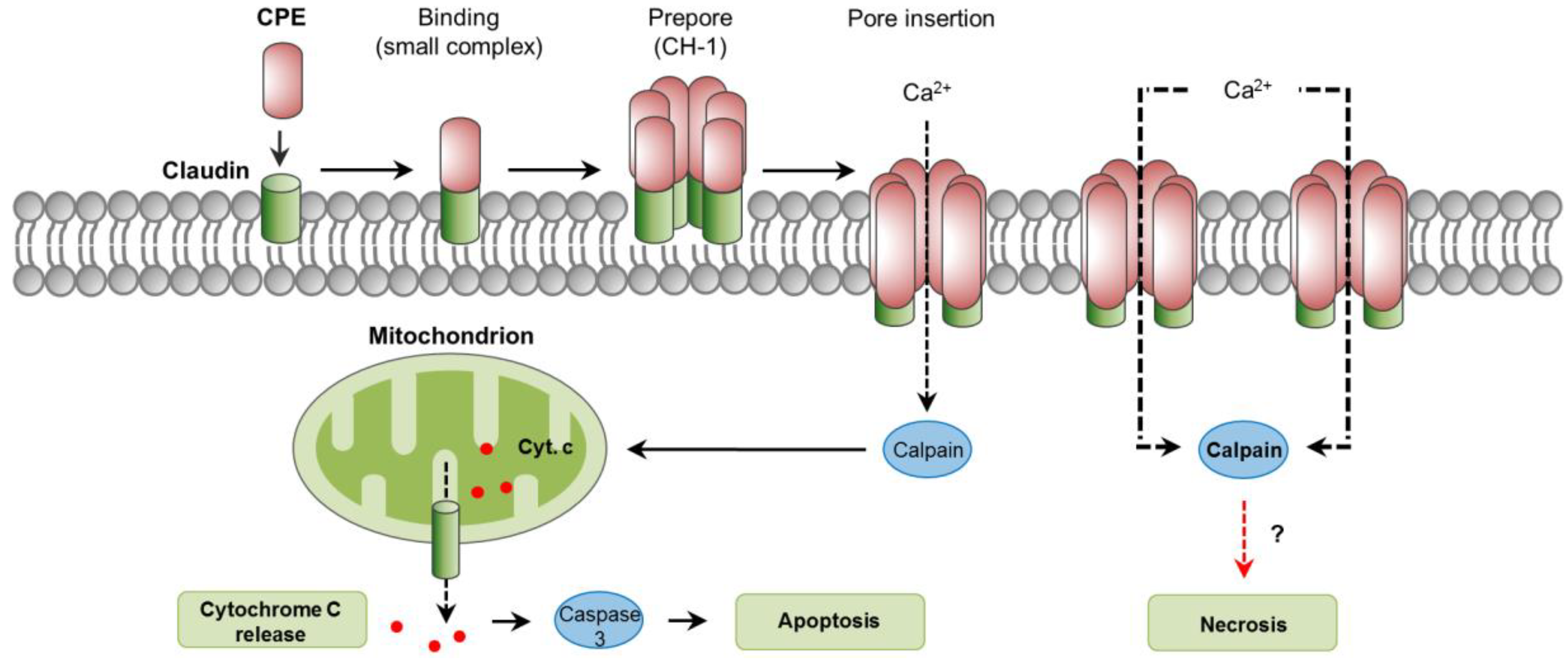
6. C. perfringens Enterotoxin (CPE)
6.1. Toxin Genetics, Structure and Role in Disease
6.2. General Mechanism of Action
6.3. Mechanisms of Cell Death
7. C. perfringens Necrotic Enteritis B-Like Toxin (NetB)
7.1. Toxin Genetics, Structure and Role in Disease
7.2. General Mechanism of Action
7.3. Mechanisms of Cell Death
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Uzal, F.A.; Freedman, J.C.; Shrestha, A.; Theoret, J.R.; Garcia, J.; Awad, M.M.; Adams, V.; Moore, R.J.; Rood, J.I.; McClane, B.A. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 2014, 9, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Revitt-Mills, S.A.; Rood, J.I.; Adams, V. Clostridium Perfringens Extracellular Toxins and Enzymes: 20 and Counting. Microbiol. Aust. 2015, 36, 114–117. [Google Scholar] [CrossRef]
- Rood, J.I.; Adams, V.; Lacey, J.; Lyras, D.; McClane, B.A.; Melville, S.B.; Moore, R.J.; Popoff, M.R.; Sarker, M.R.; Songer, J.G.; et al. Expansion of the Clostridium perfringens toxin-based typing scheme. Anaerobe 2018. [Google Scholar] [CrossRef]
- Uzal, F.A.; Vidal, J.E.; McClane, B.A.; Gurjar, A.A. Clostridium Perfringens Toxins Involved in Mammalian Veterinary Diseases. Open Toxinol. J. 2010, 2, 24–42. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 2010, 8, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Mimuro, H.; Ogawa, M.; Kobayashi, T.; Sanada, T.; Kim, M.; Sasakawa, C. Cell death and infection: A double-edged sword for host and pathogen survival. J. Cell Biol. 2011, 195, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, W.; User, S.D.; El-Gazzah, M.; Jaksik, R.; Sajjadi, E.; Rzeszowska-Wolny, J.; Los, M.J. Autophagy, apoptosis, mitoptosis and necrosis: Interdependence between those pathways and effects on cancer. Arch. Immunol. Ther. Exp. 2013, 61, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Ichim, G.; Green, D.R. Die another way—Non-apoptotic mechanisms of cell death. J. Cell Sci. 2014, 127, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A. Death in the fast lane: What’s next for necroptosis? FEBS J. 2016, 283, 2616–2625. [Google Scholar] [CrossRef] [PubMed]
- Titball, R.W.; Naylor, C.E.; Basak, A.K. The Clostridium perfringens alpha-toxin. Anaerobe 1999, 5, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Jewell, S.A.; Titball, R.W.; Huyet, J.; Naylor, C.E.; Basak, A.K.; Gologan, P.; Winlove, C.P.; Petrov, P.G. Clostridium perfringens α-toxin interaction with red cells and model membranes. Soft Matter 2015, 11, 7748–7761. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Terao, Y.; Sakurai, J.; Nagahama, M. Membrane-Binding Mechanism of Clostridium perfringens Alpha-Toxin. Toxins 2015, 7, 5268–5275. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Bryant, A.E.; Stevens, D.L.; Rood, J.I. Virulence studies on chromosomal alpha-toxin and theta-toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of alpha-toxin in Clostridium perfringens-mediated gas gangrene. Mol. Microbiol. 1995, 15, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Bryant, A.E. Biology and pathogenesis of thrombosis and procoagulant activity in invasive infections caused by group A streptococci and Clostridium perfringens. Clin. Microbiol. Rev. 2003, 16, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.O.S.; Uzal, F.A.; Oliveira, C.A.; Lobato, F.C.F. Gas gangrene (malignant edema). In Clostridial Diseases of Animals; Uzal, F.A., Songer, J.G., Prescott, J.F., Popoff, M.R., Eds.; Wiley Blackwell: Ames, IA, USA, 2016; pp. 243–254. [Google Scholar]
- Heuck, A.P.; Moe, P.C.; Johnson, B.B. The cholesterol-dependent cytolysin family of gram-positive bacterial toxins. Subcell. Biochem. 2010, 51, 551–577. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Ellemor, D.M.; Boyd, R.L.; Emmins, J.J.; Rood, J.I. Synergistic effects of alpha-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect. Immun. 2001, 69, 7904–7910. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Matsuno, T.; Shiihara, R.; Ochi, S.; Yamauchi, R.; Saito, Y.; Imagawa, H.; Nagahama, M.; Nishizawa, M.; Sakurai, J. The relationship between the metabolism of sphingomyelin species and the hemolysis of sheep erythrocytes induced by Clostridium perfringens alpha-toxin. J. Lipid Res. 2008, 49, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Kabura, M.; Takagishi, T.; Suzue, A.; Tominaga, K.; Urano, S.; Nagahama, M.; Kobayashi, K.; Furukawa, K.; Furukawa, K.; et al. Clostridium perfringens alpha-toxin recognizes the GM1a-TrkA complex. J. Biol. Chem. 2012, 287, 33070–33079. [Google Scholar] [CrossRef] [PubMed]
- Flores-Díaz, M.; Alape-Girón, A.; Clark, G.; Catimel, B.; Hirabayashi, Y.; Nice, E.; Gutiérrez, J.-M.; Titball, R.; Thelestam, M. A cellular deficiency of gangliosides causes hypersensitivity to Clostridium perfringens phospholipase C. J. Biol. Chem. 2005, 280, 26680–26689. [Google Scholar] [CrossRef] [PubMed]
- Monturiol-Gross, L.; Flores-Díaz, M.; Pineda-Padilla, M.J.; Castro-Castro, A.C.; Alape-Giron, A. Clostridium perfringens phospholipase C induced ROS production and cytotoxicity require PKC, MEK1 and NFκB activation. PLoS ONE 2014, 9, e86475. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.M.; Valero, J.G.; Pérez-Cormenzana, M.; Cano, A.; Alonso, C.; Goñi, F.M. Lipidomic profile of GM95 cell death induced by Clostridium perfringens alpha-toxin. Chem. Phys. Lipids 2017, 203, 54–70. [Google Scholar] [CrossRef] [PubMed]
- Maggio, B. The surface behavior of glycosphingolipids in biomembranes: A new frontier of molecular ecology. Prog. Biophys. Mol. Biol. 1994, 62, 55–117. [Google Scholar] [CrossRef]
- Yu, R.K.; Tsai, Y.-T.; Ariga, T.; Yanagisawa, M. Structures, biosynthesis, and functions of gangliosides—An overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sayeed, S.; Robertson, S.; Chen, J.; McClane, B.A. Sialidases affect the host cell adherence and epsilon toxin-induced cytotoxicity of Clostridium perfringens type D strain CN3718. PLoS Pathog. 2011, 7, e1002429. [Google Scholar] [CrossRef] [PubMed]
- Theoret, J.R.; Li, J.; Navarro, M.A.; Garcia, J.P.; Uzal, F.A.; McClane, B.A. Native or Proteolytically Activated NanI Sialidase Enhances the Binding and Cytotoxic Activity of Clostridium perfringens Enterotoxin and Beta Toxin. Infect. Immun. 2018, 86, e00730-17. [Google Scholar] [CrossRef] [PubMed]
- Ochi, S.; Oda, M.; Nagahama, M.; Sakurai, J. Clostridium perfringens alpha-toxin-induced hemolysis of horse erythrocytes is dependent on Ca2+ uptake. Biochim. Biophys. Acta 2003, 1613, 79–86. [Google Scholar] [CrossRef]
- Ochi, S.; Oda, M.; Matsuda, H.; Ikari, S.; Sakurai, J. Clostridium perfringens alpha-toxin activates the sphingomyelin metabolism system in sheep erythrocytes. J. Biol. Chem. 2004, 279, 12181–12189. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Ikari, S.; Matsuno, T.; Morimune, Y.; Nagahama, M.; Sakurai, J. Signal transduction mechanism involved in Clostridium perfringens alpha-toxin-induced superoxide anion generation in rabbit neutrophils. Infect. Immun. 2006, 74, 2876–2886. [Google Scholar] [CrossRef] [PubMed]
- Monturiol-Gross, L.; Flores-Díaz, M.; Campos-Rodríguez, D.; Mora, R.; Rodríguez-Vega, M.; Marks, D.L.; Alape-Girón, A. Internalization of Clostridium perfringens α-toxin leads to ERK activation and is involved on its cytotoxic effect. Cell. Microbiol. 2014, 16, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Takehara, M.; Takagishi, T.; Seike, S.; Ohtani, K.; Kobayashi, K.; Miyamoto, K.; Shimizu, T.; Nagahama, M. Clostridium perfringens α-Toxin Impairs Innate Immunity via Inhibition of Neutrophil Differentiation. Sci. Rep. 2016, 6, 28192. [Google Scholar] [CrossRef] [PubMed]
- Takehara, M.; Takagishi, T.; Seike, S.; Oishi, K.; Fujihara, Y.; Miyamoto, K.; Kobayashi, K.; Nagahama, M. Clostridium perfringens α-Toxin Impairs Lipid Raft Integrity in Neutrophils. Biol. Pharm. Bull. 2016, 39, 1694–1700. [Google Scholar] [CrossRef] [PubMed]
- Flores-Díaz, M.; Thelestam, M.; Clark, G.C.; Titball, R.W.; Alape-Girón, A. Effects of Clostridium perfringens phospholipase C in mammalian cells. Anaerobe 2004, 10, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.-M.; Moriwaki, K.; De Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Kolesnick, R.N.; Krönke, M. Regulation of ceramide production and apoptosis. Annu. Rev. Physiol. 1998, 60, 643–665. [Google Scholar] [CrossRef] [PubMed]
- Pettus, B.J.; Chalfant, C.E.; Hannun, Y.A. Ceramide in apoptosis: An overview and current perspectives. Biochim. Biophys. Acta 2002, 1585, 114–125. [Google Scholar] [CrossRef]
- Kågedal, K.; Zhao, M.; Svensson, I.; Brunk, U.T. Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem. J. 2001, 359, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.-C.; Appelqvist, H.; Nilsson, C.; Kågedal, K.; Roberg, K.; Ollinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis Int. J. Program. Cell Death 2010, 15, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [PubMed]
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium 2011, 50, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Blom, T.; Slotte, J.P.; Pitson, S.M.; Törnquist, K. Enhancement of intracellular sphingosine-1-phosphate production by inositol 1,4,5-trisphosphate-evoked calcium mobilisation in HEK-293 cells: Endogenous sphingosine-1-phosphate as a modulator of the calcium response. Cell Signal. 2005, 17, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Gurjar, A.; Li, J.; McClane, B.A. Characterization of toxin plasmids in Clostridium perfringens type C isolates. Infect. Immun. 2010, 78, 4860–4869. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, S.; Li, J.; McClane, B.A. Characterization of virulence plasmid diversity among Clostridium perfringens type B isolates. Infect. Immun. 2010, 78, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.E.; Brown, J.E.; Oyston, P.C.; Sakurai, J.; Titball, R.W. Molecular genetic analysis of beta-toxin of Clostridium perfringens reveals sequence homology with alpha-toxin, gamma-toxin, and leukocidin of Staphylococcus aureus. Infect. Immun. 1993, 61, 3958–3965. [Google Scholar] [PubMed]
- Sakurai, J.; Duncan, C.L. Some properties of beta-toxin produced by Clostridium perfringens type C. Infect. Immun. 1978, 21, 678–680. [Google Scholar] [PubMed]
- Popoff, M.R. Clostridial pore-forming toxins: Powerful virulence factors. Anaerobe 2014, 30, 220–238. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, J.; Fujii, Y. Purification and characterization of Clostridium perfringens beta toxin. Toxicon 1987, 25, 1301–1310. [Google Scholar] [CrossRef]
- Theoret, J.R.; Uzal, F.A.; McClane, B.A. Identification and characterization of Clostridium perfringens beta toxin variants with differing trypsin sensitivity and in vitro cytotoxicity activity. Infect. Immun. 2015, 83, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.P.; Anderson, M.; Blanchard, P.; Mete, A.; Uzal, F.A. The pathology of enterotoxemia by Clostridium perfringens type C in calves. J. Vet. Diagn. Investig. 2013, 25, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, S.; Uzal, F.A.; Fisher, D.J.; Saputo, J.; Vidal, J.E.; Chen, Y.; Gupta, P.; Rood, J.I.; McClane, B.A. Beta toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol. Microbiol. 2008, 67, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.E.; McClane, B.A.; Saputo, J.; Parker, J.; Uzal, F.A. Effects of Clostridium perfringens beta-toxin on the rabbit small intestine and colon. Infect. Immun. 2008, 76, 4396–4404. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.P.; Beingesser, J.; Fisher, D.J.; Sayeed, S.; McClane, B.A.; Posthaus, H.; Uzal, F.A. The effect of Clostridium perfringens type C strain CN3685 and its isogenic beta toxin null mutant in goats. Vet. Microbiol. 2012, 157, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A.; Saputo, J.; Sayeed, S.; Vidal, J.E.; Fisher, D.J.; Poon, R.; Adams, V.; Fernandez-Miyakawa, M.E.; Rood, J.I.; McClane, B.A. Development and application of new mouse models to study the pathogenesis of Clostridium perfringens type C enterotoxemias. Infect. Immun. 2009, 77, 5291–5299. [Google Scholar] [CrossRef] [PubMed]
- Miclard, J.; Jäggi, M.; Sutter, E.; Wyder, M.; Grabscheid, B.; Posthaus, H. Clostridium perfringens beta-toxin targets endothelial cells in necrotizing enteritis in piglets. Vet. Microbiol. 2009, 137, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Miclard, J.; van Baarlen, J.; Wyder, M.; Grabscheid, B.; Posthaus, H. Clostridium perfringens beta-toxin binding to vascular endothelial cells in a human case of enteritis necroticans. J. Med. Microbiol. 2009, 58, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, V.L.; Martel, A.; Pasmans, F.; Van Immerseel, F.; Posthaus, H. Endothelial binding of beta toxin to small intestinal mucosal endothelial cells in early stages of experimentally induced Clostridium perfringens type C enteritis in pigs. Vet. Pathol. 2013, 50, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Roos, S.; Wyder, M.; Candi, A.; Regenscheit, N.; Nathues, C.; van Immerseel, F.; Posthaus, H. Binding studies on isolated porcine small intestinal mucosa and in vitro toxicity studies reveal lack of effect of C. perfringens beta-toxin on the porcine intestinal epithelium. Toxins 2015, 7, 1235–1252. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Morimitsu, S.; Kihara, A.; Akita, M.; Setsu, K.; Sakurai, J. Involvement of tachykinin receptors in Clostridium perfringens beta-toxin-induced plasma extravasation. Br. J. Pharmacol. 2003, 138, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Kihara, A.; Kintoh, H.; Oda, M.; Sakurai, J. Involvement of tumour necrosis factor-alpha in Clostridium perfringens beta-toxin-induced plasma extravasation in mice. Br. J. Pharmacol. 2008, 153, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Hayashi, S.; Morimitsu, S.; Sakurai, J. Biological activities and pore formation of Clostridium perfringens beta toxin in HL 60 cells. J. Biol. Chem. 2003, 278, 36934–36941. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Ochi, S.; Oda, M.; Miyamoto, K.; Takehara, M.; Kobayashi, K. Recent insights into Clostridium perfringens beta-toxin. Toxins 2015, 7, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Shibutani, M.; Seike, S.; Yonezaki, M.; Takagishi, T.; Oda, M.; Kobayashi, K.; Sakurai, J. The p38 MAPK and JNK pathways protect host cells against Clostridium perfringens beta-toxin. Infect. Immun. 2013, 81, 3703–3708. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, C.; Popescu, F.; Wyder, M.; Sutter, E.; Zeeh, F.; Frey, J.; von Schubert, C.; Posthaus, H. Rapid cytopathic effects of Clostridium perfringens beta-toxin on porcine endothelial cells. Infect. Immun. 2010, 78, 2966–2973. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Seike, S.; Shirai, H.; Takagishi, T.; Kobayashi, K.; Takehara, M.; Sakurai, J. Role of P2X7 receptor in Clostridium perfringens beta-toxin-mediated cellular injury. Biochim. Biophys. Acta 2015, 1850, 2159–2167. [Google Scholar] [CrossRef] [PubMed]
- Seike, S.; Takehara, M.; Kobayashi, K.; Nagahama, M. Role of pannexin 1 in Clostridium perfringens beta-toxin-caused cell death. Biochim. Biophys. Acta 2016, 1858, 3150–3156. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.I.; Griffiths, E.J.; Rutter, G.A. Regulation of ATP production by mitochondrial Ca2+. Cell Calcium 2012, 52, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Ralevic, V. P2X receptors in the cardiovascular system. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1, 663–674. [Google Scholar] [CrossRef]
- Penuela, S.; Harland, L.; Simek, J.; Laird, D.W. Pannexin channels and their links to human disease. Biochem. J. 2014, 461, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Autheman, D.; Wyder, M.; Popoff, M.; D’Herde, K.; Christen, S.; Posthaus, H. Clostridium perfringens beta-toxin induces necrostatin-inhibitable, calpain-dependent necrosis in primary porcine endothelial cells. PLoS ONE 2013, 8, e64644. [Google Scholar] [CrossRef] [PubMed]
- Humphries, F.; Yang, S.; Wang, B.; Moynagh, P.N. RIP kinases: Key decision makers in cell death and innate immunity. Cell Death Differ. 2015, 22, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, S.; Li, J.; McClane, B.A. Virulence plasmid diversity in Clostridium perfringens type D isolates. Infect. Immun. 2007, 75, 2391–2398. [Google Scholar] [CrossRef] [PubMed]
- Minami, J.; Katayama, S.; Matsushita, O.; Matsushita, C.; Okabe, A. Lambda-toxin of Clostridium perfringens activates the precursor of epsilon-toxin by releasing its N- and C-terminal peptides. Microbiol. Immunol. 1997, 41, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Harkness, J.M.; Li, J.; McClane, B.A. Identification of a lambda toxin-negative Clostridium perfringens strain that processes and activates epsilon prototoxin intracellularly. Anaerobe 2012, 18, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.C.; McClane, B.A.; Uzal, F.A. New insights into Clostridium perfringens epsilon toxin activation and action on the brain during enterotoxemia. Anaerobe 2016, 41, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Bokori-Brown, M.; Savva, C.G.; Fernandes da Costa, S.P.; Naylor, C.E.; Basak, A.K.; Titball, R.W. Molecular basis of toxicity of Clostridium perfringens epsilon toxin. FEBS J. 2011, 278, 4589–4601. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.C.; Li, J.; Uzal, F.A.; McClane, B.A. Proteolytic processing and activation of Clostridium perfringens epsilon toxin by caprine small intestinal contents. mBio 2014, 5, e01994-14. [Google Scholar] [CrossRef] [PubMed]
- Filho, E.J.F.; Carvalho, A.U.; Assis, R.A.; Lobato, F.F.; Rachid, M.A.; Carvalho, A.A.; Ferreira, P.M.; Nascimento, R.A.; Fernandes, A.A.; Vidal, J.E.; et al. Clinicopathologic features of experimental Clostridium perfringens type D enterotoxemia in cattle. Vet. Pathol. 2009, 46, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R. Epsilon toxin: A fascinating pore-forming toxin. FEBS J. 2011, 278, 4602–4615. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.P.; Adams, V.; Beingesser, J.; Hughes, M.L.; Poon, R.; Lyras, D.; Hill, A.; McClane, B.A.; Rood, J.I.; Uzal, F.A. Epsilon toxin is essential for the virulence of Clostridium perfringens type D infection in sheep, goats, and mice. Infect. Immun. 2013, 81, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.P.; Giannitti, F.; Finnie, J.W.; Manavis, J.; Beingesser, J.; Adams, V.; Rood, J.I.; Uzal, F.A. Comparative neuropathology of ovine enterotoxemia produced by Clostridium perfringens type D wild-type strain CN1020 and its genetically modified derivatives. Vet. Pathol. 2015, 52, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A.; Giannitti, F.; Finnie, J.W.; Garcia, J.P. Diseases produced by Clostridium perfringens type D. In Clostridial Diseases of Animals; Wiley Blackwell: Ames, IA, USA, 2016; pp. 157–172. [Google Scholar]
- Goldstein, J.; Morris, W.E.; Loidl, C.F.; Tironi-Farinati, C.; Tironi-Farinatti, C.; McClane, B.A.; Uzal, F.A.; Fernandez Miyakawa, M.E. Clostridium perfringens epsilon toxin increases the small intestinal permeability in mice and rats. PLoS ONE 2009, 4, e7065. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A.; Songer, J.G. Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. J. Vet. Diagn. Investig. 2008, 20, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Soler-Jover, A.; Dorca, J.; Popoff, M.R.; Gibert, M.; Saura, J.; Tusell, J.M.; Serratosa, J.; Blasi, J.; Martín-Satué, M. Distribution of Clostridium perfringens epsilon toxin in the brains of acutely intoxicated mice and its effect upon glial cells. Toxicon 2007, 50, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A. Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. Anaerobe 2004, 10, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Finnie, J.W.; Manavis, J.; Blumbergs, P.C. Aquaporin-4 in acute cerebral edema produced by Clostridium perfringens type D epsilon toxin. Vet. Pathol. 2008, 45, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Dorca-Arévalo, J.; Soler-Jover, A.; Gibert, M.; Popoff, M.R.; Martín-Satué, M.; Blasi, J. Binding of epsilon-toxin from Clostridium perfringens in the nervous system. Vet. Microbiol. 2008, 131, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Wioland, L.; Dupont, J.-L.; Bossu, J.-L.; Popoff, M.R.; Poulain, B. Attack of the nervous system by Clostridium perfringens Epsilon toxin: From disease to mode of action on neural cells. Toxicon 2013, 75, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Wioland, L.; Dupont, J.-L.; Doussau, F.; Gaillard, S.; Heid, F.; Isope, P.; Pauillac, S.; Popoff, M.R.; Bossu, J.-L.; Poulain, B. Epsilon toxin from Clostridium perfringens acts on oligodendrocytes without forming pores, and causes demyelination. Cell. Microbiol. 2015, 17, 369–388. [Google Scholar] [CrossRef] [PubMed]
- Soler-Jover, A.; Blasi, J.; Gómez de Aranda, I.; Navarro, P.; Gibert, M.; Popoff, M.R.; Martín-Satué, M. Effect of epsilon toxin-GFP on MDCK cells and renal tubules in vivo. J. Histochem. Cytochem. 2004, 52, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.W.; Williamson, E.D.; Havard, H.; Modi, N.; Brown, J. Evaluation of a new cytotoxicity assay for Clostridium perfringens type D epsilon toxin. FEMS Microbiol. Lett. 1994, 116, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Gibert, M.; Gillet, D.; Laurent-Winter, C.; Boquet, P.; Popoff, M.R. Clostridium perfringens epsilon-toxin acts on MDCK cells by forming a large membrane complex. J. Bacteriol. 1997, 179, 6480–6487. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Matsushita, O.; Minami, J.; Katayama, S.; Shimamoto, S.; Okabe, A. Cleavage of a C-terminal peptide is essential for heptamerization of Clostridium perfringens epsilon-toxin in the synaptosomal membrane. J. Biol. Chem. 2001, 276, 13778–13783. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.L.; Li, J.; Uzal, F.A.; McClane, B.A. Evidence for a prepore stage in the action of Clostridium perfringens epsilon toxin. PLoS ONE 2011, 6, e22053. [Google Scholar] [CrossRef] [PubMed]
- Takagishi, T.; Oda, M.; Takehara, M.; Kobayashi, K.; Nagahama, M. Oligomer formation of Clostridium perfringens epsilon-toxin is induced by activation of neutral sphingomyelinase. Biochim. Biophys. Acta 2016, 1858, 2681–2688. [Google Scholar] [CrossRef] [PubMed]
- Ivie, S.E.; Fennessey, C.M.; Sheng, J.; Rubin, D.H.; McClain, M.S. Gene-trap mutagenesis identifies mammalian genes contributing to intoxication by Clostridium perfringens ε-toxin. PLoS ONE 2011, 6, e17787. [Google Scholar] [CrossRef] [PubMed]
- Rumah, K.R.; Ma, Y.; Linden, J.R.; Oo, M.L.; Anrather, J.; Schaeren-Wiemers, N.; Alonso, M.A.; Fischetti, V.A.; McClain, M.S.; Vartanian, T. The Myelin and Lymphocyte Protein MAL Is Required for Binding and Activity of Clostridium perfringens ε-Toxin. PLoS Pathog. 2015, 11, e1004896. [Google Scholar] [CrossRef] [PubMed]
- Schaeren-Wiemers, N.; Valenzuela, D.M.; Frank, M.; Schwab, M.E. Characterization of a rat gene, rMAL, encoding a protein with four hydrophobic domains in central and peripheral myelin. J. Neurosci. 1995, 15, 5753–5764. [Google Scholar] [CrossRef] [PubMed]
- Khalili, S.; Jahangiri, A.; Hashemi, Z.S.; Khalesi, B.; Mard-Soltani, M.; Amani, J. Structural pierce into molecular mechanism underlying Clostridium perfringens Epsilon toxin function. Toxicon 2017, 127, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Maier, E.; Gibert, M.; Popoff, M.R.; Benz, R. Clostridium perfringens epsilon toxin induces a rapid change of cell membrane permeability to ions and forms channels in artificial lipid bilayers. J. Biol. Chem. 2001, 276, 15736–15740. [Google Scholar] [CrossRef] [PubMed]
- Chassin, C.; Bens, M.; de Barry, J.; Courjaret, R.; Bossu, J.L.; Cluzeaud, F.; Ben Mkaddem, S.; Gibert, M.; Poulain, B.; Popoff, M.R.; et al. Pore-forming epsilon toxin causes membrane permeabilization and rapid ATP depletion-mediated cell death in renal collecting duct cells. Am. J. Physiol. Renal Physiol. 2007, 293, F927–F937. [Google Scholar] [CrossRef] [PubMed]
- Fennessey, C.M.; Ivie, S.E.; McClain, M.S. Coenzyme depletion by members of the aerolysin family of pore-forming toxins leads to diminished ATP levels and cell death. Mol. Biosyst. 2012, 8, 2097–2105. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Gibert, M.; Gourch, A.; Bens, M.; Vandewalle, A.; Popoff, M.R. Clostridium perfringens epsilon toxin rapidly decreases membrane barrier permeability of polarized MDCK cells. Cell. Microbiol. 2003, 5, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Lonchamp, E.; Dupont, J.-L.; Wioland, L.; Courjaret, R.; Mbebi-Liegeois, C.; Jover, E.; Doussau, F.; Popoff, M.R.; Bossu, J.-L.; de Barry, J.; et al. Clostridium perfringens epsilon toxin targets granule cells in the mouse cerebellum and stimulates glutamate release. PLoS ONE 2010, 5, e13046. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, J.; Nagahama, M.; Oda, M.; Tsuge, H.; Kobayashi, K. Clostridium perfringens iota-toxin: Structure and function. Toxins 2009, 1, 208–228. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Miyamoto, K.; McClane, B.A. Comparison of virulence plasmids among Clostridium perfringens type E isolates. Infect. Immun. 2007, 75, 1811–1819. [Google Scholar] [CrossRef] [PubMed]
- Gibert, M.; Petit, L.; Raffestin, S.; Okabe, A.; Popoff, M.R. Clostridium perfringens iota-toxin requires activation of both binding and enzymatic components for cytopathic activity. Infect. Immun. 2000, 68, 3848–3853. [Google Scholar] [CrossRef] [PubMed]
- Songer, J.G. Infections by Clostridium perfringens type E. In Clostridial Diseases of Animals; Uzal, F.A., Songer, J.G., Prescott, J.F., Popoff, M.R., Eds.; Wiley Blackwell: Ames, IA, USA, 2016; pp. 173–176. [Google Scholar]
- Schmidt, G.; Papatheodorou, P.; Aktories, K. Novel receptors for bacterial protein toxins. Curr. Opin. Microbiol. 2015, 23, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Wigelsworth, D.J.; Ruthel, G.; Schnell, L.; Herrlich, P.; Blonder, J.; Veenstra, T.D.; Carman, R.J.; Wilkins, T.D.; Van Nhieu, G.T.; Pauillac, S.; et al. CD44 Promotes intoxication by the clostridial iota-family toxins. PLoS ONE 2012, 7, e51356. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.F.; Mainguy, G.; Gibert, M.; Marvaud, J.C.; Stiles, B.G.; Popoff, M.R. Transcytosis of iota-toxin across polarized CaCo-2 cells. Mol. Microbiol. 2002, 43, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Nagayasu, K.; Kobayashi, K.; Sakurai, J. Binding component of Clostridium perfringens iota-toxin induces endocytosis in Vero cells. Infect. Immun. 2002, 70, 1909–1914. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Umezaki, M.; Tashiro, R.; Oda, M.; Kobayashi, K.; Shibutani, M.; Takagishi, T.; Ishidoh, K.; Fukuda, M.; Sakurai, J. Intracellular trafficking of Clostridium perfringens iota-toxin b. Infect. Immun. 2012, 80, 3410–3416. [Google Scholar] [CrossRef] [PubMed]
- Knapp, O.; Benz, R.; Popoff, M.R. Pore-forming activity of clostridial binary toxins. Biochim. Biophys. Acta 2016, 1858, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, H.; Nagahama, M.; Nishimura, H.; Hisatsune, J.; Sakaguchi, Y.; Itogawa, Y.; Katunuma, N.; Sakurai, J. Crystal structure and site-directed mutagenesis of enzymatic components from Clostridium perfringens iota-toxin. J. Mol. Biol. 2003, 325, 471–483. [Google Scholar] [CrossRef]
- Tsuge, H.; Nagahama, M.; Oda, M.; Iwamoto, S.; Utsunomiya, H.; Marquez, V.E.; Katunuma, N.; Nishizawa, M.; Sakurai, J. Structural basis of actin recognition and arginine ADP-ribosylation by Clostridium perfringens-toxin. Proc. Natl. Acad. Sci. USA 2008, 105, 7399–7404. [Google Scholar] [CrossRef] [PubMed]
- Gibert, M.; Monier, M.-N.; Ruez, R.; Hale, M.L.; Stiles, B.G.; Benmerah, A.; Johannes, L.; Lamaze, C.; Popoff, M.R. Endocytosis and toxicity of clostridial binary toxins depend on a clathrin-independent pathway regulated by Rho-GDI. Cell. Microbiol. 2011, 13, 154–170. [Google Scholar] [CrossRef] [PubMed]
- Takehara, M.; Takagishi, T.; Seike, S.; Oda, M.; Sakaguchi, Y.; Hisatsune, J.; Ochi, S.; Kobayashi, K.; Nagahama, M. Cellular Entry of Clostridium perfringens Iota-Toxin and Clostridium botulinum C2 Toxin. Toxins 2017, 9, 247. [Google Scholar] [CrossRef] [PubMed]
- Gibert, M.; Marvaud, J.C.; Pereira, Y.; Hale, M.L.; Stiles, B.G.; Boquet, P.; Lamaze, C.; Popoff, M.R. Differential requirement for the translocation of clostridial binary toxins: Iota toxin requires a membrane potential gradient. FEBS Lett. 2007, 581, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Umezaki, M.; Oda, M.; Kobayashi, K.; Tone, S.; Suda, T.; Ishidoh, K.; Sakurai, J. Clostridium perfringens iota-toxin b induces rapid cell necrosis. Infect. Immun. 2011, 79, 4353–4360. [Google Scholar] [CrossRef] [PubMed]
- Hilger, H.; Pust, S.; von Figura, G.; Kaiser, E.; Stiles, B.G.; Popoff, M.R.; Barth, H. The long-lived nature of Clostridium perfringens iota toxin in mammalian cells induces delayed apoptosis. Infect. Immun. 2009, 77, 5593–5601. [Google Scholar] [CrossRef] [PubMed]
- McClane, B.A.; Robertson, S.L.; Li, J. Clostridium perfringens. In Food Microbiology: Fundamentals and Frontiers; Doyle, M.P., Buchanan, R.L., Eds.; ASM Press: Washington, DC, USA, 2013; pp. 465–489. [Google Scholar]
- McClane, B.A.; Lyerly, D.M.; Wilkins, T.D. Enterotoxic clostridia: Clostridium perfringens type A and Clostridium difficile. In Gram-Positive Pathogens; Fischetti, V.A., Novick, R.P., Ferretti, R.P., Portnoy, D.A., Rood, J.I., Eds.; ASM Press: Washington, DC, USA, 2006; pp. 703–714. [Google Scholar]
- Freedman, J.C.; Shrestha, A.; McClane, B.A. Clostridium perfringens Enterotoxin: Action, Genetics, and Translational Applications. Toxins 2016, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Hanna, P.C.; Mietzner, T.A.; Schoolnik, G.K.; McClane, B.A. Localization of the receptor-binding region of Clostridium perfringens enterotoxin utilizing cloned toxin fragments and synthetic peptides. The 30 C-terminal amino acids define a functional binding region. J. Biol. Chem. 1991, 266, 11037–11043. [Google Scholar] [PubMed]
- Harada, M.; Kondoh, M.; Ebihara, C.; Takahashi, A.; Komiya, E.; Fujii, M.; Mizuguchi, H.; Tsunoda, S.-I.; Horiguchi, Y.; Yagi, K.; et al. Role of tyrosine residues in modulation of claudin-4 by the C-terminal fragment of Clostridium perfringens enterotoxin. Biochem. Pharmacol. 2007, 73, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Komiya, E.; Kakutani, H.; Yoshida, T.; Fujii, M.; Horiguchi, Y.; Mizuguchi, H.; Tsutsumi, Y.; Tsunoda, S.; Koizumi, N.; et al. Domain mapping of a claudin-4 modulator, the C-terminal region of C-terminal fragment of Clostridium perfringens enterotoxin, by site-directed mutagenesis. Biochem. Pharmacol. 2008, 75, 1639–1648. [Google Scholar] [CrossRef] [PubMed]
- Smedley, J.G.; McClane, B.A. Fine mapping of the N-terminal cytotoxicity region of Clostridium perfringens enterotoxin by site-directed mutagenesis. Infect. Immun. 2004, 72, 6914–6923. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Miyamoto, K.; Sayeed, S.; McClane, B.A. Organization of the cpe locus in CPE-positive Clostridium perfringens type C and D isolates. PLoS ONE 2010, 5, e10932. [Google Scholar] [CrossRef] [PubMed]
- Billington, S.J.; Wieckowski, E.U.; Sarker, M.R.; Bueschel, D.; Songer, J.G.; McClane, B.A. Clostridium perfringens type E animal enteritis isolates with highly conserved, silent enterotoxin gene sequences. Infect. Immun. 1998, 66, 4531–4536. [Google Scholar] [PubMed]
- Miyamoto, K.; Yumine, N.; Mimura, K.; Nagahama, M.; Li, J.; McClane, B.A.; Akimoto, S. Identification of novel Clostridium perfringens type E strains that carry an iota toxin plasmid with a functional enterotoxin gene. PLoS ONE 2011, 6, e20376. [Google Scholar] [CrossRef] [PubMed]
- Sarker, M.R.; Carman, R.J.; McClane, B.A. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol. Microbiol. 1999, 33, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Carman, R.J. Clostridium perfringens in spontaneous and antibiotic associated diarrhoea of man and other animals. Rev. Med. Microbiol. 1997, 8, S43–S45. [Google Scholar] [CrossRef]
- Bos, J.; Smithee, L.; McClane, B.; Distefano, R.F.; Uzal, F.; Songer, J.G.; Mallonee, S.; Crutcher, J.M. Fatal necrotizing colitis following a foodborne outbreak of enterotoxigenic Clostridium perfringens type A infection. Clin. Infect. Dis. 2005, 40, e78–e83. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Fatal foodborne Clostridium perfringens illness at a state psychiatric hospital—Louisiana, 2010. MMWR Morb. Mortal. Wkly. Rep. 2012, 61, 605–608. [Google Scholar]
- Caserta, J.A.; Robertson, S.L.; Saputo, J.; Shrestha, A.; McClane, B.A.; Uzal, F.A. Development and application of a mouse intestinal loop model to study the in vivo action of Clostridium perfringens enterotoxin. Infect. Immun. 2011, 79, 3020–3027. [Google Scholar] [CrossRef] [PubMed]
- Katahira, J.; Inoue, N.; Horiguchi, Y.; Matsuda, M.; Sugimoto, N. Molecular cloning and functional characterization of the receptor for Clostridium perfringens enterotoxin. J. Cell Biol. 1997, 136, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Katahira, J.; Horiguchi, Y.; Sonoda, N.; Furuse, M.; Tsukita, S. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction integral membrane protein. FEBS Lett. 2000, 476, 258–261. [Google Scholar] [CrossRef]
- Shrestha, A.; McClane, B.A. Human claudin-8 and -14 are receptors capable of conveying the cytotoxic effects of Clostridium perfringens enterotoxin. mBio 2013, 4, e00594-12. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.L.; Smedley, J.G.; Singh, U.; Chakrabarti, G.; Van Itallie, C.M.; Anderson, J.M.; McClane, B.A. Compositional and stoichiometric analysis of Clostridium perfringens enterotoxin complexes in Caco-2 cells and claudin 4 fibroblast transfectants. Cell. Microbiol. 2007, 9, 2734–2755. [Google Scholar] [CrossRef] [PubMed]
- Smedley, J.G.; Uzal, F.A.; McClane, B.A. Identification of a prepore large-complex stage in the mechanism of action of Clostridium perfringens enterotoxin. Infect. Immun. 2007, 75, 2381–2390. [Google Scholar] [CrossRef] [PubMed]
- McClane, B.A.; Uzal, F.A.; Miyakawa, M.F.; Lyerly, D.; Wilkins, T.D. The enterotoxic clostridia. In The Prokaryotes; Dworkin, M., Falkow, S., Rosenburg, E., Schleifer, H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 688–752. [Google Scholar]
- McClane, B.A.; Wnek, A.P.; Hulkower, K.I.; Hanna, P.C. Divalent cation involvement in the action of Clostridium perfringens type A enterotoxin. Early events in enterotoxin action are divalent cation-independent. J. Biol. Chem. 1988, 263, 2423–2435. [Google Scholar] [PubMed]
- McClane, B.A. Clostridium perfringens enterotoxin acts by producing small molecule permeability alterations in plasma membranes. Toxicology 1994, 87, 43–67. [Google Scholar] [CrossRef]
- Chakrabarti, G.; McClane, B.A. The importance of calcium influx, calpain and calmodulin for the activation of CaCo-2 cell death pathways by Clostridium perfringens enterotoxin. Cell. Microbiol. 2005, 7, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, G.; Zhou, X.; McClane, B.A. Death pathways activated in CaCo-2 cells by Clostridium perfringens enterotoxin. Infect. Immun. 2003, 71, 4260–4270. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.; Van Itallie, C.M.; Mitic, L.L.; Anderson, J.M.; McClane, B.A. CaCo-2 cells treated with Clostridium perfringens enterotoxin form multiple large complex species, one of which contains the tight junction protein occludin. J. Biol. Chem. 2000, 275, 18407–18417. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.; Klein, E.; McClane, B.A. Clostridium perfringens type A enterotoxin induces concurrent development of tissue damage and fluid accumulation in the rabbit ileum. J. Diarrheal Dis. 1994, 12, 200–207. [Google Scholar]
- Garcia, J.P.; Li, J.; Shrestha, A.; Freedman, J.C.; Beingesser, J.; McClane, B.A.; Uzal, F.A. Clostridium perfringens type A enterotoxin damages the rabbit colon. Infect. Immun. 2014, 82, 2211–2218. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.C.; Navarro, M.A.; Morrell, E.; Beingesser, J.; Shrestha, A.; McClane, B.A.; Uzal, F.A. Evidence that Clostridium perfringens Enterotoxin-Induced Intestinal Damage and Enterotoxemic Death in Mice Can Occur Independently of Intestinal Caspase-3 Activation. Infect. Immun. 2018, IAI.00931-17. [Google Scholar] [CrossRef] [PubMed]
- Lepp, D.; Roxas, B.; Parreira, V.R.; Marri, P.R.; Rosey, E.L.; Gong, J.; Songer, J.G.; Vedantam, G.; Prescott, J.F. Identification of novel pathogenicity loci in Clostridium perfringens strains that cause avian necrotic enteritis. PLoS ONE 2010, 5, e10795. [Google Scholar] [CrossRef]
- Bannam, T.L.; Yan, X.-X.; Harrison, P.F.; Seemann, T.; Keyburn, A.L.; Stubenrauch, C.; Weeramantri, L.H.; Cheung, J.K.; McClane, B.A.; Boyce, J.D.; et al. Necrotic enteritis-derived Clostridium perfringens strain with three closely related independently conjugative toxin and antibiotic resistance plasmids. mBio 2011, 2, e00190-11. [Google Scholar] [CrossRef] [PubMed]
- Parreira, V.R.; Costa, M.; Eikmeyer, F.; Blom, J.; Prescott, J.F. Sequence of two plasmids from Clostridium perfringens chicken necrotic enteritis isolates and comparison with C. perfringens conjugative plasmids. PLoS ONE 2012, 7, e49753. [Google Scholar] [CrossRef] [PubMed]
- Lacey, J.A.; Keyburn, A.L.; Ford, M.E.; Portela, R.W.; Johanesen, P.A.; Lyras, D.; Moore, R.J. Conjugation-mediated horizontal gene transfer of Clostridium perfringens plasmids in the chicken gastrointestinal tract results in the formation of new virulent strains. Appl. Environ. Microbiol. 2017, 83, e01814-17. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-X.; Porter, C.J.; Hardy, S.P.; Steer, D.; Smith, A.I.; Quinsey, N.S.; Hughes, V.; Cheung, J.K.; Keyburn, A.L.; Kaldhusdal, M.; et al. Structural and functional analysis of the pore-forming toxin NetB from Clostridium perfringens. mBio 2013, 4, e00019-13. [Google Scholar] [CrossRef] [PubMed]
- Keyburn, A.L.; Yan, X.-X.; Bannam, T.L.; Van Immerseel, F.; Rood, J.I.; Moore, R.J. Association between avian necrotic enteritis and Clostridium perfringens strains expressing NetB toxin. Vet. Res. 2010, 41, 21. [Google Scholar] [CrossRef] [PubMed]
- Rood, J.I.; Keyburn, A.L.; Moore, R.J. NetB and necrotic enteritis: The hole movable story. Avian Pathol. J. WVPA 2016, 45, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.F.; Parreira, V.R.; Mehdizadeh Gohari, I.; Lepp, D.; Gong, J. The pathogenesis of necrotic enteritis in chickens: What we know and what we need to know: A review. Avian Pathol. 2016, 45, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Olkowski, A.A.; Wojnarowicz, C.; Chirino-Trejo, M.; Laarveld, B.; Sawicki, G. Sub-clinical necrotic enteritis in broiler chickens: Novel etiological consideration based on ultra-structural and molecular changes in the intestinal tissue. Res. Vet. Sci. 2008, 85, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Savva, C.G.; Fernandes da Costa, S.P.; Bokori-Brown, M.; Naylor, C.E.; Cole, A.R.; Moss, D.S.; Titball, R.W.; Basak, A.K. Molecular architecture and functional analysis of NetB, a pore-forming toxin from Clostridium perfringens. J. Biol. Chem. 2013, 288, 3512–3522. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.C.; Hendricks, M.R.; McClane, B.A. The Potential Therapeutic Agent Mepacrine Protects Caco-2 Cells against Clostridium perfringens Enterotoxin Action. mSphere 2017, 2, e00352-17. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Toxin Produced | |||||
|---|---|---|---|---|---|---|
| α (CPA) | β (CPB) | ε (ETX) | ι (ITX) | CPE | NetB | |
| A | + | − | − | − | − | − |
| B | + | + | + | − | − | − |
| C | + | + | − | − | +/− | − |
| D | + | − | + | − | +/− | − |
| E | + | − | − | + | +/− | − |
| F | + | − | − | − | + | − |
| G | + | − | − | − | − | + |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navarro, M.A.; McClane, B.A.; Uzal, F.A. Mechanisms of Action and Cell Death Associated with Clostridium perfringens Toxins. Toxins 2018, 10, 212. https://doi.org/10.3390/toxins10050212
Navarro MA, McClane BA, Uzal FA. Mechanisms of Action and Cell Death Associated with Clostridium perfringens Toxins. Toxins. 2018; 10(5):212. https://doi.org/10.3390/toxins10050212
Chicago/Turabian StyleNavarro, Mauricio A., Bruce A. McClane, and Francisco A. Uzal. 2018. "Mechanisms of Action and Cell Death Associated with Clostridium perfringens Toxins" Toxins 10, no. 5: 212. https://doi.org/10.3390/toxins10050212
APA StyleNavarro, M. A., McClane, B. A., & Uzal, F. A. (2018). Mechanisms of Action and Cell Death Associated with Clostridium perfringens Toxins. Toxins, 10(5), 212. https://doi.org/10.3390/toxins10050212