Activation of Aflatoxin Biosynthesis Alleviates Total ROS in Aspergillus parasiticus

Abstract
:1. Introduction
2. Results
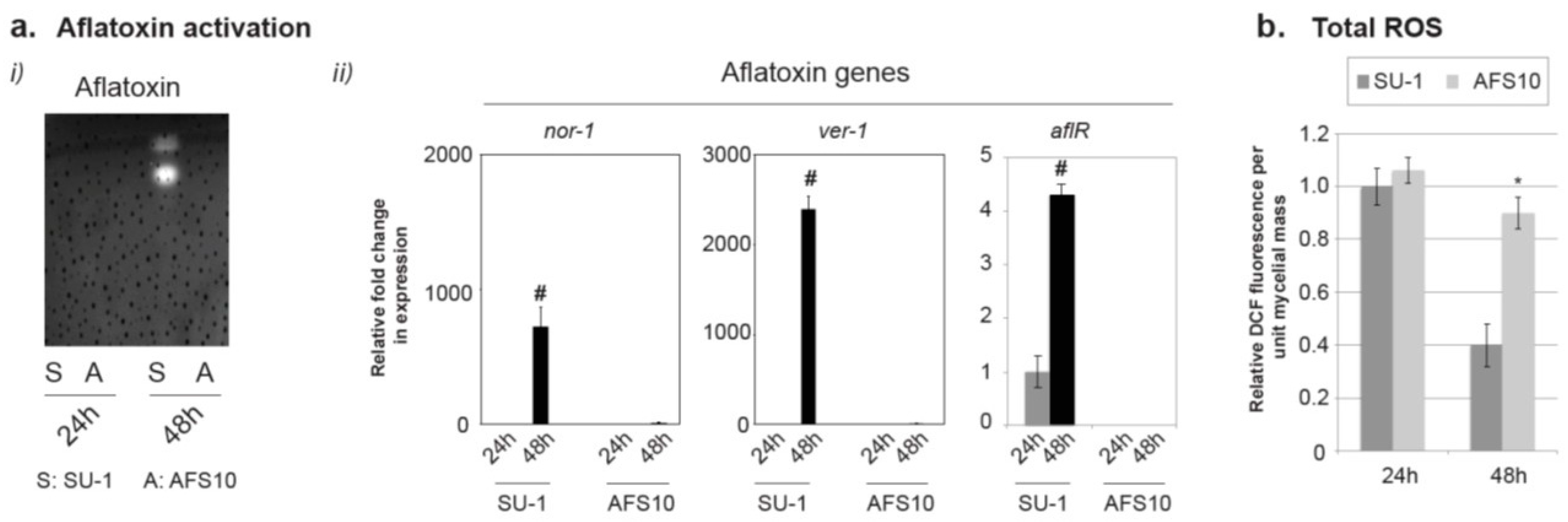
2.1. SU-1 Demonstrates a Significantly Larger Decrease in Total ROS Compared to AFS10 between 24 h and 48 h
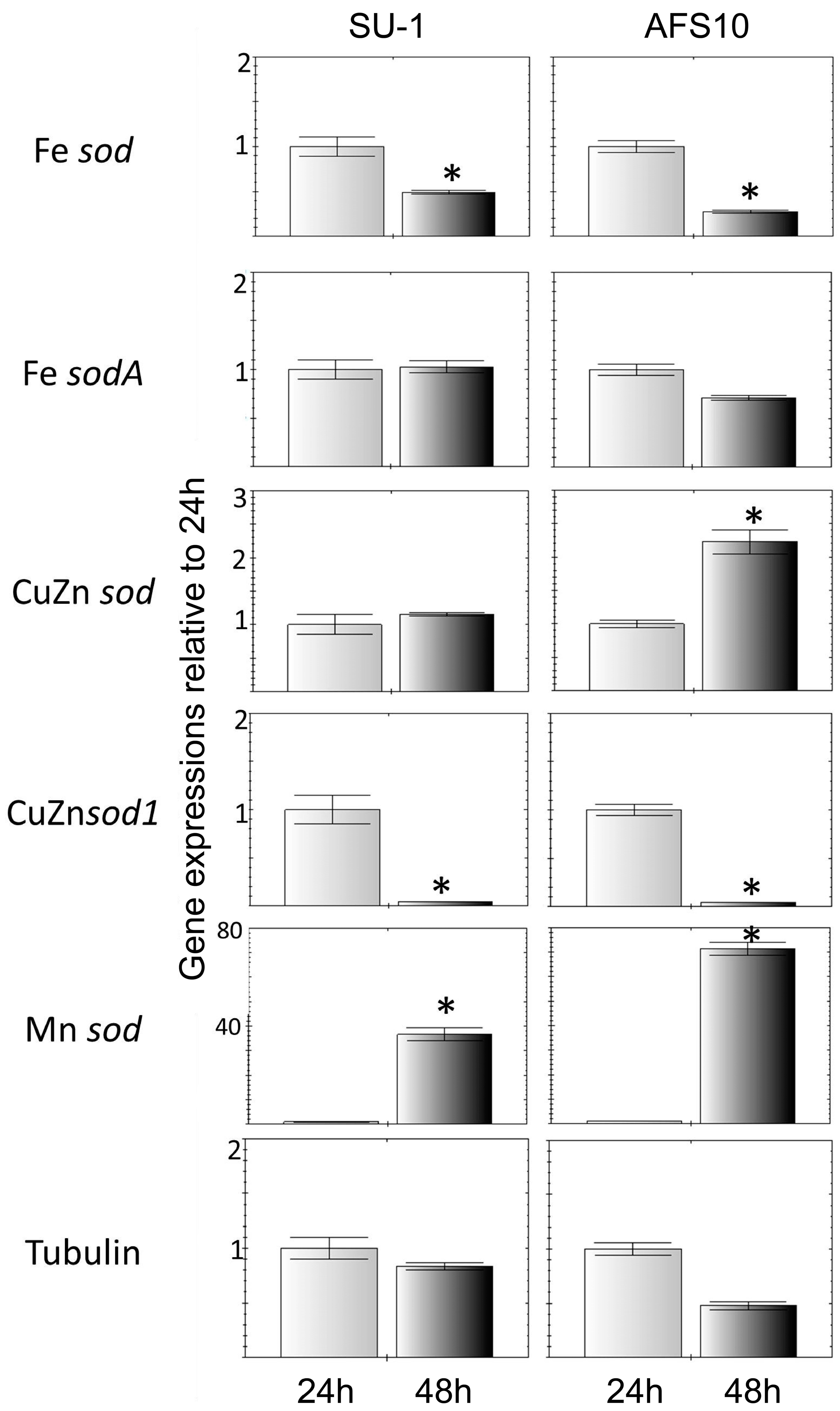
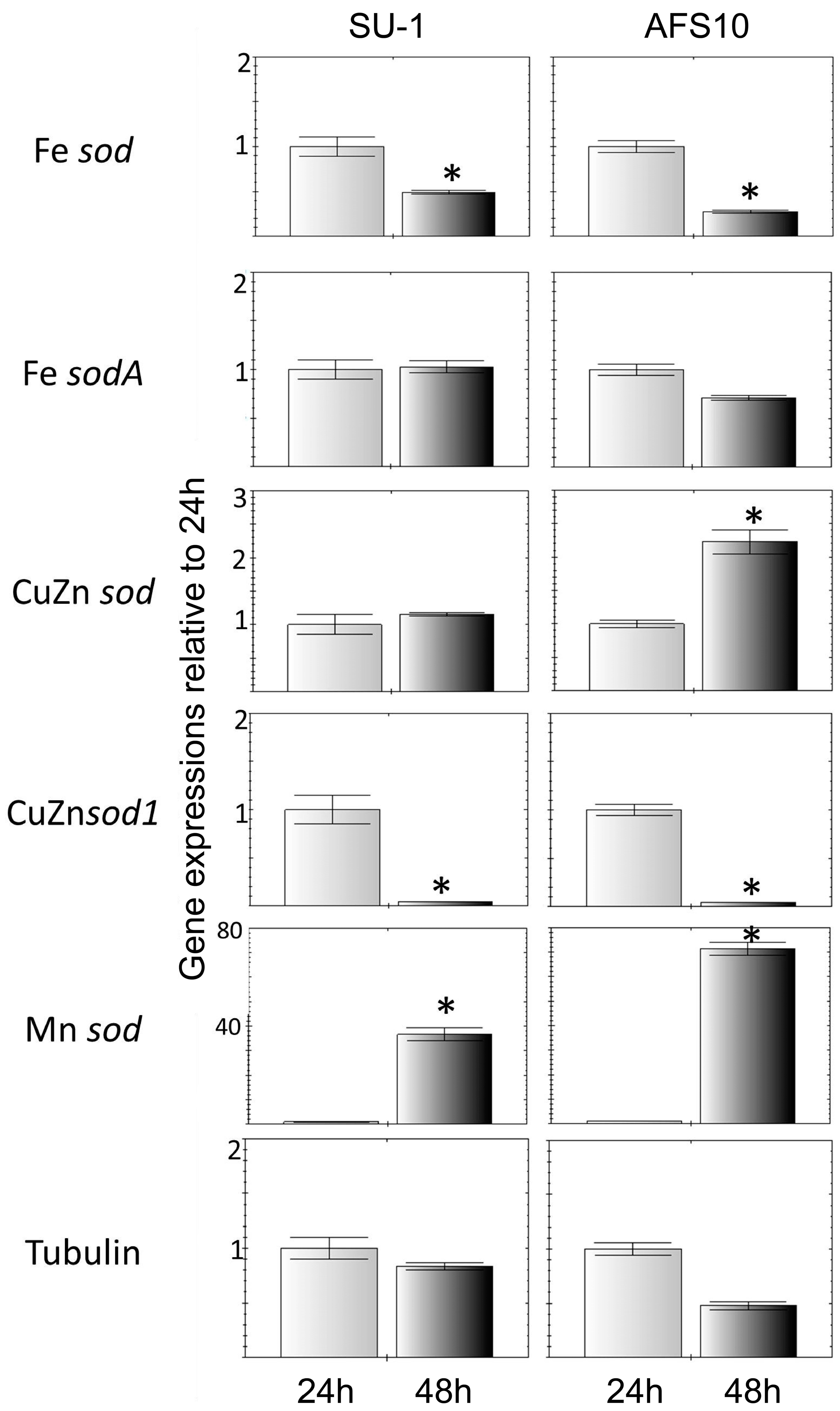
2.2. Higher Total ROS in AFS10 Compared to SU-1 at 48 h Associates with Significant Differences in SOD Gene Expression
2.2.1. Bioinformatics Analyses of SOD Genes
2.2.2. Expression Profiles of SOD Genes
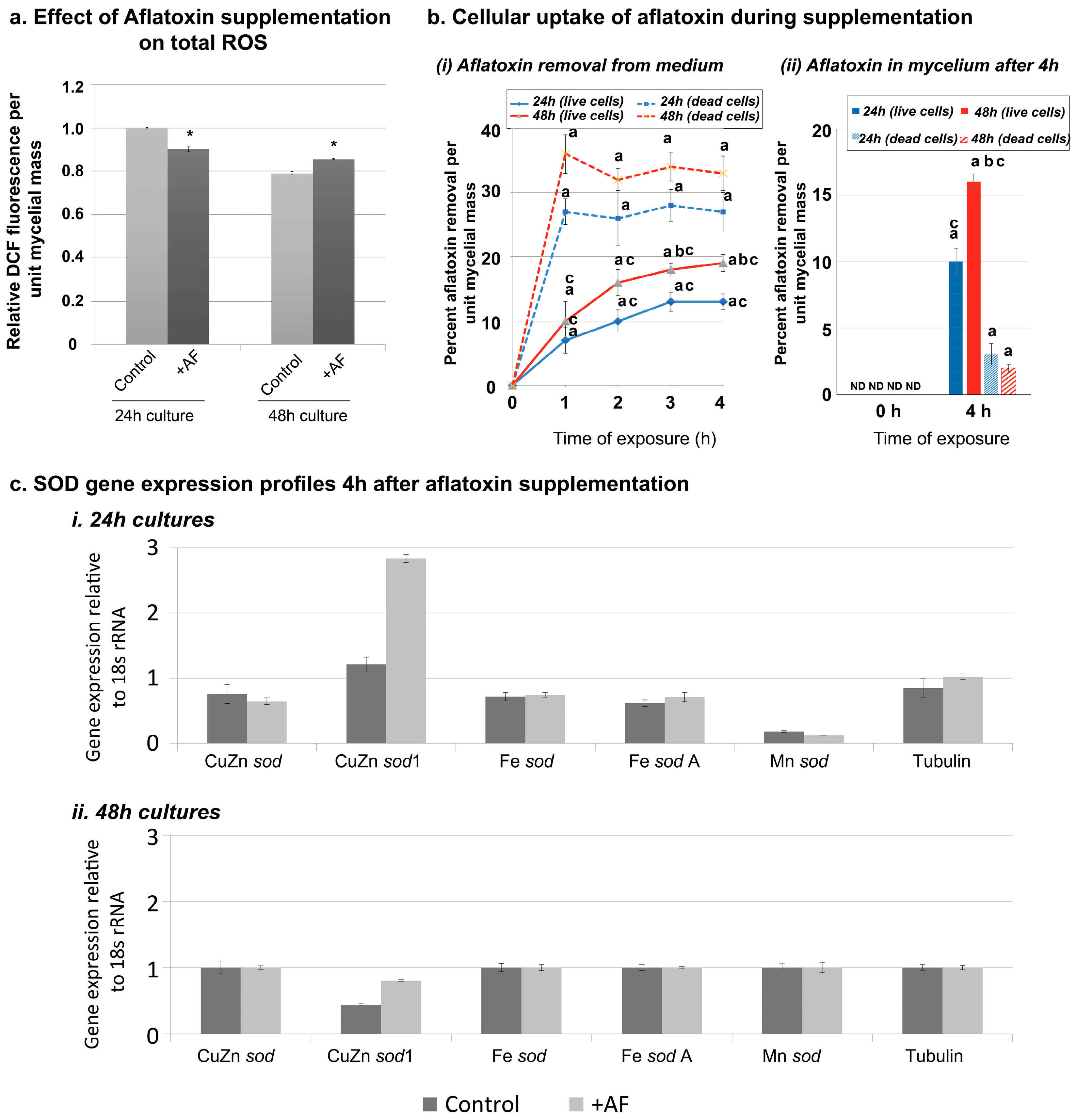
2.3. Aflatoxin Supplementation to AFS10 Growth Medium Changes Total ROS Output without Changing the SOD Transcript Levels
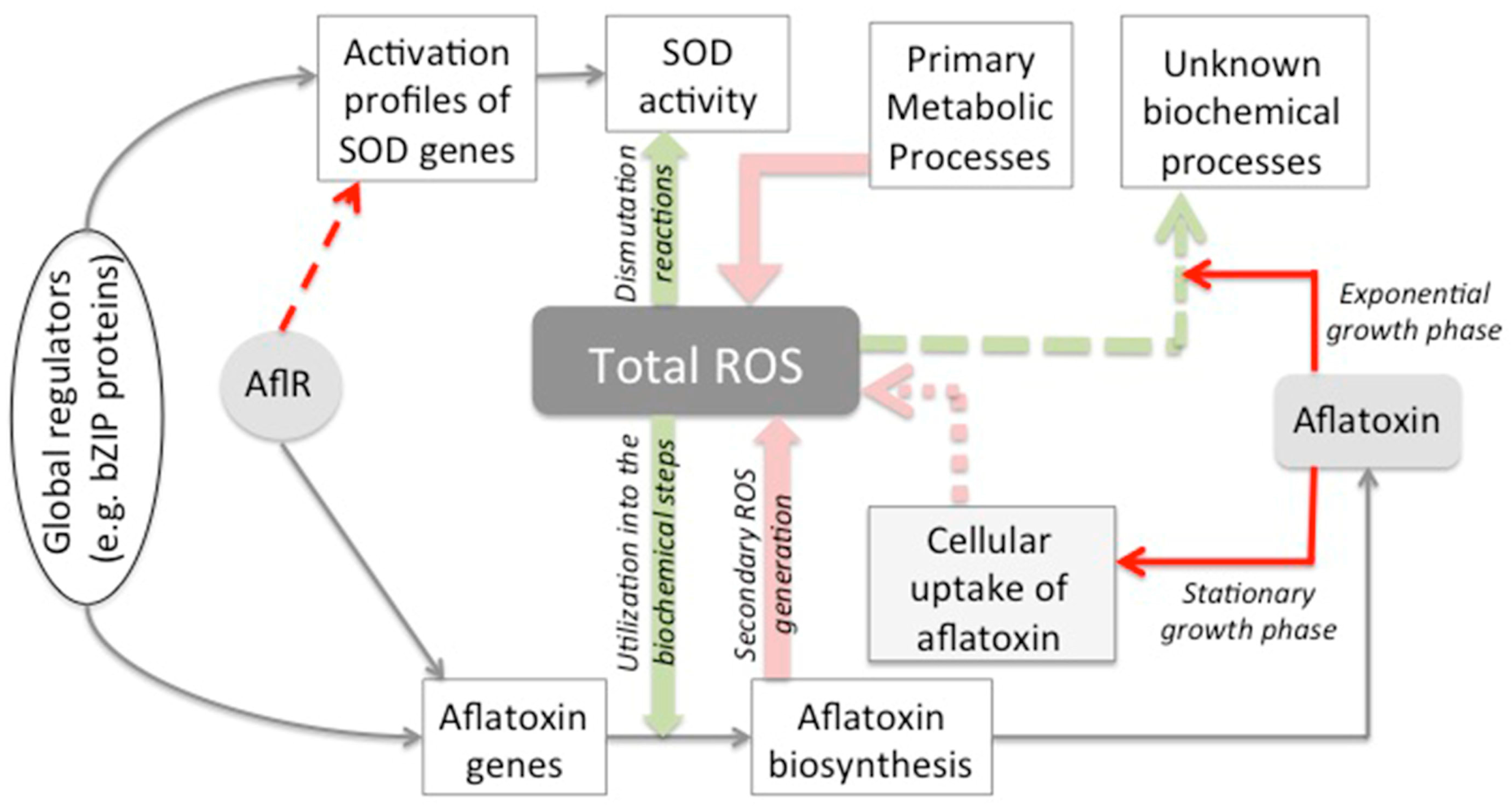
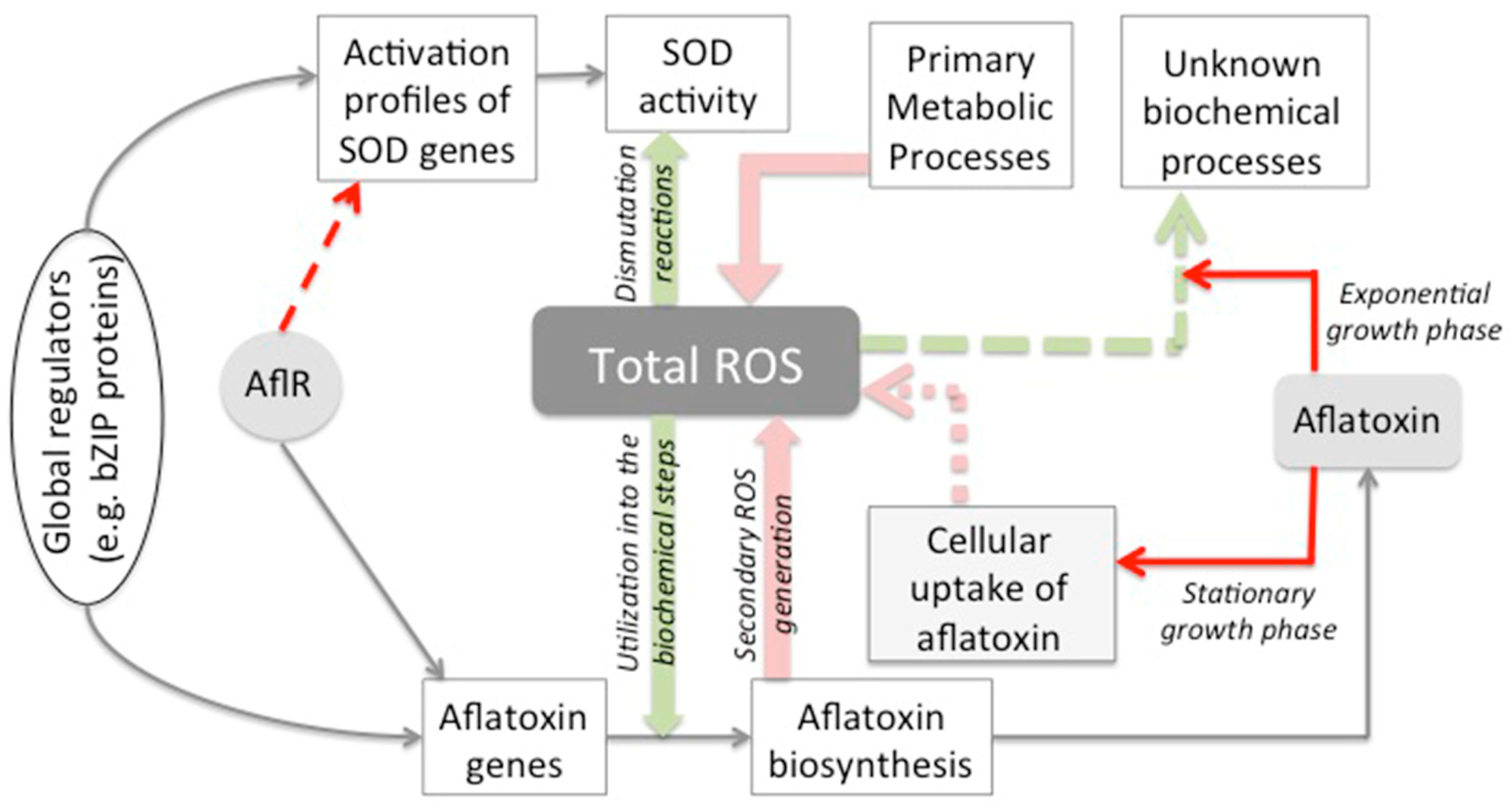
3. Discussion
4. Materials and Methods
4.1. Strains, Media, and Growth Conditions
4.2. Quantification of ROS
4.3. Identification of Superoxide Dismutase Genes
4.4. RNA Extraction, Purification and cDNA Synthesis
4.5. Quantitative PCR Assays
4.6. Aflatoxin Supplementation Experiments
4.7. Aflatoxin Quantification
4.8. Statistical Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Keller, N.P.; Turner, G.; Bennett, J.W. Fungal secondary metabolism—From biochemistry to genomics. Nat. Rev. Microbiol. 2005, 3, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Magnussen, A.; Parsi, M.A. Aflatoxins, hepatocellular carcinoma and public health. World J. Gastroenterol. 2013, 19, 1508–1512. [Google Scholar] [CrossRef] [PubMed]
- Roze, L.V.; Chanda, A.; Wee, J.; Awad, D.; Linz, J.E. Stress-related transcription factor AtfB integrates secondary metabolism with oxidative stress response in aspergilli. J. Biol. Chem. 2011, 286, 35137–35148. [Google Scholar] [CrossRef] [PubMed]
- Brakhage, A.A. Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 2013, 11, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Keller, N.P. Transcriptional regulatory elements in fungal secondary metabolism. J. Microbiol. 2011, 49, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chang, P.-K.; Ehrlich, K.C.; Cary, J.W.; Bhatnagar, D.; Cleveland, T.E.; Payne, G.A.; Linz, J.E.; Woloshuk, C.P.; Bennett, J.W. Clustered Pathway Genes in Aflatoxin Biosynthesis. Appl. Environ. Microbiol. 2004, 70, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Chanda, A.; Roze, L.V.; Kang, S.; Artymovich, K.A.; Hicks, G.R.; Raikhel, N.V.; Calvo, A.M.; Linz, J.E. A key role for vesicles in fungal secondary metabolism. Proc. Natl. Acad. Sci. USA 2009, 106, 19533–19538. [Google Scholar] [CrossRef] [PubMed]
- Kale, S.P.; Milde, L.; Trapp, M.K.; Frisvad, J.C.; Keller, N.P.; Bok, J.W. Requirement of LaeA for secondary metabolism and sclerotial production in Aspergillus flavus. Fungal Genet. Biol. 2008, 45, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Bayram, O.; Krappmann, S.; Ni, M.; Bok, J.W.; Helmstaedt, K.; Valerius, O.; Braus-Stromeyer, S.; Kwon, N.J.; Keller, N.P.; Yu, J.H.; et al. VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science 2008, 320, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Calvo, A.M.; Bok, J.; Brooks, W.; Keller, N.P. veA is required for toxin and sclerotial production in Aspergillus parasiticus. Appl. Environ. Microbiol. 2004, 70, 4733–4739. [Google Scholar] [CrossRef] [PubMed]
- Calvo, A.M. The VeA regulatory system and its role in morphological and chemical development in fungi. Fungal Genet. Biol. 2008, 45, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Duran, R.M.; Cary, J.W.; Calvo, A.M. Production of cyclopiazonic acid, aflatrem, and aflatoxin by Aspergillus flavus is regulated by veA, a gene necessary for sclerotial formation. Appl. Microbiol. Biotechnol. 2007, 73, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Cary, J.W.; Ehrlich, K.C.; Kale, S.P.; Calvo, A.M.; Bhatnagar, D.; Cleveland, T.E. Regulatory elements in aflatoxin biosynthesis. Mycotoxin Res. 2006, 22, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Scharf, D.H.; Heinekamp, T.; Remme, N.; Hortschansky, P.; Brakhage, A.A.; Hertweck, C. Biosynthesis and function of gliotoxin in Aspergillus fumigatus. Appl. Microbiol. Biotechnol. 2012, 93, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Bok, J.W.; Chung, D.; Balajee, S.A.; Marr, K.A.; Andes, D.; Nielsen, K.F.; Frisvad, J.C.; Kirby, K.A.; Keller, N.P. GliZ, a Transcriptional Regulator of Gliotoxin Biosynthesis, Contributes to Aspergillus fumigatus Virulence. Infect. Immun. 2006, 74, 6761–6768. [Google Scholar] [CrossRef] [PubMed]
- Chanda, A.; Roze, L.V.; Linz, J.E. A possible role for exocytosis in aflatoxin export in Aspergillus parasiticus. Eukaryot. Cell 2010, 9, 1724–1727. [Google Scholar] [CrossRef] [PubMed]
- Roze, L.V.; Chanda, A.; Linz, J.E. Compartmentalization and molecular traffic in secondary metabolism: A new understanding of established cellular processes. Fungal Genet. Biol. 2011, 48, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Lim, F.Y.; Keller, N.P. Spatial and temporal control of fungal natural product synthesis. Nat. Prod. Rep. 2014, 31, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Menke, J.; Weber, J.; Broz, K.; Kistler, H.C. Cellular development associated with induced mycotoxin synthesis in the filamentous fungus Fusarium graminearum. PLoS ONE 2013, 8, e63077. [Google Scholar] [CrossRef] [PubMed]
- Van Krimpen, P.C.; Van Bennekom, W.P.; Bult, A. Penicillins and cephalosporins. Physicochemical properties and analysis in pharmaceutical and biological matrices. Pharm. Weekbl. Sci. 1987, 9, 1–23. [Google Scholar] [PubMed]
- Rohlfs, M.; Albert, M.; Keller, N.P.; Kempken, F. Secondary chemicals protect mould from fungivory. Biol. Lett. 2007, 3, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Linz, J.E.; Chanda, A.; Hong, S.Y.; Whitten, D.A.; Wilkerson, C.; Roze, L.V. Proteomic and biochemical evidence support a role for transport vesicles and endosomes in stress response and secondary metabolism in Aspergillus parasiticus. J. Proteome Res. 2012, 11, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Roze, L.V.; Chanda, A.; Laivenieks, M.; Beaudry, R.M.; Artymovich, K.A.; Koptina, A.V.; Awad, D.W.; Valeeva, D.; Jones, A.D.; Linz, J.E. Volatile profiling reveals intracellular metabolic changes in Aspergillus parasiticus: veA regulates branched chain amino acid and ethanol metabolism. BMC Biochem. 2010, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Baidya, S.; Duran, R.M.; Lohmar, J.M.; Harris-Coward, P.Y.; Cary, J.W.; Hong, S.Y.; Roze, L.V.; Linz, J.E.; Calvo, A.M. VeA is associated with the response to oxidative stress in the aflatoxin producer Aspergillus flavus. Eukaryot. Cell 2014, 13, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Roze, L.V.; Wee, J.; Linz, J.E. Evidence that a transcription factor regulatory network coordinates oxidative stress response and secondary metabolism in aspergilli. Microbiologyopen 2013, 2, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Roze, L.V.; Linz, J.E. Oxidative stress-related transcription factors in the regulation of secondary metabolism. Toxins (Basel) 2013, 5, 683–702. [Google Scholar] [CrossRef] [PubMed]
- Montibus, M.; Ducos, C.; Bonnin-Verdal, M.N.; Bormann, J.; Ponts, N.; Richard-Forget, F.; Barreau, C. The bZIP transcription factor Fgap1 mediates oxidative stress response and trichothecene biosynthesis but not virulence in Fusarium graminearum. PLoS ONE 2013, 8, e83377. [Google Scholar] [CrossRef] [PubMed]
- Reverberi, M.; Zjalic, S.; Punelli, F.; Ricelli, A.; Fabbri, A.A.; Fanelli, C. Apyap1 affects aflatoxin biosynthesis during Aspergillus parasiticus growth in maize seeds. Food Addit. Contam. 2007, 24, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Reverberi, M.; Gazzetti, K.; Punelli, F.; Scarpari, M.; Zjalic, S.; Ricelli, A.; Fabbri, A.A.; Fanelli, C. Aoyap1 regulates OTA synthesis by controlling cell redox balance in Aspergillus ochraceus. Appl. Microbiol. Biotechnol. 2012, 95, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Montibus, M.; Pinson-Gadais, L.; Richard-Forget, F.; Barreau, C.; Ponts, N. Coupling of transcriptional response to oxidative stress and secondary metabolism regulation in filamentous fungi. Crit. Rev. Microbiol. 2015, 41, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.B.; Reinke, A.W.; Szilágyi, M.; Emri, T.; Chiang, Y.M.; Keating, A.E.; Pócsi, I.; Wang, C.C.; Keller, N.P. bZIP transcription factors affecting secondary metabolism, sexual development and stress responses in Aspergillus nidulans. Microbiology 2013, 159, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Jayashree, T.; Subramanyam, C. Oxidative stress as a prerequisite for aflatoxin production by Aspergillus parasiticus. Free Radic. Biol. Med. 2000, 29, 981–985. [Google Scholar] [CrossRef]
- Narasaiah, K.V.; Sashidhar, R.B.; Subramanyam, C. Biochemical analysis of oxidative stress in the production of aflatoxin and its precursor intermediates. Mycopathologia 2006, 162, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Reverberi, M.; Zjalic, S.; Ricelli, A.; Fabbri, A.A.; Fanelli, C. Oxidant/antioxidant balance in Aspergillus parasiticus affects aflatoxin biosynthesis. Mycotoxin Res. 2006, 22, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Reverberi, M.; Zjalic, S.; Ricelli, A.; Punelli, F.; Camera, E.; Fabbri, C.; Picardo, M.; Fanelli, C.; Fabbri, A.A. Modulation of antioxidant defense in Aspergillus parasiticus is involved in aflatoxin biosynthesis: A role for the ApyapA gene. Eukaryot. Cell 2008, 7, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Cary, J.W.; Dyer, J.M.; Ehrlich, K.C.; Wright, M.S.; Liang, S.H.; Linz, J.E. Molecular and functional characterization of a second copy of the aflatoxin regulatory gene, aflR-2, from Aspergillus parasiticus. Biochem. Biophys. Acta 2002, 1576, 316–323. [Google Scholar] [CrossRef]
- Ehrlich, K.C.; Montalbano, B.G.; Cary, J.W. Binding of the C6-zinc cluster protein, AFLR, to the promoters of aflatoxin pathway biosynthesis genes in Aspergillus parasiticus. Gene 1999, 230, 249–257. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide dismutases. Annu. Rev. Biochem. 1975, 44, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Skory, C.D.; Chang, P.-K.; Linz, J.E. Regulated Expression of the nor-1 and ver-1 Genes Associated with Aflatoxin Biosynthesis. Appl. Environ. Microbiol. 1993, 59, 1642–1646. [Google Scholar] [PubMed]
- Roze, L.V.; Arthur, A.E.; Hong, S.Y.; Chanda, A.; Linz, J.E. The initiation and pattern of spread of histone H4 acetylation parallel the order of transcriptional activation of genes in the aflatoxin cluster. Mol. Microbiol. 2007, 66, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Payne, G.A.; Nierman, W.C.; Machida, M.; Bennett, J.W.; Campbell, B.C.; Robens, J.F.; Bhatnagar, D.; Dean, R.A.; Cleveland, T.E. Aspergillus flavus genomics as a tool for studying the mechanism of aflatoxin formation. Food Addit. Contam. Part A 2008, 25, 1152–1157. [Google Scholar]
- Sigrist, C.J.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucleic Acids Res. 2013, 41, D344–D347. [Google Scholar] [CrossRef] [PubMed]
- Price, M.S.; Yu, J.; Nierman, W.C.; Kim, H.S.; Pritchard, B.; Jacobus, C.A.; Bhatnagar, D.; Cleveland, T.E.; Payne, G.A. The aflatoxin pathway regulator AflR induces gene transcription inside and outside of the aflatoxin biosynthetic cluster. FEMS Microbiol. Lett. 2006, 255, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.B.; Amaike, S.; Wohlbach, D.J.; Gasch, A.P.; Chiang, Y.M.; Wang, C.C.; Bok, J.W.; Rohlfs, M.; Keller, N.P. An Aspergillus nidulans bZIP response pathway hardwired for defensive secondary metabolism operates through aflR. Mol. Microbiol. 2012, 83, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Roze, L.V.; Laivenieks, M.; Hong, S.Y.; Wee, J.; Wong, S.S.; Vanos, B.; Awad, D.; Ehrlich, K.C.; Linz, J.E. Aflatoxin Biosynthesis Is a Novel Source of Reactive Oxygen Species—A Potential Redox Signal to Initiate Resistance to Oxidative Stress? Toxins 2015, 7, 1411–1430. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Gummadidala, P.M.; Rima, R.A.; Ahmed, R.U.; Kenne, G.J.; Mitra, C.; Gomaa, O.M.; Hill, J.; McFadden, S.; Banaszek, N.; et al. Quantitative acoustic contrast tomography reveals unique multiscale physical fluctuations during aflatoxin synthesis in Aspergillus parasiticus. Fungal Genet. Biol. 2014, 73, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Wee, J.; Hong, S.Y.; Roze, L.V.; Day, D.M.; Chanda, A.; Linz, J.E. The Fungal bZIP Transcription Factor AtfB Controls Virulence-Associated Processes in Aspergillus parasiticus. Toxins (Basel) 2017, 9, 287. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Roze, L.V.; Trail, F.; Linz, J.E. Role of cis-acting sites NorL, a TATA box, and AflR1 in nor-1 transcriptional activation in Aspergillus parasiticus. Appl. Environ. Microbiol. 2005, 71, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Roze, L.V.; Miller, M.J.; Rarick, M.; Mahanti, N.; Linz, J.E. A novel cAMP-response element, CRE1, modulates expression of nor-1 in Aspergillus parasiticus. J. Biol. Chem. 2004, 279, 27428–27439. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.K.; Scharfenstein, L.L.; Luo, M.; Mahoney, N.; Molyneux, R.J.; Yu, J.; Brown, R.L.; Campbell, B.C. Loss of msnA, a putative stress regulatory gene, in Aspergillus parasiticus and Aspergillus flavus increased production of conidia, aflatoxins and kojic acid. Toxins (Basel) 2011, 3, 82–104. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J.; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Gummadidala, P.M.; Holder, M.E.; O’Brien, J.L.; Ajami, N.J.; Petrosino, J.F.; Mitra, C.; Chen, Y.P.; Decho, A.W.; Chanda, A. Complete Genome Sequence of Vibrio gazogenes ATCC 43942. Genome Announc. 2017, 5, e00733-17. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Linz, J.E. Functional expression and subcellular localization of the aflatoxin pathway enzyme Ver-1 fused to enhanced green fluorescent protein. Appl. Environ. Microbiol. 2008, 74, 6385–6396. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FeSOD (Gene bank accession number EED58116.1) |
| MLPRFLRPQSTLRAVSSLTQKPASALPRFQTRGLHRVPQLTHDTHFKNNGIQELLSPEAFDFAWTQYQTLLIDKLNLLTQDTVDADAKPGELLVKYSRRPEMASVFNYASMAHNNHFFFNCLSPTPTQIPDKFAKDIVDTCSSIESLKLDFLATANAMFGPGFVWL AKNLEREGLMHIFCTYNAGSPYPAAHSRRQPVDMATHSPDAPLGNQFAGAMGAHSANQKSLAPGAVDVQPILCVNTWEHVWMMDYGIGGKAEYLERWWDRINWEVVFDNYNAVS SMKGTRHAANRNRSLSML |
| FeSODA (Gene bank accession number EED55486.1) |
| MAASLIRTSARTALRAGASATPKAAGVAGLTFARGKATLPDLAYDYGALEPSISGKIMELHHKNHHQTYVNSYNTAIEQLQEAVAKEDITTQINLKPLINFHGGGHINHTLFWENLAPKSQGGGEPPSGALAKAIDESFGSLGEFQSKMNAALAGIQGSGWAWLVK DKQTGNIGIKTYAVSSSLTRTLSLVSSSLFSVLMLGSTPTTFNTRTARLSTSAPSGTSSTGRRLRSASRKRAKVGWITCSRSIPAGSGKLIFLVWDPLRPLRIFFPHLTSQAINSLEMSAESPGEKRGGFRAFFAGALRPKKSRQVLRKASTPNLKEGLQSKDDVPA MPSLTPLEAHRLKYREVNLQKDTQLGETHDHTAMLHSIGVGELDPSDPHAQLHEFDNRPPGEPMIASLTSDLWAKVTEYLNPAERASLAFSSRTLYARLGREPWITINLPENHDYKADFLISQDRLLPHHLLCFPCGKYHRRTQEGYEKLQPADIINPLFDCPNARNN ALPAPRHRITHGRVLYFTFHQLVMRAYRFGPRYGISADSLSRRWRRDGWSHQTRYHIHQGRLLMRVVSTCFAEPGLSASQQRLLLYSRDDYWPYFSVCAHWRDGELMNVCKCALGHIPVPRTTNGLQGLEHRAKDMYHRREHNPNALASLCGKCRPMRRCPECPSEYL VEVKLTEDRSGSHRNLFRHAIVVTRWSDLGDGRSPRLSKEWAAINGDEAGEGYDSFEKIGKRAISGIFESAITDDTLPGQRILSMNPKERSWVRLGIIGIEVPYLYFALGVICGGKLGVLSGVIFCIILYYTRVGVWVGWVGWVGWVGWVGWVGWVGWVGWIGLCGFI |
| CuZnSOD1 (Gene bank accession number EED46237.1) |
| MVKAVAVLRGDSKISGTVTFEQADANAPTTVSWNITGHDANAERAFHVHQFGDNTNGCTSAGPHFNPFGKEHGAPEDENRHVGDLGNFKTDAEGNAVGSKQDKLIKLIGAESVLGRTLVIHAGTDDLGRSEHPESKKTGNAGARPACGVIGIAA |
| Cytosolic CuZn SOD (Gene bank accession number EED49986.1) |
| MLTKSLFAGAALGLSLSSAVAHEAPVVEGNEPQTVYEAVLQDKDNTTVRGTFTTHGAEDGIGIQFRVALTGVPKDTFLNYHIHDNPVPKDGNCYATGGHLDPYKRGDQPPCNTTVPQTCQVGDISGKHGPVWTADGNFEVLYRDFFLSNVEDTIAFFGNRSVVV HLPDNKRINCGNFHLVSDGEEKKKKEEAKEDQGC |
| MnSOD (Gene bank accession number EED56070.1) |
| MATTFSLPPLPYAYDALEPVICKQIMEIHHQKHHQTYITNLNAALSAQSTALAANNIPQLINLQQKIKFNGGGHINHSLFWKNLAPHASPETNIDQAAPVLKAAIEAQYGSVEKFKEAFGATLLGLQGSGWGWLVANGPGGKLEIVSTKDQDPVTDKVPVFGVD MWEHAYYLQYFNNKASYVEGIWKVLNWRTAEDRFKNGVEGSALLKL |
| Gene | Conserved Domain | Conserved Sequence |
|---|---|---|
| FeSOD | None | None |
| FeSOD A | None | None |
| CuZnSOD1 | CuZn superoxide dismutase signature 1 | AFHVHQfGDnT |
| CuZn superoxide dismutase signature 2 | GNAGaRpACgvI | |
| CuZnSOD cytosolic | None | None |
| MnSOD | Manganese and iron superoxide dismutases signature | DmWEHAYY |
| (i). Frequency data for the presence of frequency patterns in the 390 SODs within the PROSITE database | |||||||
| ID | Patterns | Sites corresponding to the Patterns | Frequency | ||||
| PS00001 | ASN_GLYCOSYLATION (pattern) | N-glycosylation site | 302 | ||||
| PS00004 | CAMP_PHOSPHO_SITE (pattern) | cAMP- and cGMP-dependent protein kinase phosphorylation site | 47 | ||||
| PS00005 | PKC_PHOSPHO_SITE (pattern) | Protein kinase C phosphorylation site | 273 | ||||
| PS00006 | CK2_PHOSPHO_SITE (pattern) | Casein kinase II phosphorylation site | 331 | ||||
| PS00007 | TYR_PHOSPHO_SITE (pattern) | Tyrosine kinase phosphorylation site | 37 | ||||
| PS00008 | MYRISTYL (pattern) | N-myristoylation site | 387 | ||||
| PS00009 | AMIDATION (pattern) | Amidation site | 35 | ||||
| PS00016 | RGD (pattern) | Cell attachment sequence | 38 | ||||
| PS00017 | ATP_GTP_A (pattern) | ATP/GTP-binding site motif A (P-loop) | 14 | ||||
| PS00342 | MICROBODIES_CTER (pattern) | Microbodies C-terminal targeting signal | 34 | ||||
| PS50310 | ALA_RICH (profile) | Alanine-rich region profile | 2 | ||||
| PS50321 | ASN_RICH (profile) | Asparagine-rich region profile | 1 | ||||
| PS50324 | SER_RICH (profile) | Serine-rich region profile | 1 | ||||
| (ii). Frequency of occurrence of the most frequent patterns in FeSOD, FeSOD A and CuZnSOD cytosolic | |||||||
| 4 most frequent sites in SODs (from table above) | Consensus Pattern | Frequency of occurrence in | |||||
| CuZnSOD cytosolic | FeSOD | FeSODA | |||||
| N-myristoylation site | G-{EDRKHPFYW}-x(2) -[STAGCN]-{P} | 5 | 4 | 15 | |||
| CK-2 phosphorylation site | [ST]-x(2)-[DE] | 3 | 1 | 7 | |||
| N-glycosylation site | N-{P}-[ST]-{P} | 3 | 1 | 1 | |||
| Protein kinase C phosphorylation site | [ST]-x-[RK] | 2 | 6 | 11 | |||
| NO. | Genes | Primer Sequences |
|---|---|---|
| 1 | nor-1 | F 5′-CACTTAGCCAGCACGATCAA-3′ |
| R 5′-ATGATCATCCGACTGCCTTC-3′ | ||
| 2 | ver-1 | F 5′-AACACTCGTGGCCAGTTCTT-3’ |
| R 5′-ATATACTCCCGCGACACAGC-3’ | ||
| 3 | β-tubulin | F 5′-TCTCCAAGATCCGTGAGGAG-3′ |
| R 5′-TTCAGGTCACCGTAAGAGGG- 3′ | ||
| 4 | FeSOD | F 5′-GAGATGGCCTCCGTATTCAA-3′ |
| R 5′-CATCAATCCTTCCCTCTCCA-3′ | ||
| 5 | FeSOD A | F 5′-CCAAGGAGGACATCACCACT-3′ |
| R 5′-GCATAGGTCTTGATGCCGAT-3′ | ||
| 6 | CuZnSOD1 | F 5′-CACCAGTTCGGTGACAACAC-3′ |
| R 5′-GTGTTCACTACGGCCAAGGT-3′ | ||
| 7 | CuZnSOD cytosolic | F 5′-CCTCCTTGCAATACAACCGT-3′ |
| R 5′-GTCTTCCTTCGCCTCTTCCT-3′ | ||
| 8 | MnSOD | F 5′-CCACATCAACCACTCCCTCT-3′ |
| R 5′-TCCTGATCCTTCGTCGAAAC-3′ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kenne, G.J.; Gummadidala, P.M.; Omebeyinje, M.H.; Mondal, A.M.; Bett, D.K.; McFadden, S.; Bromfield, S.; Banaszek, N.; Velez-Martinez, M.; Mitra, C.; et al. Activation of Aflatoxin Biosynthesis Alleviates Total ROS in Aspergillus parasiticus. Toxins 2018, 10, 57. https://doi.org/10.3390/toxins10020057
Kenne GJ, Gummadidala PM, Omebeyinje MH, Mondal AM, Bett DK, McFadden S, Bromfield S, Banaszek N, Velez-Martinez M, Mitra C, et al. Activation of Aflatoxin Biosynthesis Alleviates Total ROS in Aspergillus parasiticus. Toxins. 2018; 10(2):57. https://doi.org/10.3390/toxins10020057
Chicago/Turabian StyleKenne, Gabriel J., Phani M. Gummadidala, Mayomi H. Omebeyinje, Ananda M. Mondal, Dominic K. Bett, Sandra McFadden, Sydney Bromfield, Nora Banaszek, Michelle Velez-Martinez, Chandrani Mitra, and et al. 2018. "Activation of Aflatoxin Biosynthesis Alleviates Total ROS in Aspergillus parasiticus" Toxins 10, no. 2: 57. https://doi.org/10.3390/toxins10020057
APA StyleKenne, G. J., Gummadidala, P. M., Omebeyinje, M. H., Mondal, A. M., Bett, D. K., McFadden, S., Bromfield, S., Banaszek, N., Velez-Martinez, M., Mitra, C., Mikell, I., Chatterjee, S., Wee, J., & Chanda, A. (2018). Activation of Aflatoxin Biosynthesis Alleviates Total ROS in Aspergillus parasiticus. Toxins, 10(2), 57. https://doi.org/10.3390/toxins10020057