Selective Closed-State Nav1.7 Blocker JZTX-34 Exhibits Analgesic Effects against Pain


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Synthesis and Refolding of JZTX-34
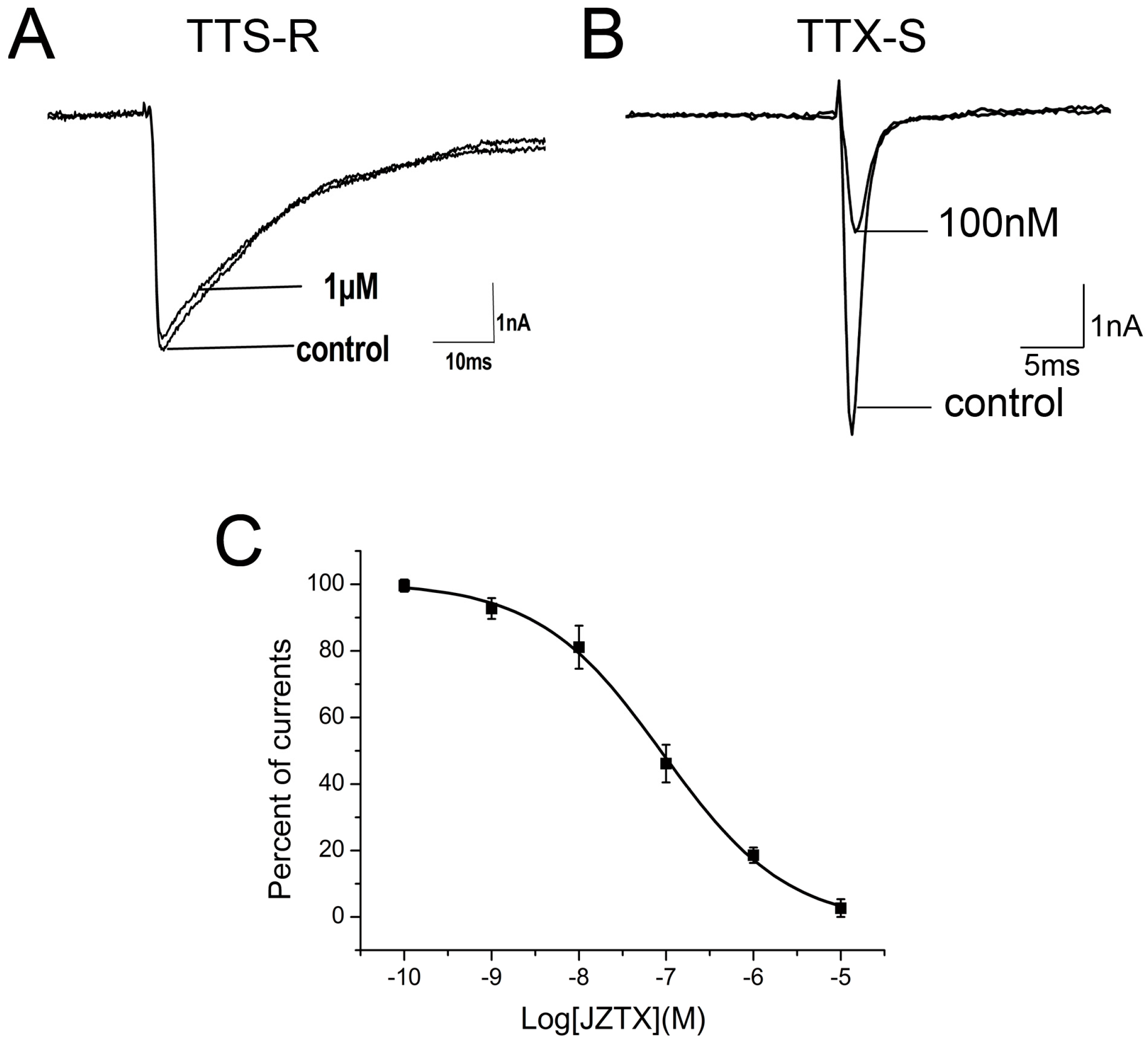
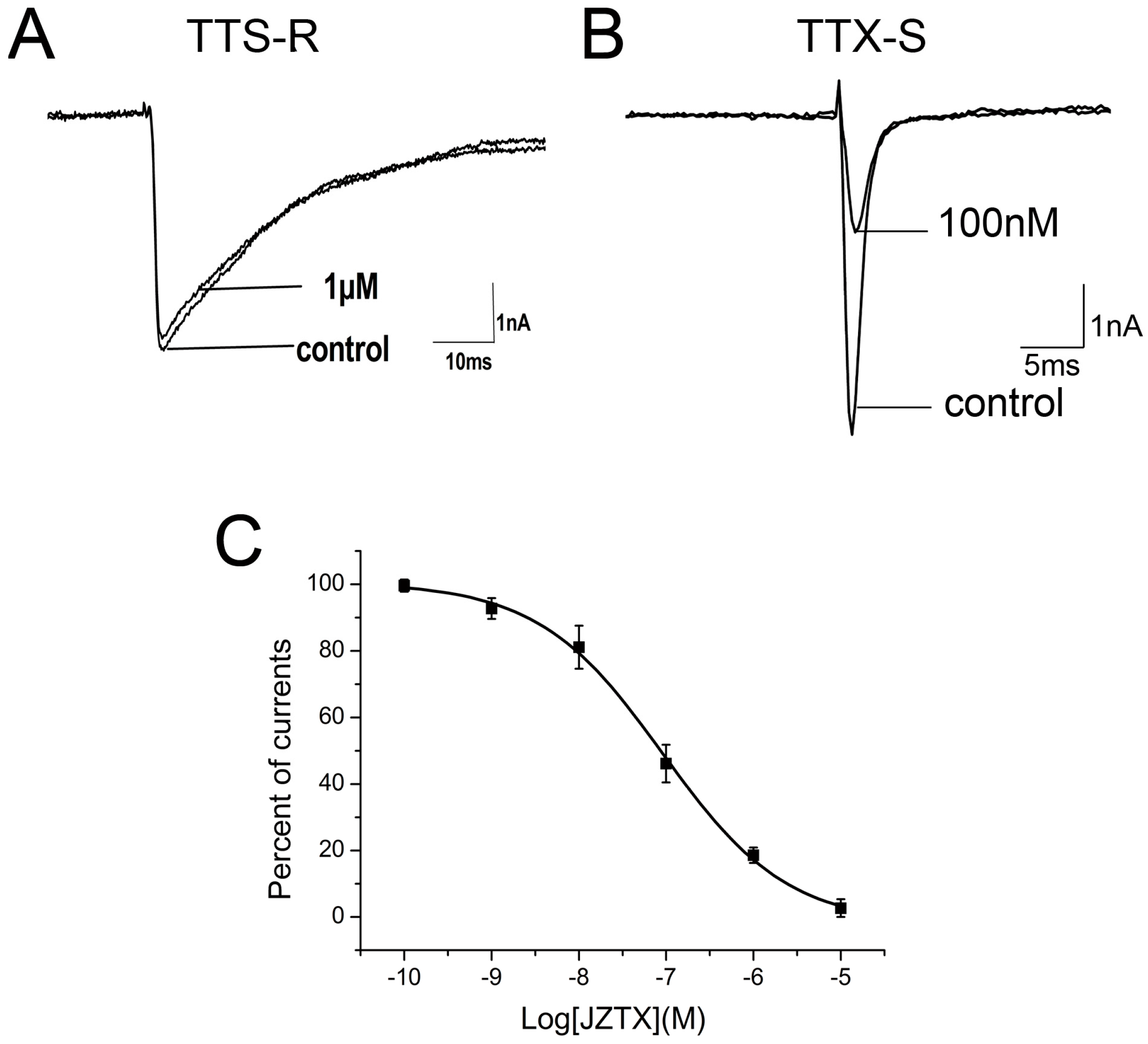
2.2. Inhibition of JZTX-34 on Rat DRG Sodium Channels
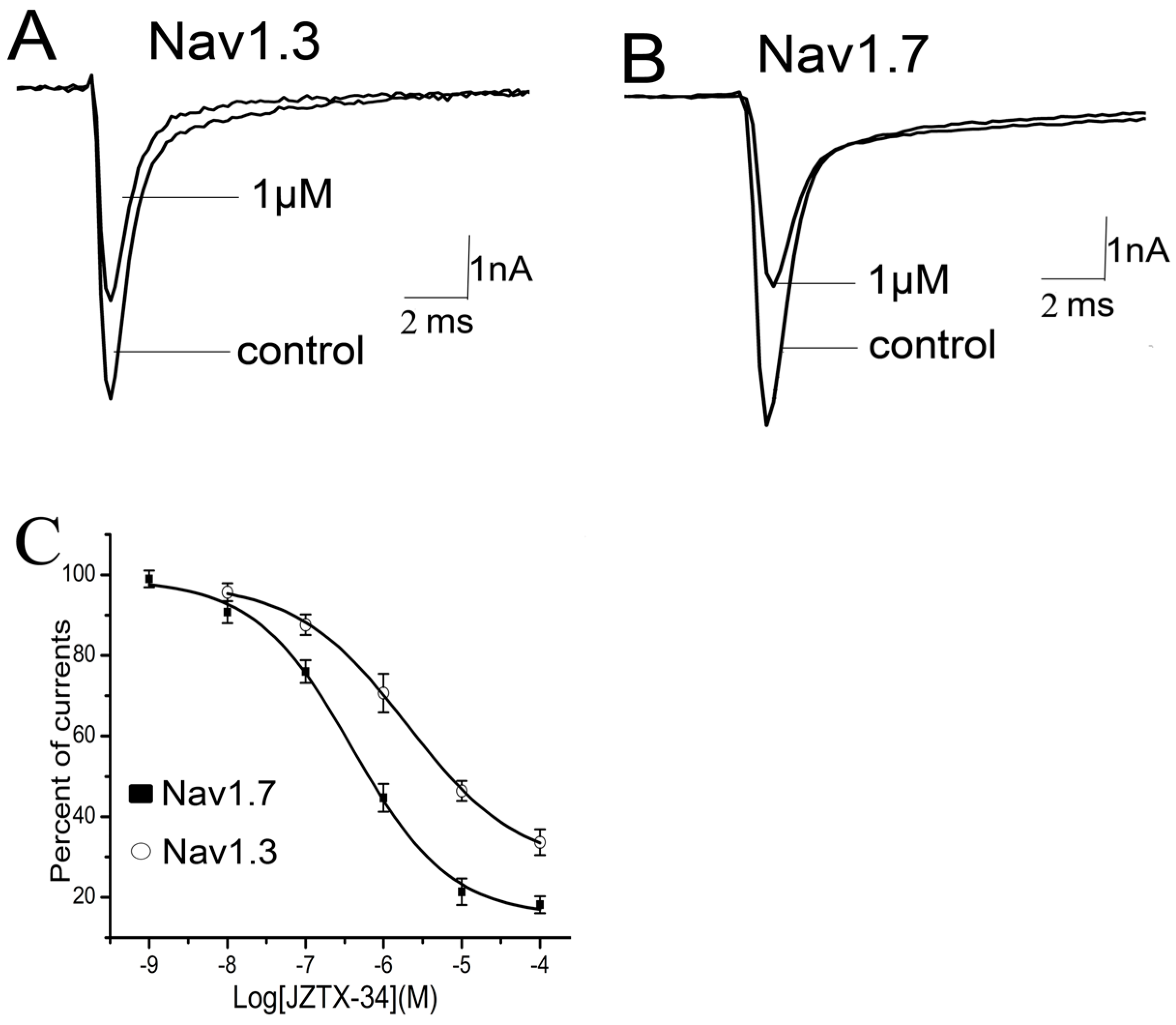
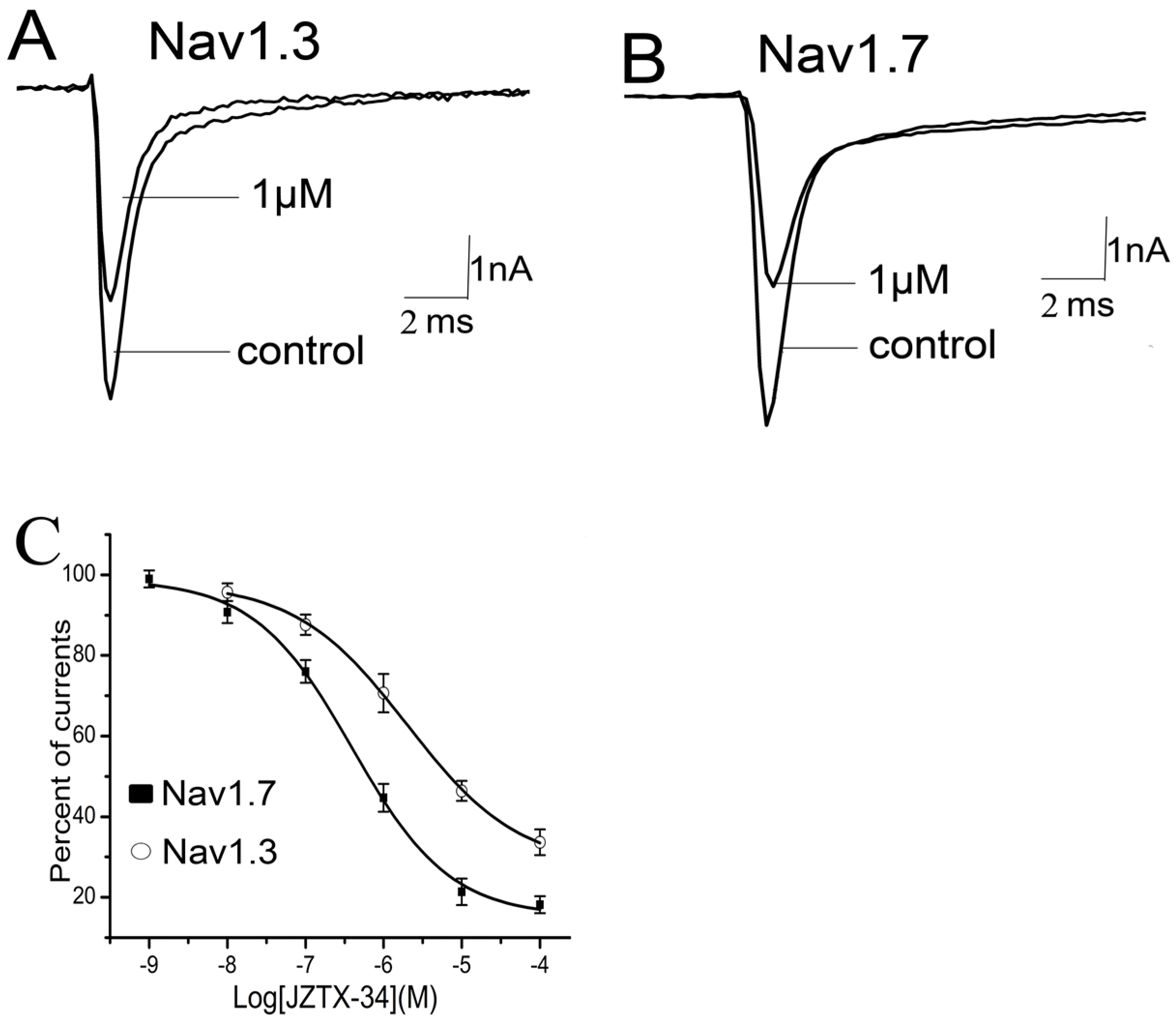
2.3. Subtype Selectivity of JZTX-34 Interaction with Sodium Channels
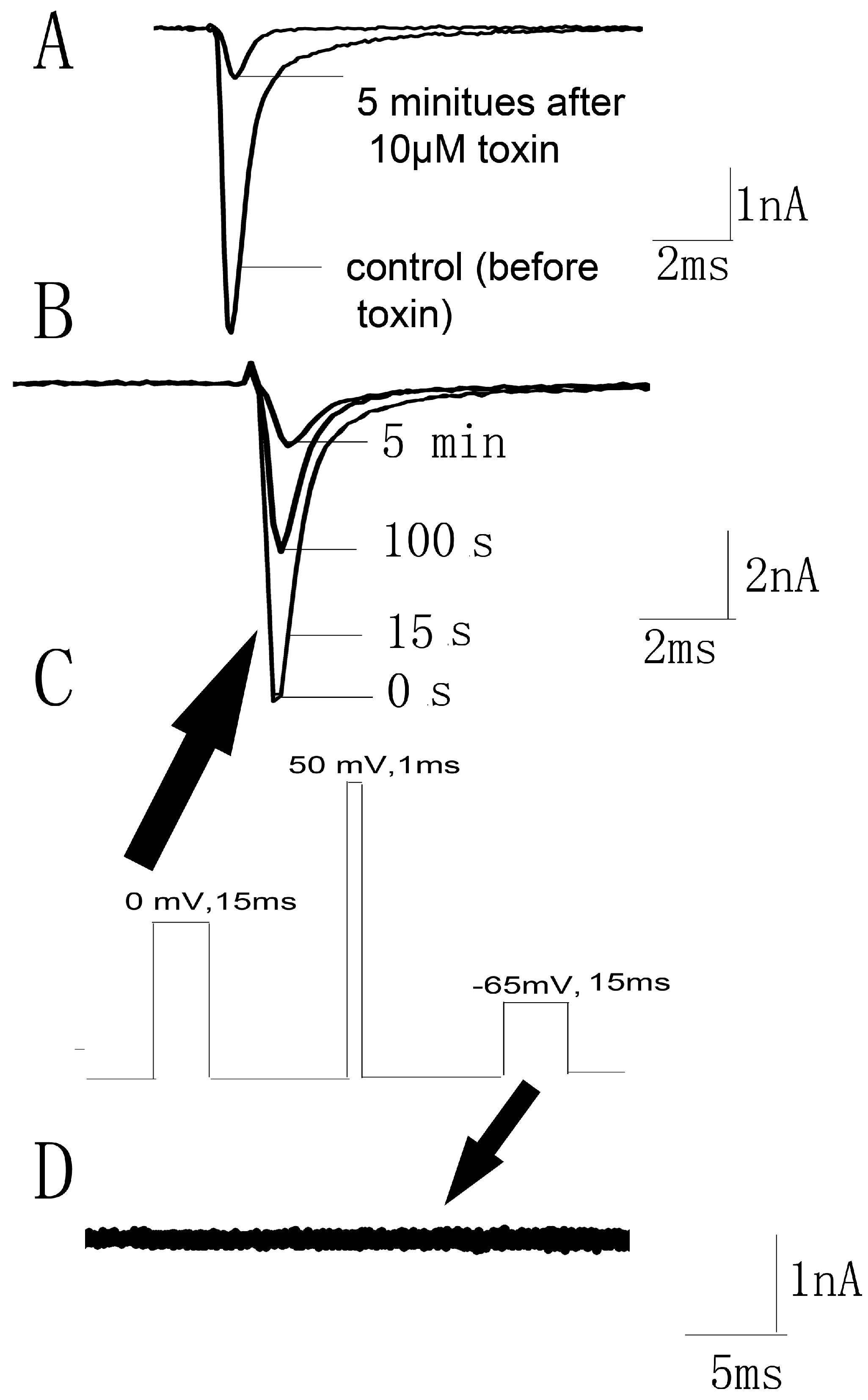
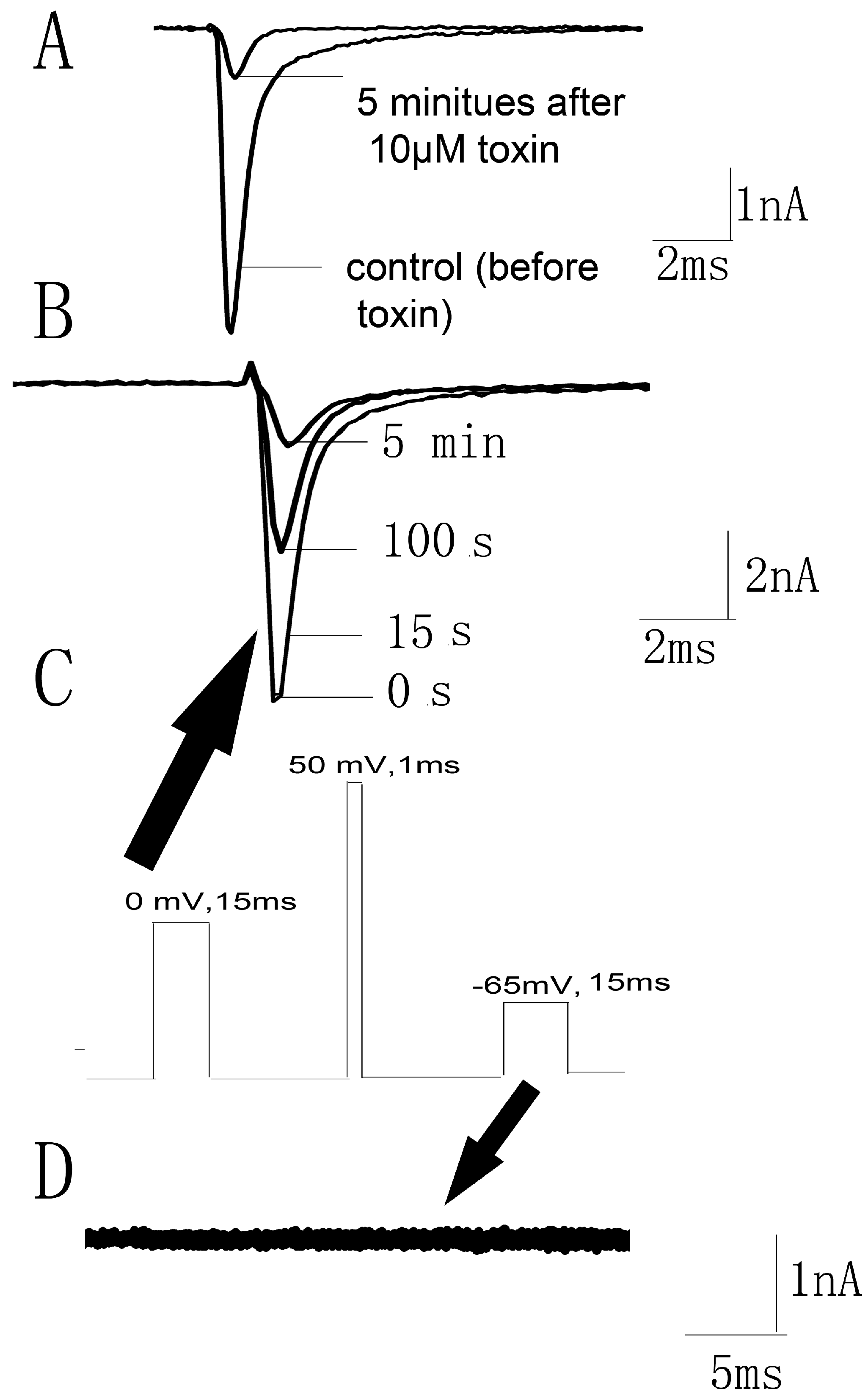
2.4. Kinetics Effects of JZTX-34 on Nav1.7
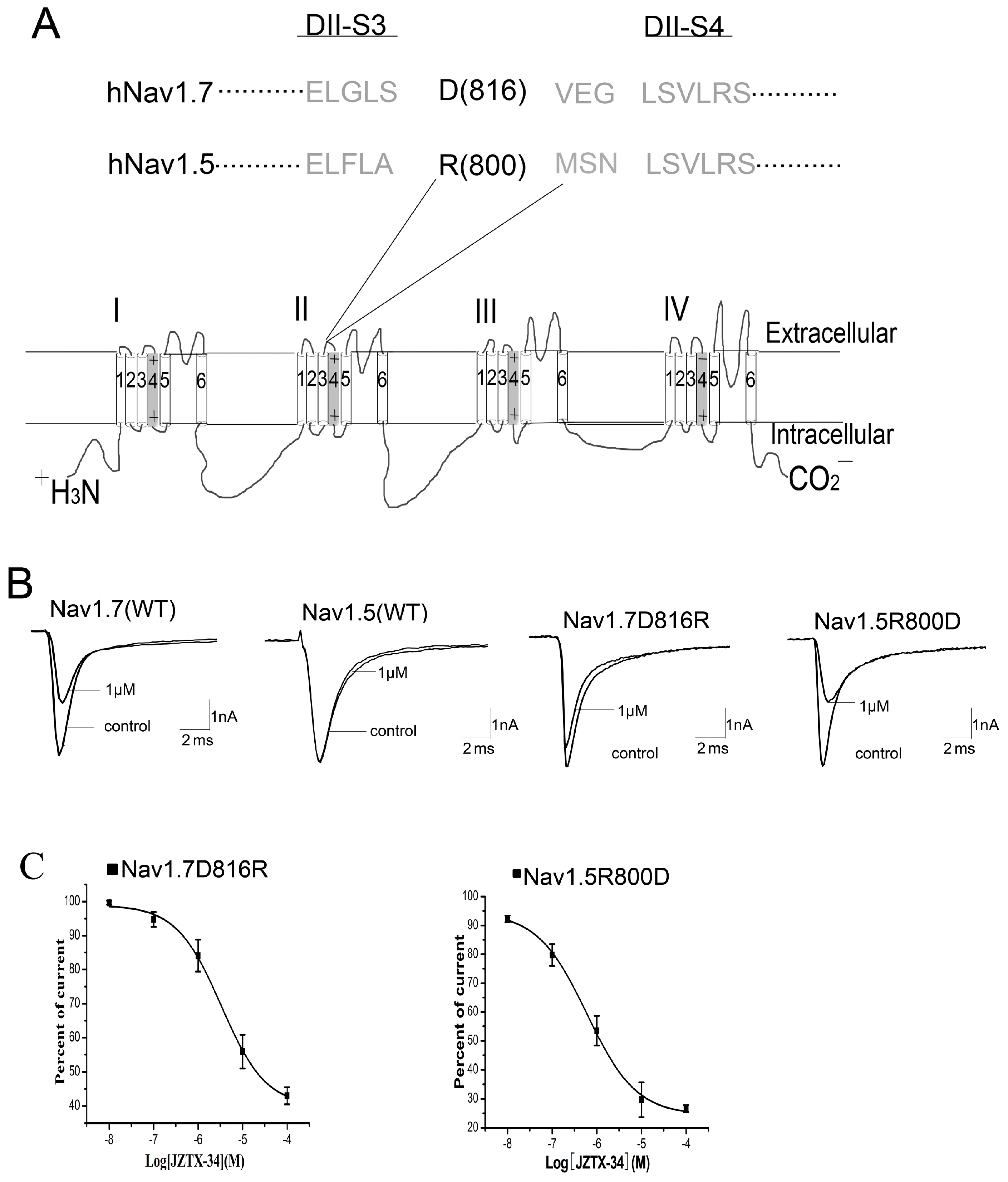
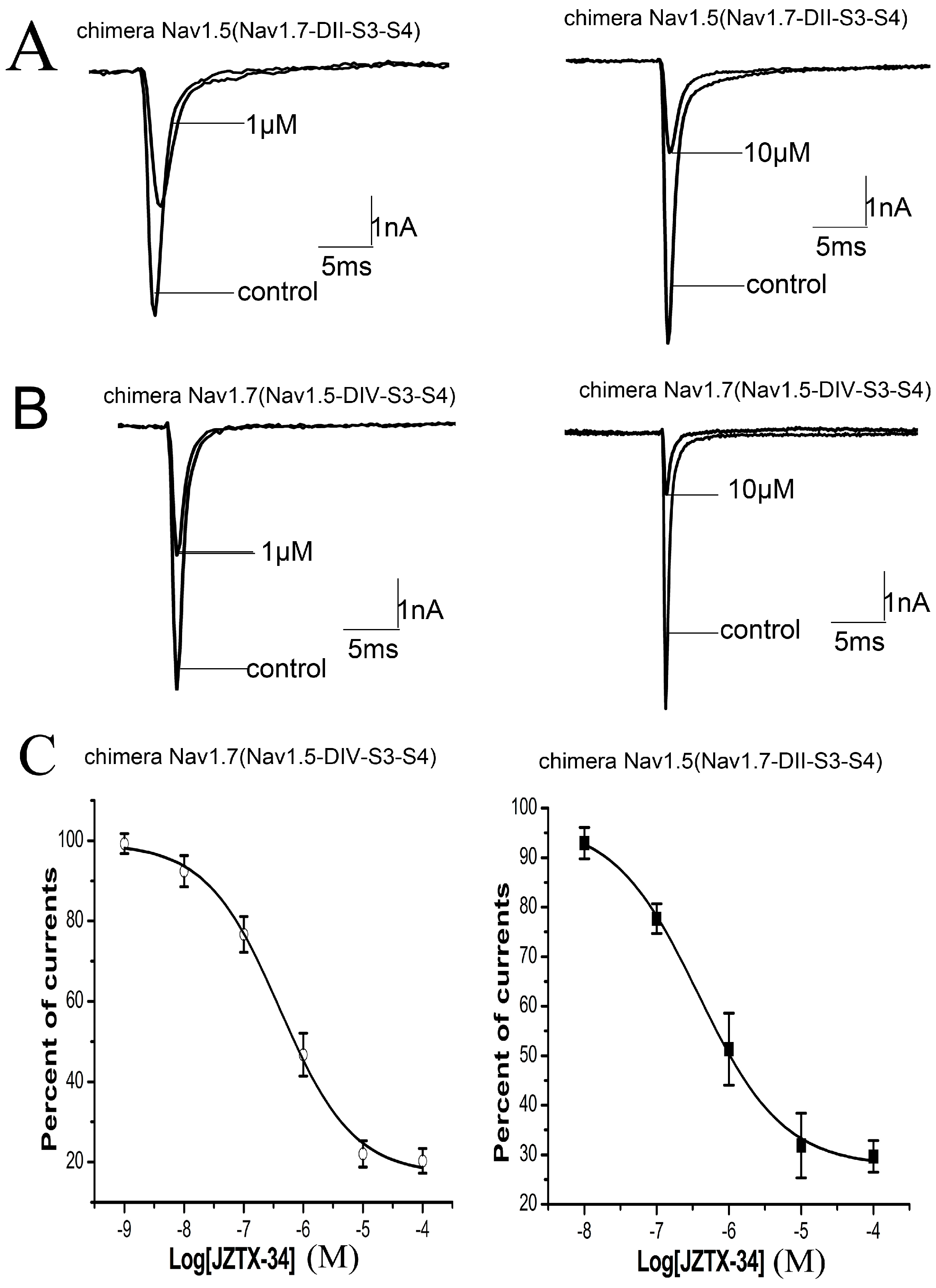
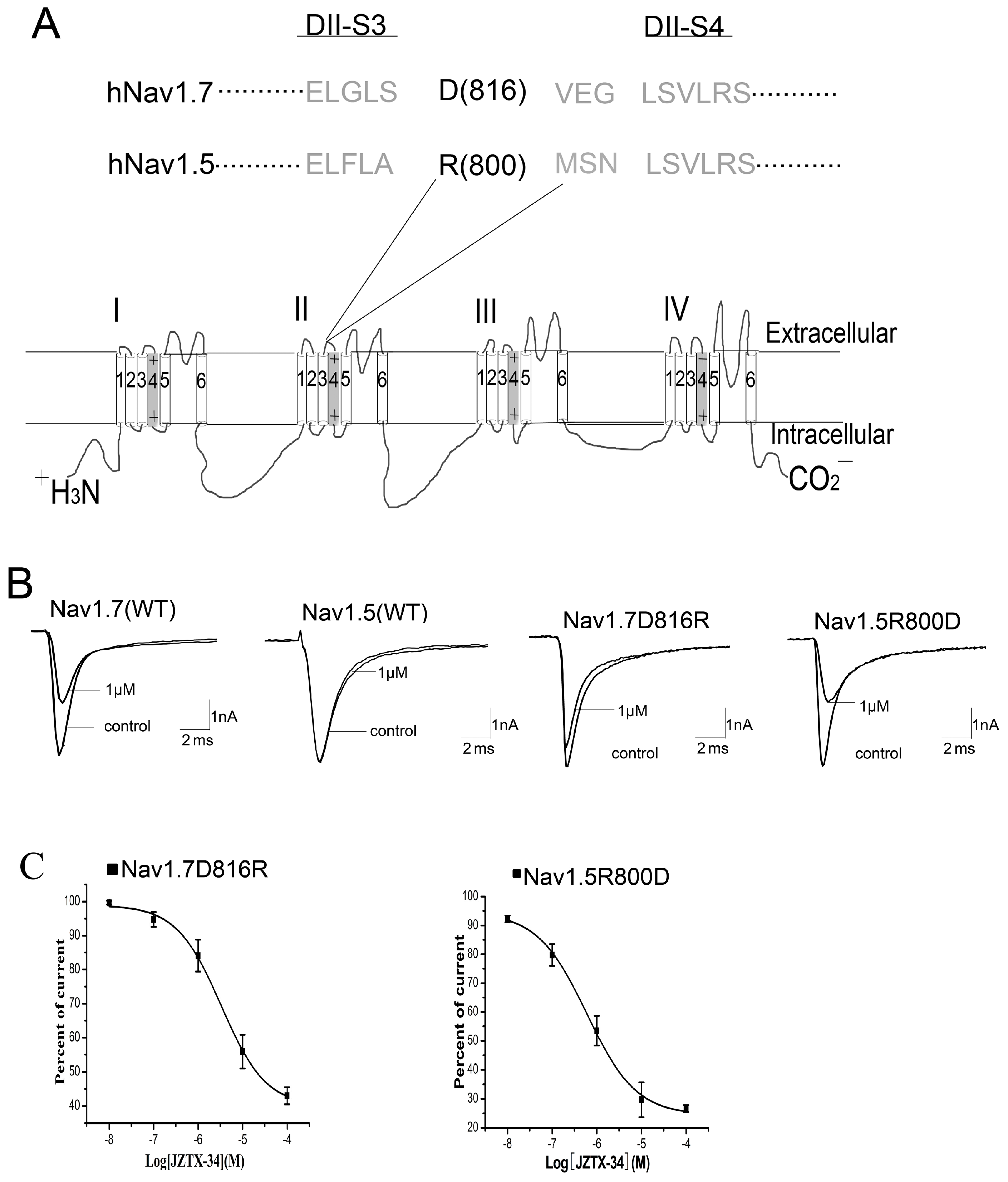
2.5. Binding Site of Toxin on Nav1.7
2.6. JZTX-34 Binds to Voltage Sensor of Domain Ii in A Closed Configuration
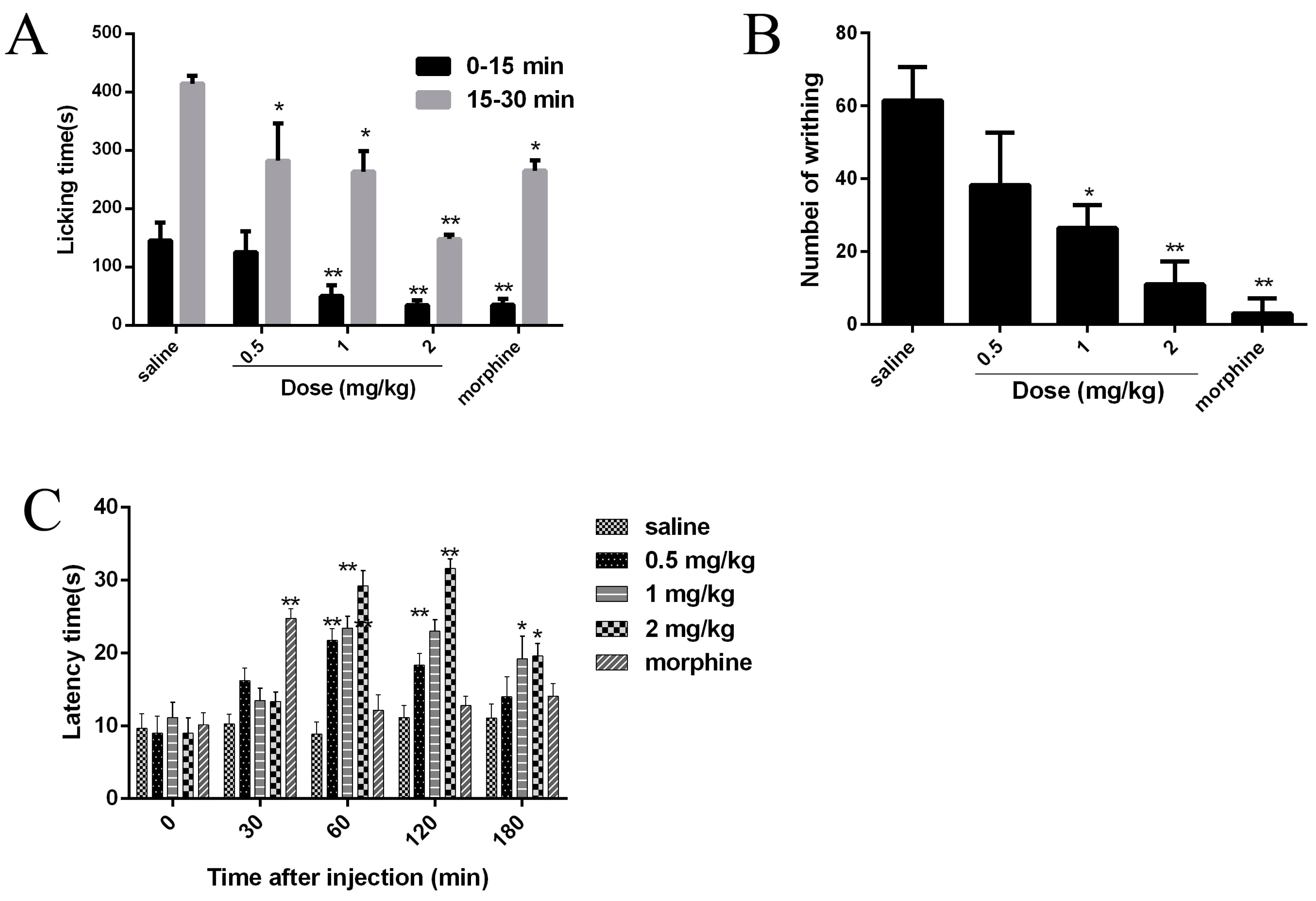
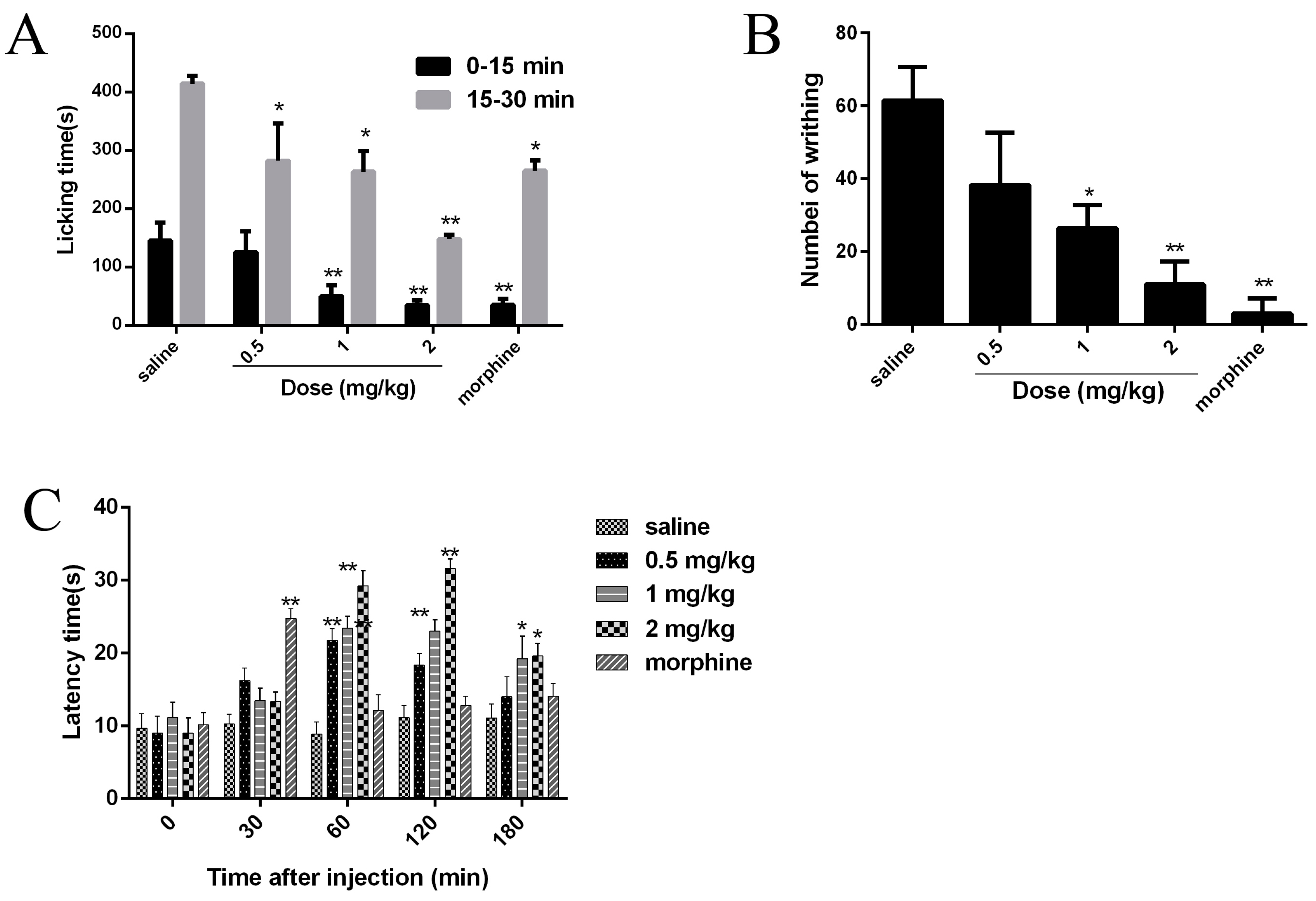
2.7. Analgesic Effects of JZTX-34 on Pain
3. Discussion
4. Experimental Section
4.1. Toxin and Animals
4.2. Peptide Synthesis and Refolding
4.3. Mass Spectrometric Analysis
4.4. Acute Isolation of DRG Neurons and Sodium Channel Recording
4.5. Construction of Chimeric and Mutants of Nav1.5 and Nav1.7
4.6. Transient Expression of Sodium Channel Subtypes
4.7. Whole-Cell Patch-Clamp Recordings
4.8. Paw Licking Induced by Formalin
4.9. Abdominal Writhing Response Caused by Acetic Acid
4.10 Hot Plate
4.11 Data Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflict of Interest
References
- Hargus, N.J.; Patel, M.K. Voltage-gated Na+ channels in neuropathic pain. Expert Opin. Investig. Drugs 2007, 16, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Zhao, J.; Malinowski, S.; Chahine, M.; O’Leary, M.E. Differential expression of sodium channel beta subunits in dorsal root ganglion sensory neurons. J. Biol. Chem. 2012, 287, 15044–15053. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International union of pharmacology. Xlvii. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.L.; Barchi, R.L.; Caldwell, J.H.; Hofmann, F.; Howe, J.R.; Hunter, J.C.; Kallen, R.G.; Mandel, G.; Meisler, M.H.; Netter, Y.B.; et al. Nomenclature of voltage-gated sodium channels. Neuron 2000, 28, 365–368. [Google Scholar] [CrossRef]
- Cregg, R.; Momin, A.; Rugiero, F.; Wood, J.N.; Zhao, J. Pain channelopathies. J. Physiol. 2010, 588, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Meisler, M.H.; Kearney, J.A. Sodium channel mutations in epilepsy and other neurological disorders. J. Clin. Investig. 2005, 115, 2010–2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shen, J.; Li, Z.; Timothy, K.; Vincent, G.M.; Priori, S.G.; Schwartz, P.J.; Keating, M.T. Cardiac sodium channel mutations in patients with long qt syndrome, an inherited cardiac arrhythmia. Hum. Mol. Genet. 1995, 4, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Emery, E.C.; Luiz, A.P.; Wood, J.N. Nav1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin. Ther. Targets 2016, 20, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Y.; Li, S.; Xu, Z.; Li, H.; Ma, L.; Fan, J.; Bu, D.; Liu, B.; Fan, Z.; et al. Mutations in scn9a, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J. Med. Genet. 2004, 41, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Cestele, S.; Catterall, W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar] [CrossRef]
- King, G.F.; Vetter, I. No gain, no pain: Nav1.7 as an analgesic target. ACS Chem. Neurosci. 2014, 5, 749–751. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Cohen, C.J.; Dehnhardt, C.M. Inhibitors of voltage-gated sodium channel nav1.7: Patent applications since 2010. Pharm. Pat. Anal. 2014, 3, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewska, J.M.; Palmer, J.; Morisset, V.; Giblin, G.M.; Obermann, M.; Ettlin, D.A.; Cruccu, G.; Bendtsen, L.; Estacion, M.; Derjean, D.; et al. Safety and efficacy of a nav1.7 selective sodium channel blocker in patients with trigeminal neuralgia: A double-blind, placebo-controlled, randomised withdrawal phase 2a trial. Lancet Neurol. 2017, 16, 291–300. [Google Scholar] [CrossRef]
- Xiao, Y.; Blumenthal, K.; Jackson, J.O., 2nd.; Liang, S.; Cummins, T.R. The tarantula toxins protx-ii and huwentoxin-iv differentially interact with human nav1.7 voltage sensors to inhibit channel activation and inactivation. Mol. Pharmacol. 2010, 78, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Jackson, J.O., 2nd.; Liang, S.; Cummins, T.R. Common molecular determinants of tarantula huwentoxin-iv inhibition of Na+ channel voltage sensors in domains ii and iv. J. Biol. Chem. 2011, 286, 27301–27310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Huang, P.F.; Meng, E.; Li, W.Y.; Zhou, L.; Zhu, L.Y.; Wu, L.; Li, M.J.; Liang, S.P.; Zhang, D.Y. An efficient strategy for heterologous expression and purification of active peptide hainantoxin-iv. PLoS ONE 2015, 10, e0117099. [Google Scholar] [CrossRef] [PubMed]
- Deuis, J.R.; Wingerd, J.S.; Winter, Z.; Durek, T.; Dekan, Z.; Sousa, S.R.; Zimmermann, K.; Hoffmann, T.; Weidner, C.; Nassar, M.A.; et al. Analgesic effects of gptx-1, pf-04856264 and cnv1014802 in a mouse model of nav1.7-mediated pain. Toxins 2016, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, Y.; Rong, M.; Zhao, L.; Jiang, L.; Zhang, D.; Wang, M.; Xiao, Y.; Liang, S. Expression and characterization of jingzhaotoxin-34, a novel neurotoxin from the venom of the tarantula chilobrachys jingzhao. Peptides 2009, 30, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Peigneur, S.; Gao, B.; Lu, X.; Cao, C.; Tytgat, J. Evolutionary diversification of mesobuthus alpha-scorpion toxins affecting sodium channels. Mol. Cell. Proteom. MCP 2012, 11, M111–012054. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Yarov-Yarovoy, V.; Scheuer, T.; Karbat, I.; Cohen, L.; Gordon, D.; Gurevitz, M.; Catterall, W.A. Structure-function map of the receptor site for beta-scorpion toxins in domain ii of voltage-gated sodium channels. J. Biol. Chem. 2011, 286, 33641–33651. [Google Scholar] [CrossRef] [PubMed]
- Cestele, S.; Qu, Y.; Rogers, J.C.; Rochat, H.; Scheuer, T.; Catterall, W.A. Voltage sensor-trapping: Enhanced activation of sodium channels by beta-scorpion toxin bound to the s3-s4 loop in domain ii. Neuron 1998, 21, 919–931. [Google Scholar] [CrossRef]
- Sollod, B.L.; Wilson, D.; Zhaxybayeva, O.; Gogarten, J.P.; Drinkwater, R.; King, G.F. Were arachnids the first to use combinatorial peptide libraries? Peptides 2005, 26, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Lewis, R.J. Use of venom peptides to probe ion channel structure and function. J. Biol. Chem. 2010, 285, 13315–13320. [Google Scholar] [CrossRef] [PubMed]
- Rong, M.; Chen, J.; Tao, H.; Wu, Y.; Jiang, P.; Lu, M.; Su, H.; Chi, Y.; Cai, T.; Zhao, L.; et al. Molecular basis of the tarantula toxin jingzhaotoxin-iii (beta-trtx-cj1alpha) interacting with voltage sensors in sodium channel subtype nav1.5. FASEB J. 2011, 25, 3177–3185. [Google Scholar] [CrossRef] [PubMed]
- Schmalhofer, W.A.; Calhoun, J.; Burrows, R.; Bailey, T.; Kohler, M.G.; Weinglass, A.B.; Kaczorowski, G.J.; Garcia, M.L.; Koltzenburg, M.; Priest, B.T. Protx-ii, a selective inhibitor of nav1.7 sodium channels, blocks action potential propagation in nociceptors. Mol. Pharmacol. 2008, 74, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Bingham, J.P.; Zhu, W.; Moczydlowski, E.; Liang, S.; Cummins, T.R. Tarantula huwentoxin-iv inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain ii voltage sensor in the closed configuration. J. Biol. Chem. 2008, 283, 27300–27313. [Google Scholar] [CrossRef] [PubMed]
- Flinspach, M.; Xu, Q.; Piekarz, A.D.; Fellows, R.; Hagan, R.; Gibbs, A.; Liu, Y.; Neff, R.A.; Freedman, J.; Eckert, W.A.; et al. Insensitivity to pain induced by a potent selective closed-state nav1.7 inhibitor. Sci. Rep. 2017, 7, 39662. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, Z.; Tang, D.; Xun, X.; Liu, L.; Li, X.; Nie, D.; Xiang, Y.; Yi, J.; Yi, J. Analgesic effects of huwentoxin-iv on animal models of inflammatory and neuropathic pain. Protein Pept. Lett. 2014, 21, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.K.; Long, J.; Zou, A.; Ligutti, J.; Andrews, K.L.; Poppe, L.; Biswas, K.; Moyer, B.D.; McDonough, S.I.; Miranda, L.P. Single residue substitutions that confer voltage-gated sodium ion channel subtype selectivity in the nav1.7 inhibitory peptide gptx-1. J. Med. Chem. 2016, 59, 2704–2717. [Google Scholar] [CrossRef] [PubMed]
- Shcherbatko, A.; Rossi, A.; Foletti, D.; Zhu, G.; Bogin, O.; Galindo Casas, M.; Rickert, M.; Hasa-Moreno, A.; Bartsevich, V.; Crameri, A.; et al. Engineering highly potent and selective microproteins against nav1.7 sodium channel for treatment of pain. J. Biol. Chem. 2016, 291, 13974–13986. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Luo, X.; Kuang, F.; Deng, M.; Wang, M.; Zeng, X.; Liang, S. Synthesis and characterization of huwentoxin-iv, a neurotoxin inhibiting central neuronal sodium channels. Toxicon. 2008, 51, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Liang, S. Inhibition of neuronal tetrodotoxin-sensitive na+ channels by two spider toxins: Hainantoxin-iii and hainantoxin-iv. Eur. J. Pharmacol. 2003, 477, 1–7. [Google Scholar] [CrossRef]
- Owoyele, V.B.; Adediji, J.O.; Soladoye, A.O. Anti-inflammatory activity of aqueous leaf extract of chromolaena odorata. Inflammopharmacology 2005, 13, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.A.; Arruda, A.; Silva, M.A.; Cardoso, C.A.; Vieira Mdo, C.; Kassuya, C.A.; Arena, A.C. Anti-inflammatory effects and acute toxicity of hydroethanolic extract of jacaranda decurrens roots in adult male rats. J. Ethnopharmacol. 2012, 144, 802–805. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, X.; Li, P.; Chen, B.; Huang, J.; Lai, R.; Liu, J.; Rong, M. Selective Closed-State Nav1.7 Blocker JZTX-34 Exhibits Analgesic Effects against Pain. Toxins 2018, 10, 64. https://doi.org/10.3390/toxins10020064
Zeng X, Li P, Chen B, Huang J, Lai R, Liu J, Rong M. Selective Closed-State Nav1.7 Blocker JZTX-34 Exhibits Analgesic Effects against Pain. Toxins. 2018; 10(2):64. https://doi.org/10.3390/toxins10020064
Chicago/Turabian StyleZeng, Xiongzhi, Pengpeng Li, Bo Chen, Juan Huang, Ren Lai, Jingze Liu, and Mingqiang Rong. 2018. "Selective Closed-State Nav1.7 Blocker JZTX-34 Exhibits Analgesic Effects against Pain" Toxins 10, no. 2: 64. https://doi.org/10.3390/toxins10020064
APA StyleZeng, X., Li, P., Chen, B., Huang, J., Lai, R., Liu, J., & Rong, M. (2018). Selective Closed-State Nav1.7 Blocker JZTX-34 Exhibits Analgesic Effects against Pain. Toxins, 10(2), 64. https://doi.org/10.3390/toxins10020064