Fluorescence Polarization Immunoassay of Mycotoxins: A Review

Abstract
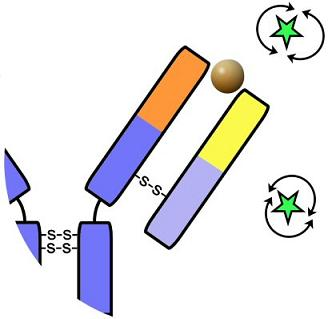
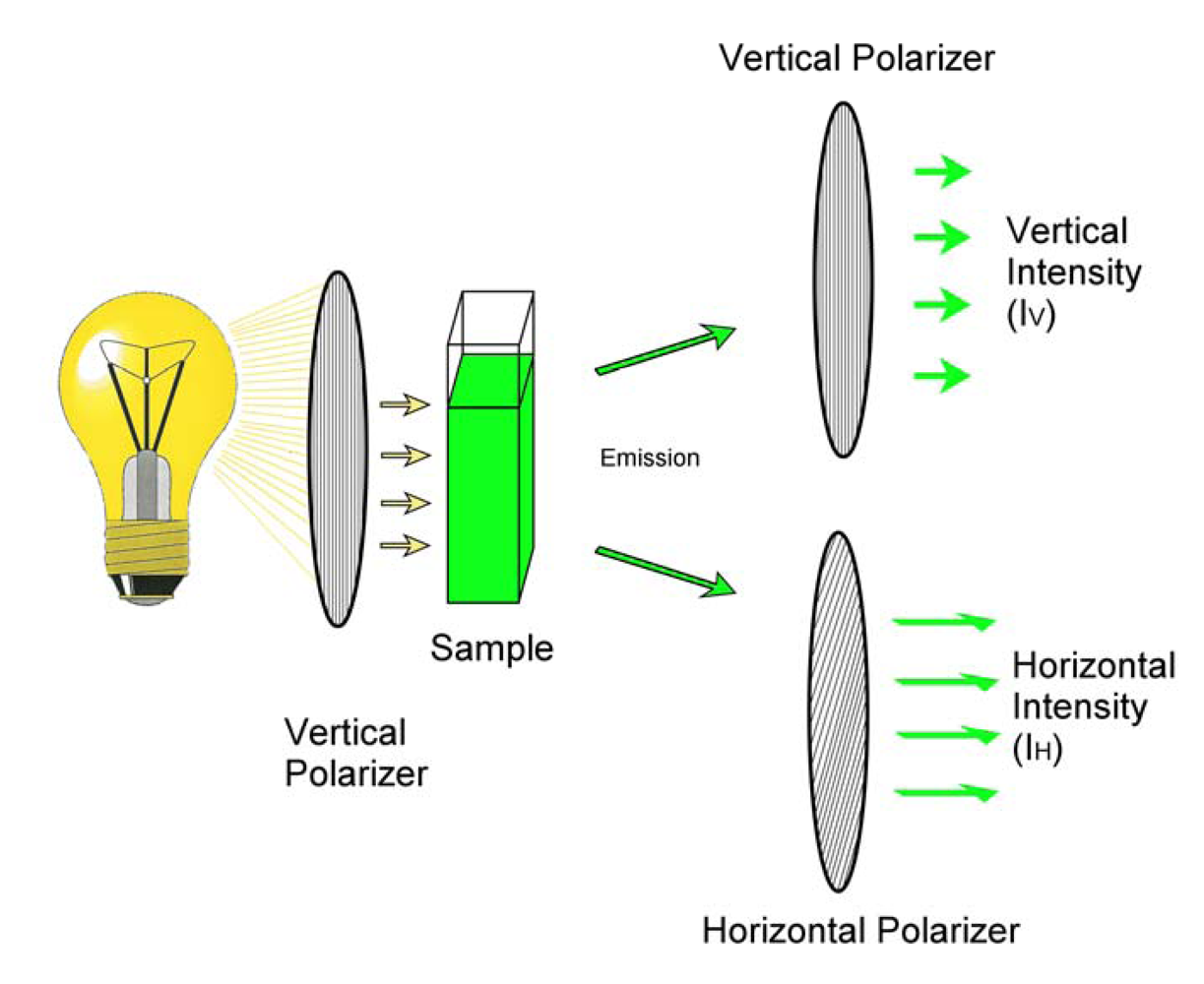
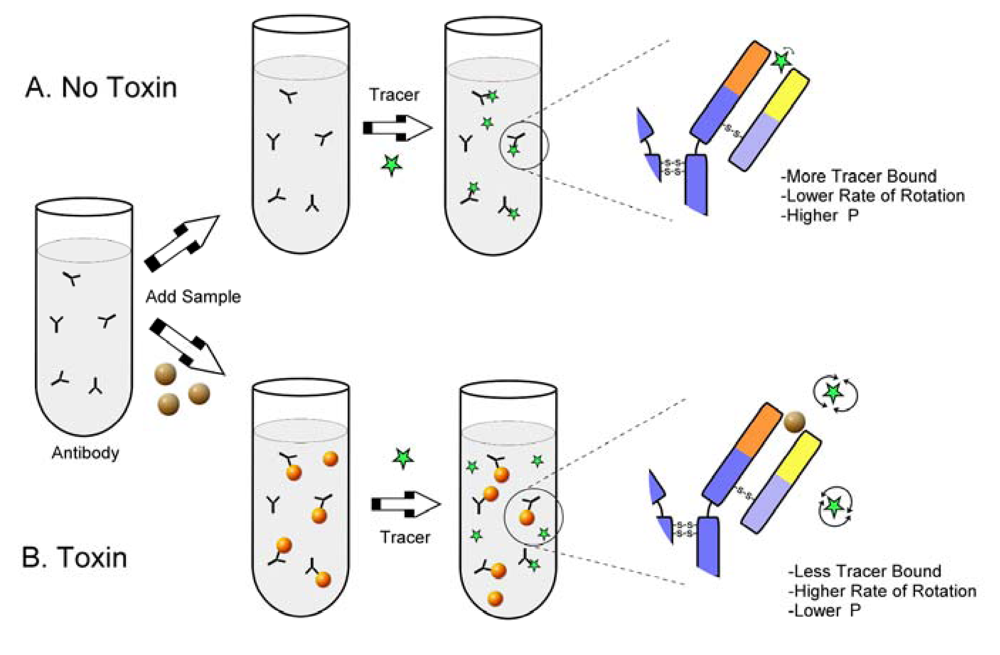
:
1. Introduction

| Toxin | Analogs tested | Tracera | Matrix | Sample Cleanup | Limit of Detection or IC50 | Ref. |
|---|---|---|---|---|---|---|
| Fumonisins | FB1, FB2, FB3 | FB1-DTAF | maize | Filtration, Dilution | LOD: 500 μg/kg in maize | [7] |
| Aflatoxins | AFB1, AFB2, AFG1, AFG2 | AFB1-FL | Maize, sorghum, peanut butter, peanut paste, popcorn | Filtration | IC50: 28 ng/mL (AFB1) in methanol/water | [8] |
| Ochratoxin A | OTA | OTA-EDF | barley | Centrifuge, Filtration, Dilution | LOD: 3 ng/mL in buffer | [9] |
| OTA | OTA-EDF | rice | Centrifuge, Dilution | LOD: 0.3 ng/mL in buffer | [10] | |
| OTA | Oligo-Fluo | buffer | none | LOD: 2 ng/mL in buffer | [12] | |
| OTA | OTA-EDF | red wine | Dilution, SPEb | LOD: 0.7 ng/mL in red wine | [11] | |
| Deoxynivalenol | 15-Ac-DON, DON, HT-2 toxin | DON-FL | wheat | Centrifuge | IC50: 30 to 1000 ng/mL in buffer (see text) | [13] |
| 3-Ac-DON, DON, 15-Ac-DON | DON-FL2 | wheat, maize | Filtration | IC50: 12 ng/mL in buffer | [14] | |
| 3-Ac-DON, DON, 15-Ac-DON | DON-FL2 | Wheat, semolina, pasta | Filtration | LOD: 0.08 μg/kg in all three matrices | [16] | |
| Zearalenone | ZEN, ZAN, α-ZAOL, α-ZEOL, β-ZEOL, β-ZAOL | ZEN-FL2 | maize | Filtration | LOD: 110 μg/kg in maize | [17] |
| ZEN, ZAN, α-ZAOL, α-ZEOL, β-ZEOL, β-ZAOL | ZEN-FL2 | buffer | none | IC50: 67 to 450 ng/mL in buffer (multiple antibodies) | [18] | |
| ZEN | ZEN-HMDF | Cereal products | Filtration, dilution | LOD: 137 μg/kg in maize | [19] |
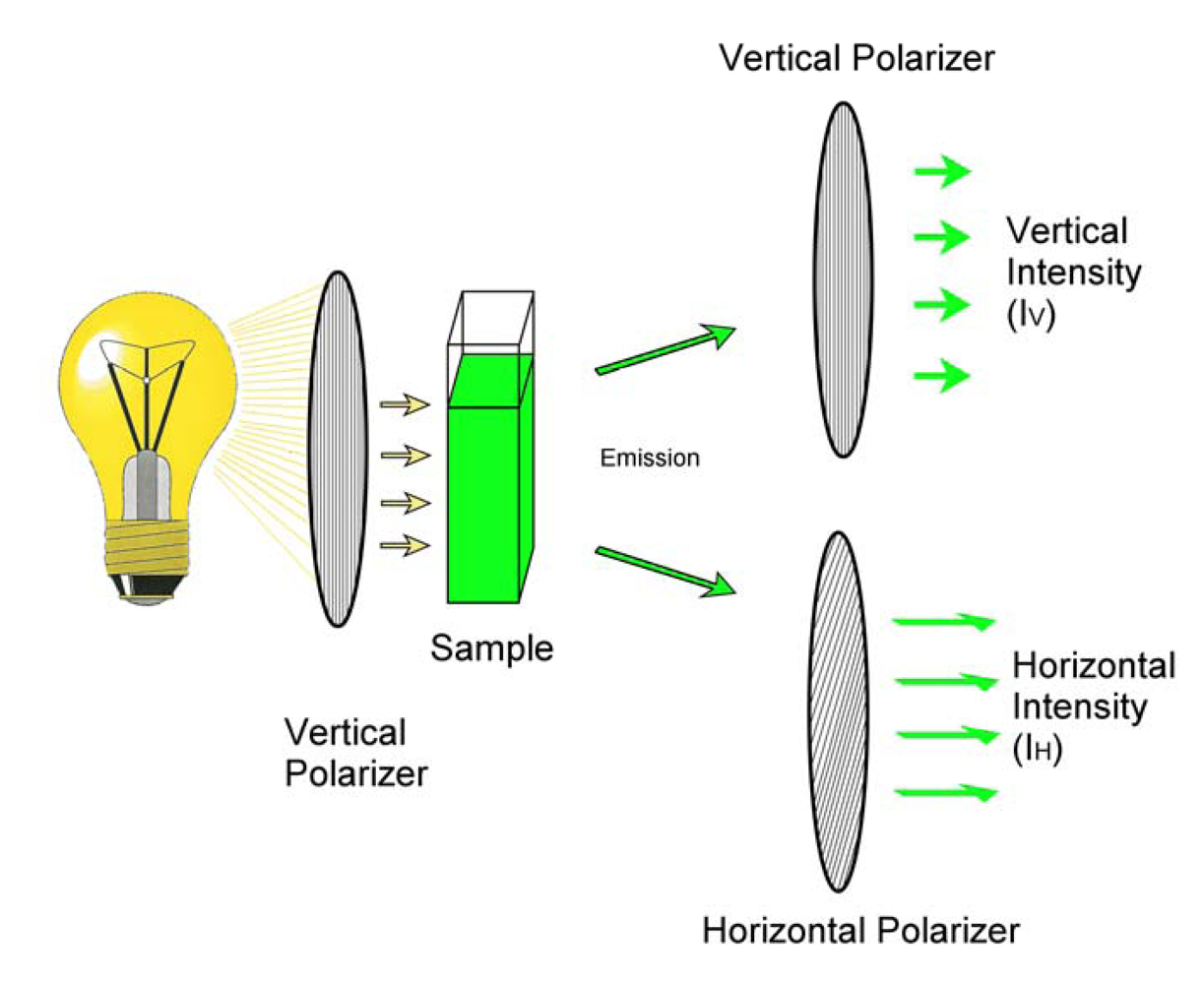
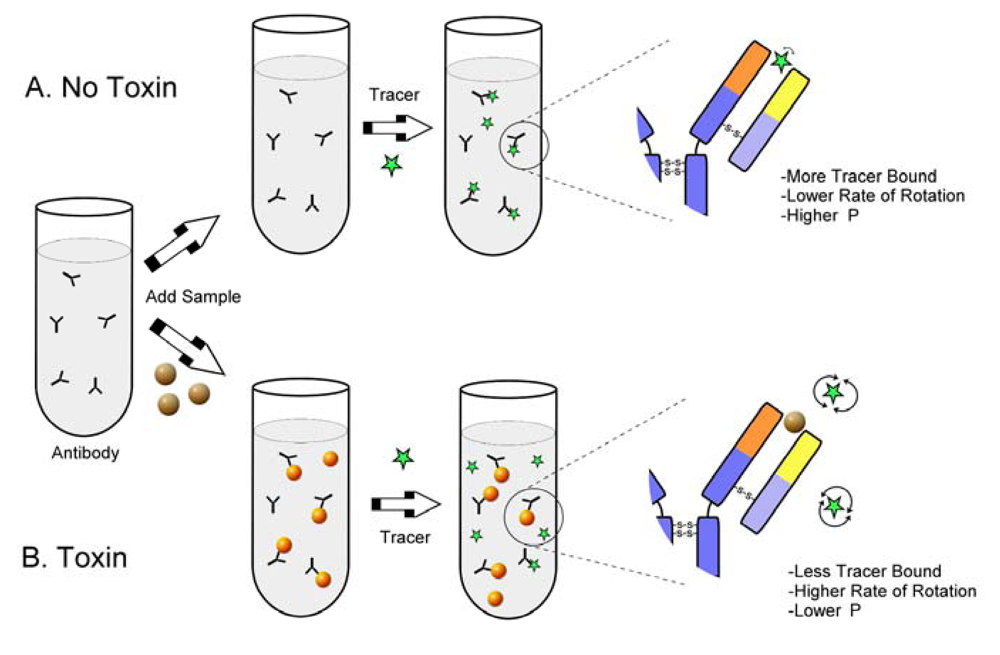
2. FPIA for Mycotoxins
2.1. Fumonisins
2.2. Aflatoxins
2.3. Ochratoxin A (OTA)
2.4. Deoxynivalenol
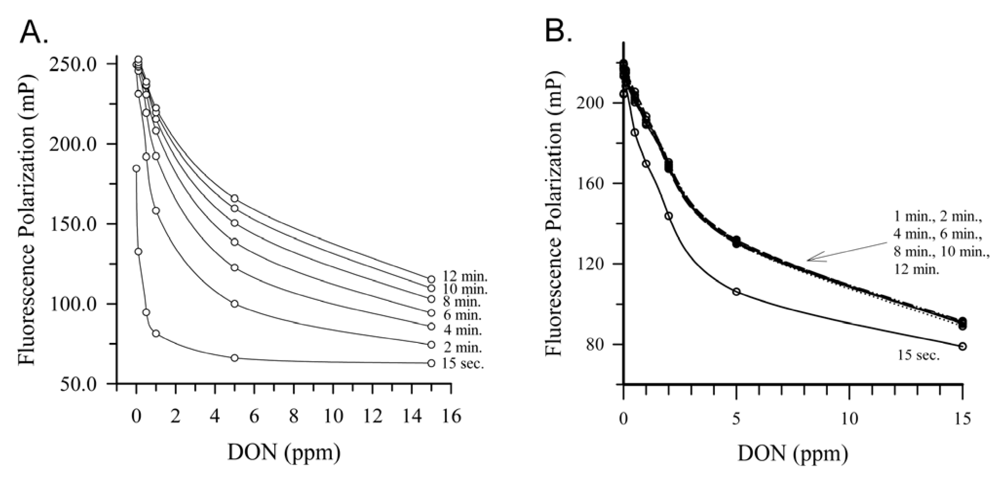
{kind=link}
{kind=link}
{kind=link}
{kind=link}
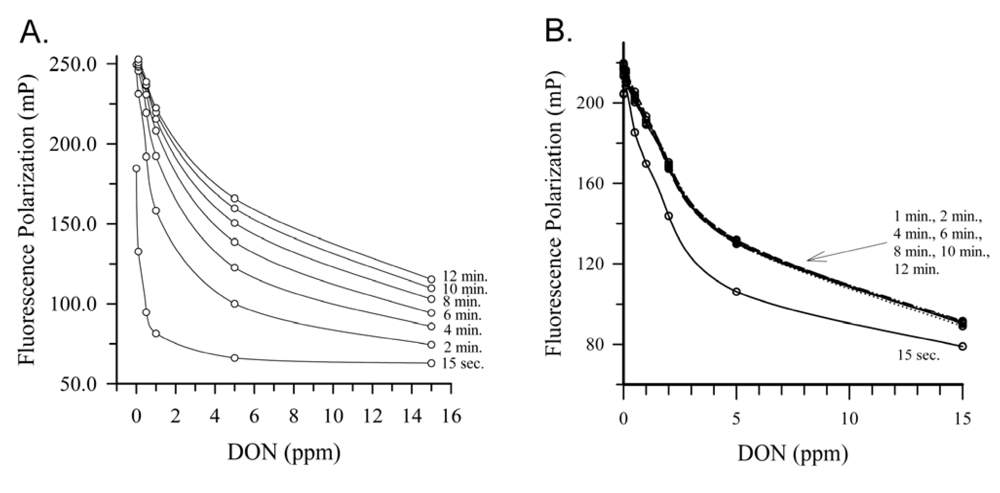{kind=link}
{kind=link}
2.5. Zearalenone
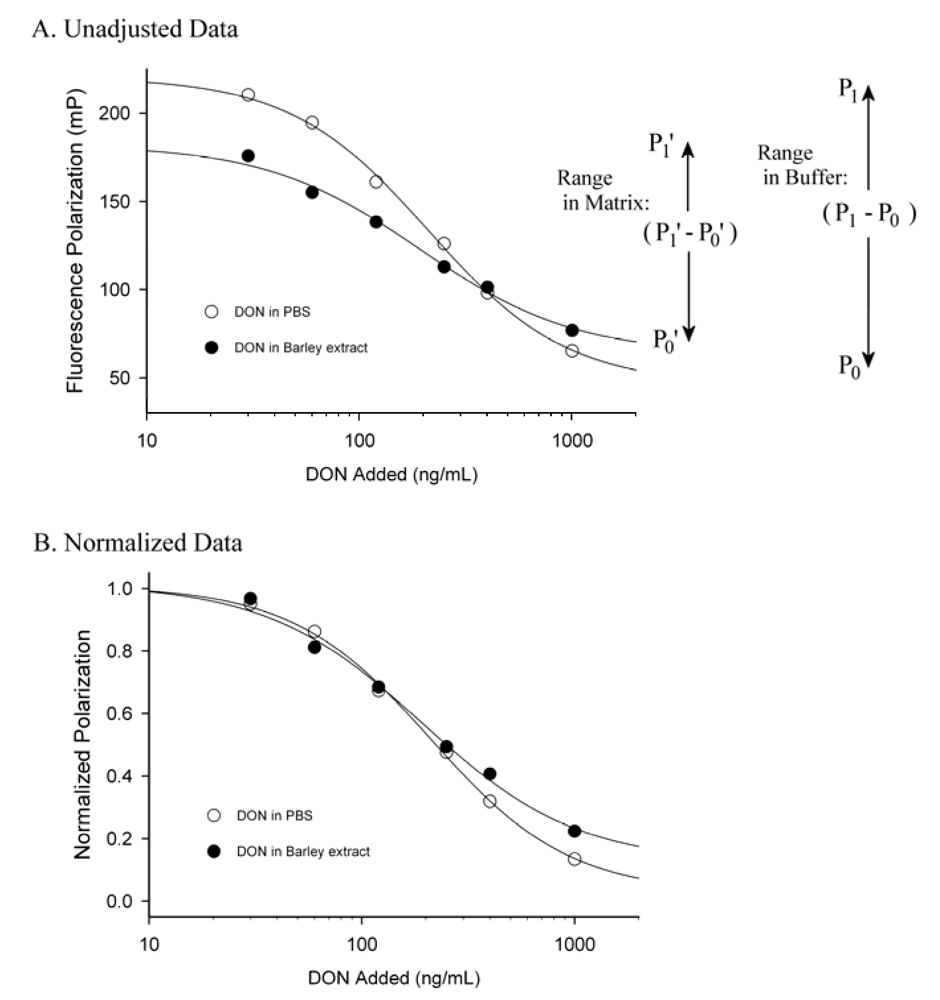
3. Matrix Effects and Data Treatment
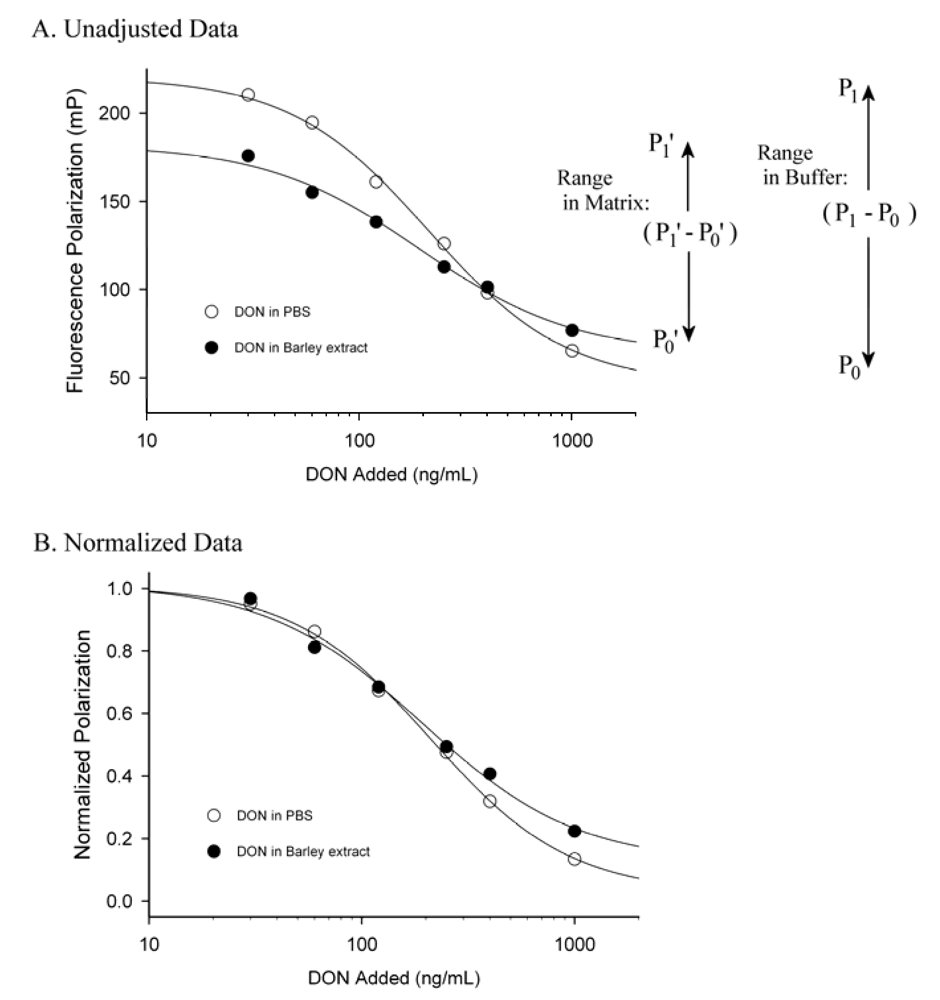
4. Conclusions
Acknowledgements
Disclaimer
References and Notes
- Council for Agricultural Science and Technology, Mycotoxins: Risks in Plant, Animal, and Human Systems, Task Force Report No. 139; Council for Agricultural Science and Technology: Ames, IA, USA, 2003.
- Maragos, C.M. Biosensors for mycotoxin analysis: Recent developments and future prospects. World Mycotoxin J. 2009, 2, 221–238. [Google Scholar] [Green Version]
- Haber, E.; Bennett, J.C. Polarization of fluorescence as a measure of antigen-antibody interaction. Proc. Nat. Acad. Sci. 1962, 48, 1935–1942. [Google Scholar] [Green Version]
- Nasir, M.S.; Jolley, M.E. Fluorescence polarization: an analytical tool for immunoassay and drug discovery. Comb. Chem. High Thr. Scr. 1999, 2, 177–190. [Google Scholar] [Green Version]
- Dandliker, W.B.; Kelly, R.J.; Dandliker, J.; Farquhar, J.; Levin, J. Fluorescence polarization immunoassay theory and experimental method. Immunochemistry 1973, 10, 219–227. [Google Scholar] [PubMed][Green Version]
- Smith, D.S.; Eremin, S.A. Fluorescence polarization immunoassays and related methods for simple, high-throughput screening of small molecules. Anal. Bioanal. Chem. 2008, 391, 1499–1507. [Google Scholar] [PubMed][Green Version]
- Maragos, C.M.; Jolley, M.E.; Plattner, R.D.; Nasir, M.S. Fluorescence polarization as a means for determination of fumonisins in maize. J. Agric. Food Chem. 2001, 49, 596–602. [Google Scholar] [PubMed][Green Version]
- Nasir, M.S.; Jolley, M.E. Development of a fluorescence polarization assay for the determination of aflatoxins in grains. J. Agric. Food Chem. 2002, 50, 3116–3121. [Google Scholar] [PubMed][Green Version]
- Shim, W.B.; Kolosova, A.Y.; Kim, Y.J.; Yang, Z.Y.; Park, S.J.; Eremin, S.A.; Lee, I.S.; Chung, D.H. Fluorescence polarization immunoassay based upon a monoclonal antibody for the detection of ochratoxin A. Intl. J. Food Sci. Technol. 2004, 39, 829–837. [Google Scholar] [CrossRef]
- Park, J.H.; Chung, D.H.; Lee, I.S. Application of fluorescence polarization immunoassay for the screening of ochratoxin A in unpolished rice. J. Life Sci. 2006, 16, 1006–1013. [Google Scholar]
- Zezza, F.; Longobardi, F.; Pascale, M.; Eremin, S.A.; Visconti, A. Fluorescence polarization immunoassay for rapid screening of ochratoxin A in red wine. Anal. Bioanal. Chem. 2009, 395, 1317–1323. [Google Scholar] [PubMed]
- Cruz-Aguado, J.A.; Penner, G. Fluorescence polarization based displacement assay for the determination of small molecules with aptamers. Anal. Chem. 2008, 80, 8853–8855. [Google Scholar] [PubMed]
- Maragos, C.M.; Jolley, M.E.; Nasir, M.S. Fluorescence polarization as a tool for the determination of deoxynivalenol in wheat. Food Addit. Contam. 2002, 19, 400–407. [Google Scholar] [PubMed]
- Maragos, C.M.; Plattner, R.D. Rapid fluorescence polarization immunoassay for the mycotoxin deoxynivalenol in wheat. J. Agric. Food Chem. 2002, 50, 1827–1832. [Google Scholar] [PubMed]
- Berthiller, F.; Schuhmacher, R.; Adam, G.; Krska, R. Formation, determination and significance of masked and other conjugated mycotoxins. Anal. Bioanal. Chem. 2009, 395, 1243–1252. [Google Scholar] [PubMed]
- Lippolis, V.; Pascale, M.; Visconti, A. Optimization of a fluorescence polarization immunoassay for rapid quantification of deoxynivalenol in Durum wheat-based products. J. Food Prot. 2006, 11, 2712–2719. [Google Scholar]
- Maragos, C.M.; Kim, E.K. Detection of zearalenone and related metabolites by fluorescence polarization immunoassay. J. Food Prot. 2004, 67, 1039–1043. [Google Scholar] [PubMed]
- Maragos, C.M.; Kim, E.K. Cross-reactivity of six zearalenone antibodies in a hand-held fluorescence polarisation immunoassay. In Proceedings of the XIth International IUPAC Symposium on Mycotoxins and Phycotoxins; Njapau, H., Trujillo, S., van Egmond, H.P., Park, D.L., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2006; pp. 91–100. [Google Scholar]
- Chun, H.S.; Choi, E.H.; Chang, H.J.; Choi, S.W.; Eremin, S.A. A fluorescence polarization immunoassay for the detection of zearalenone in corn. Anal. Chim. Acta 2009, 639, 83–89. [Google Scholar] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maragos, C. Fluorescence Polarization Immunoassay of Mycotoxins: A Review. Toxins 2009, 1, 196-207. https://doi.org/10.3390/toxins1020196
Maragos C. Fluorescence Polarization Immunoassay of Mycotoxins: A Review. Toxins. 2009; 1(2):196-207. https://doi.org/10.3390/toxins1020196
Chicago/Turabian StyleMaragos, Chris. 2009. "Fluorescence Polarization Immunoassay of Mycotoxins: A Review" Toxins 1, no. 2: 196-207. https://doi.org/10.3390/toxins1020196
APA StyleMaragos, C. (2009). Fluorescence Polarization Immunoassay of Mycotoxins: A Review. Toxins, 1(2), 196-207. https://doi.org/10.3390/toxins1020196