Bridging the Gap between Gut Microbial Dysbiosis and Cardiovascular Diseases

Abstract
1. Introduction
1.1. Definition and Introduction to the Gut Microbiota
1.2. Establishment, Development, and Changes to the Gut Microbiota
1.3. Homeostatic Functions
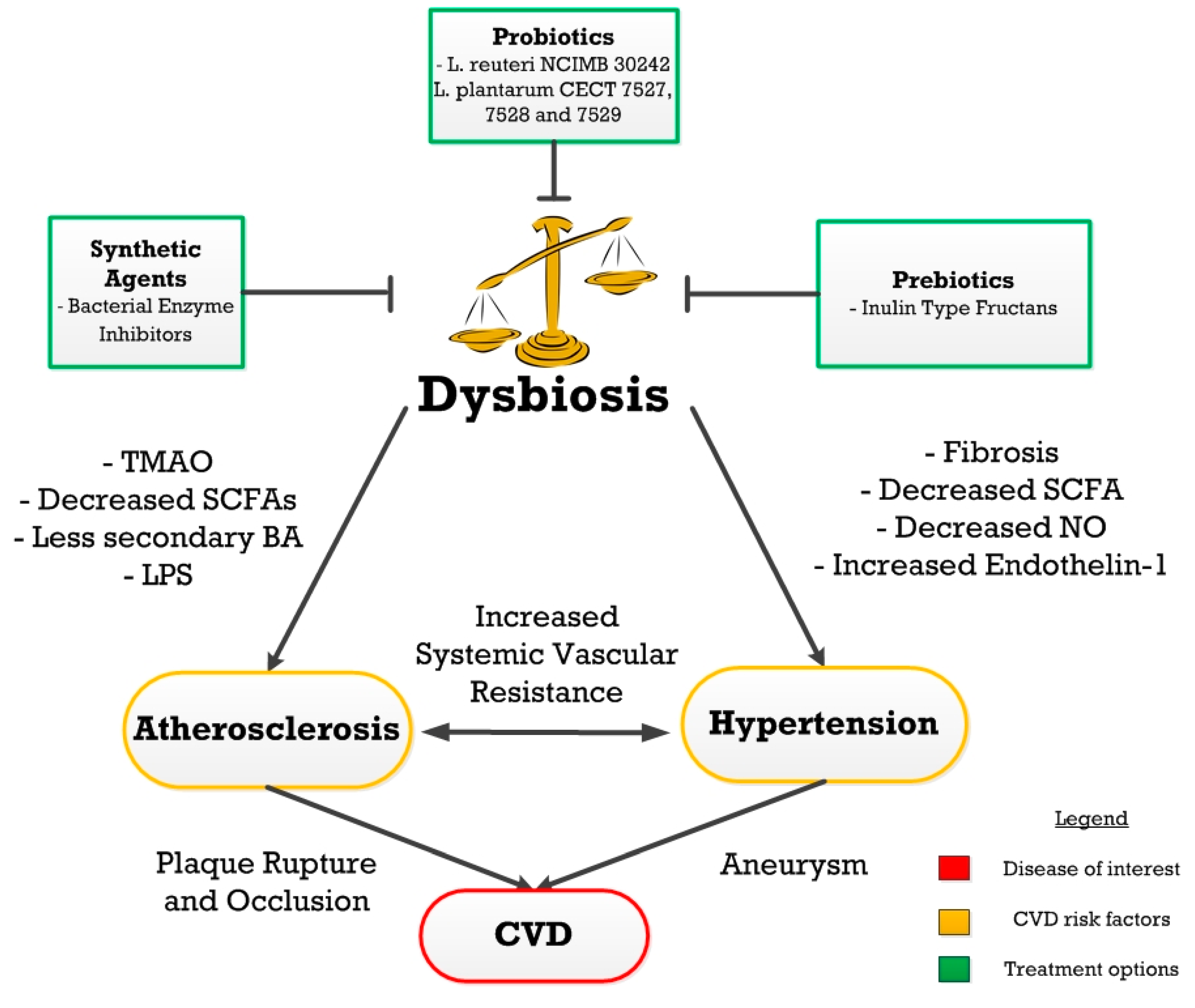
1.4. Gut Dysbiosis
2. Atherosclerosis
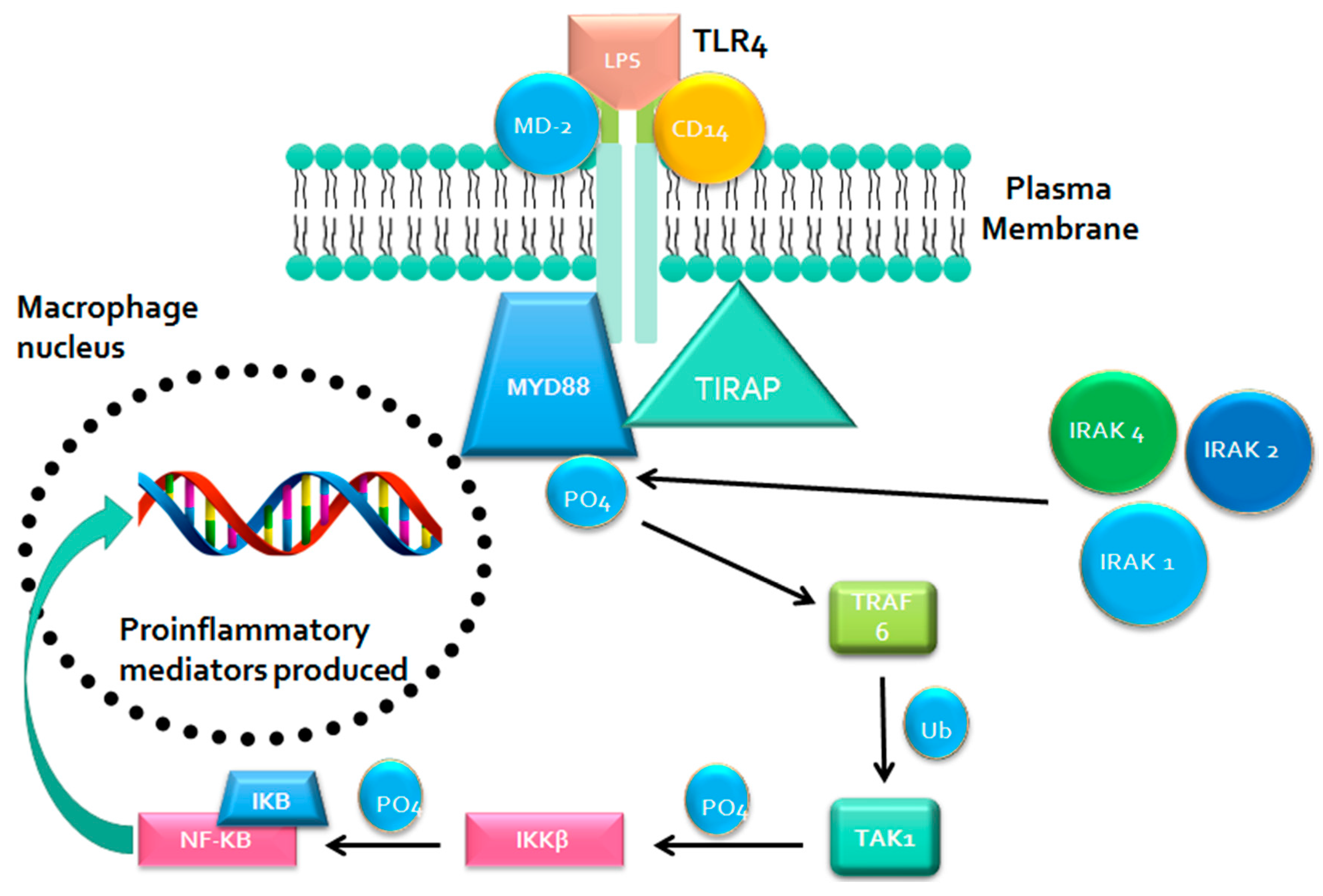
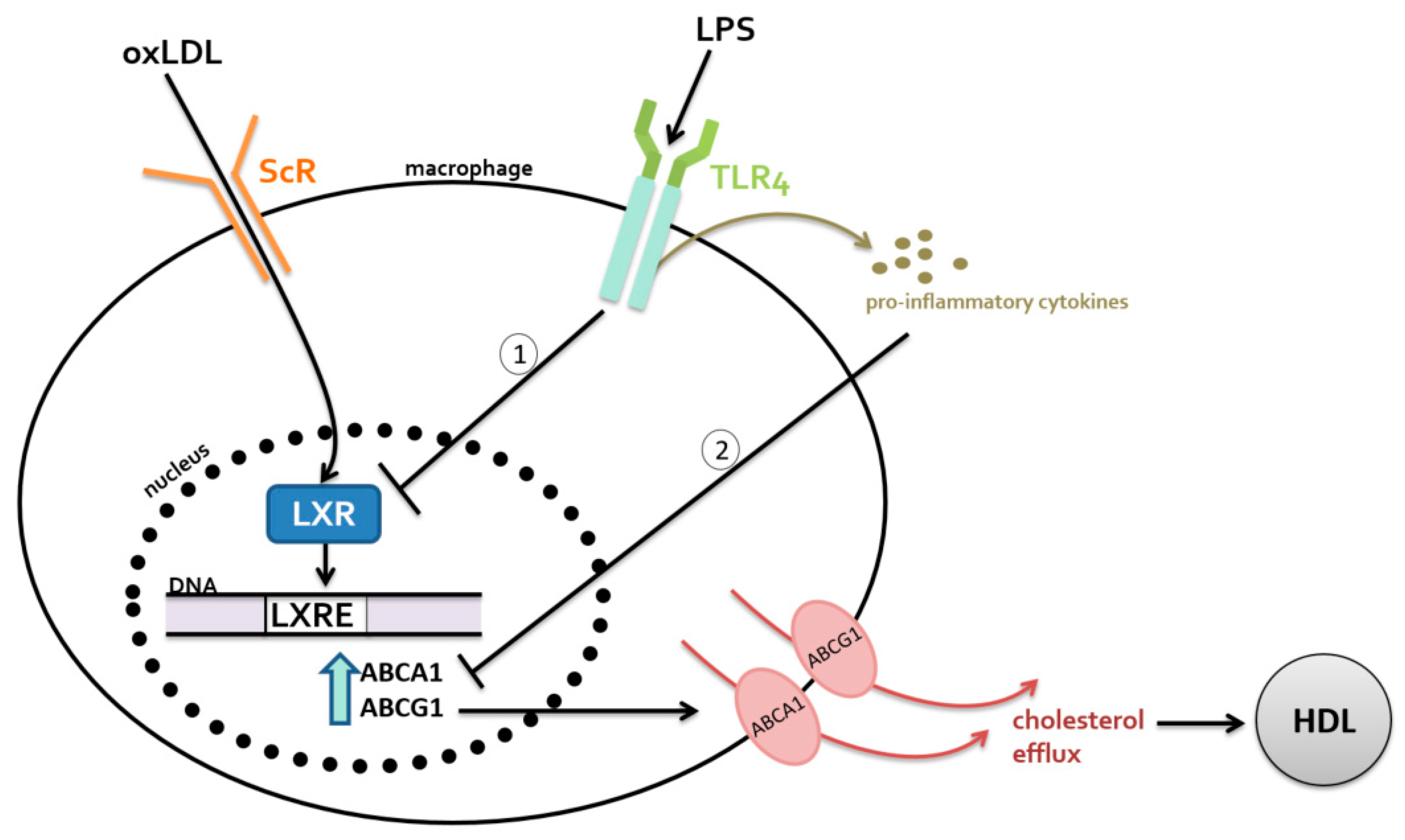
2.1. Metabolism-Independent Pathway
MYD88 Signaling
2.2. Metabolism-Dependent Pathway
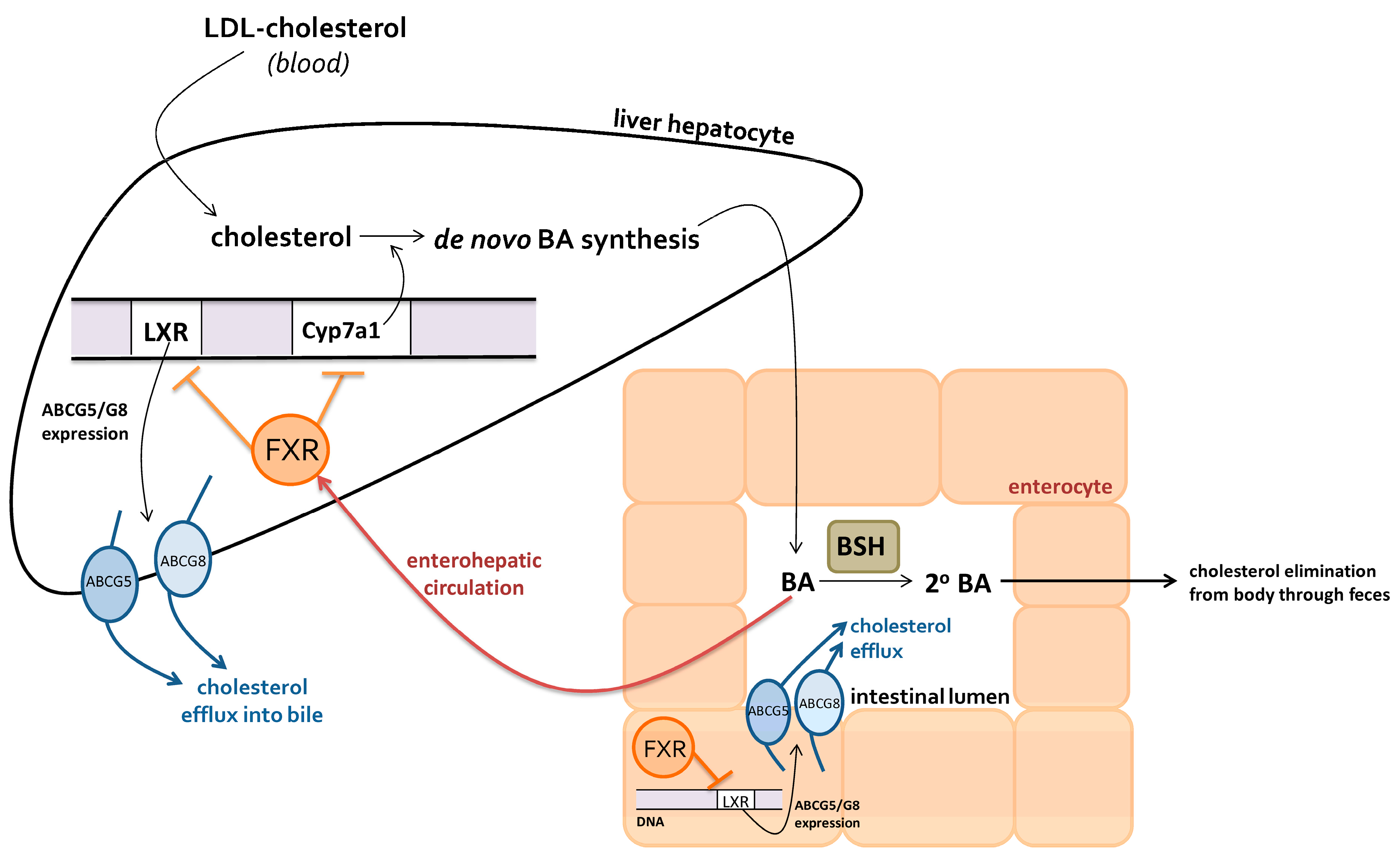
2.2.1. Bile Acids
2.2.2. Trimethylamine-N-Oxide
2.2.3. Butyrate
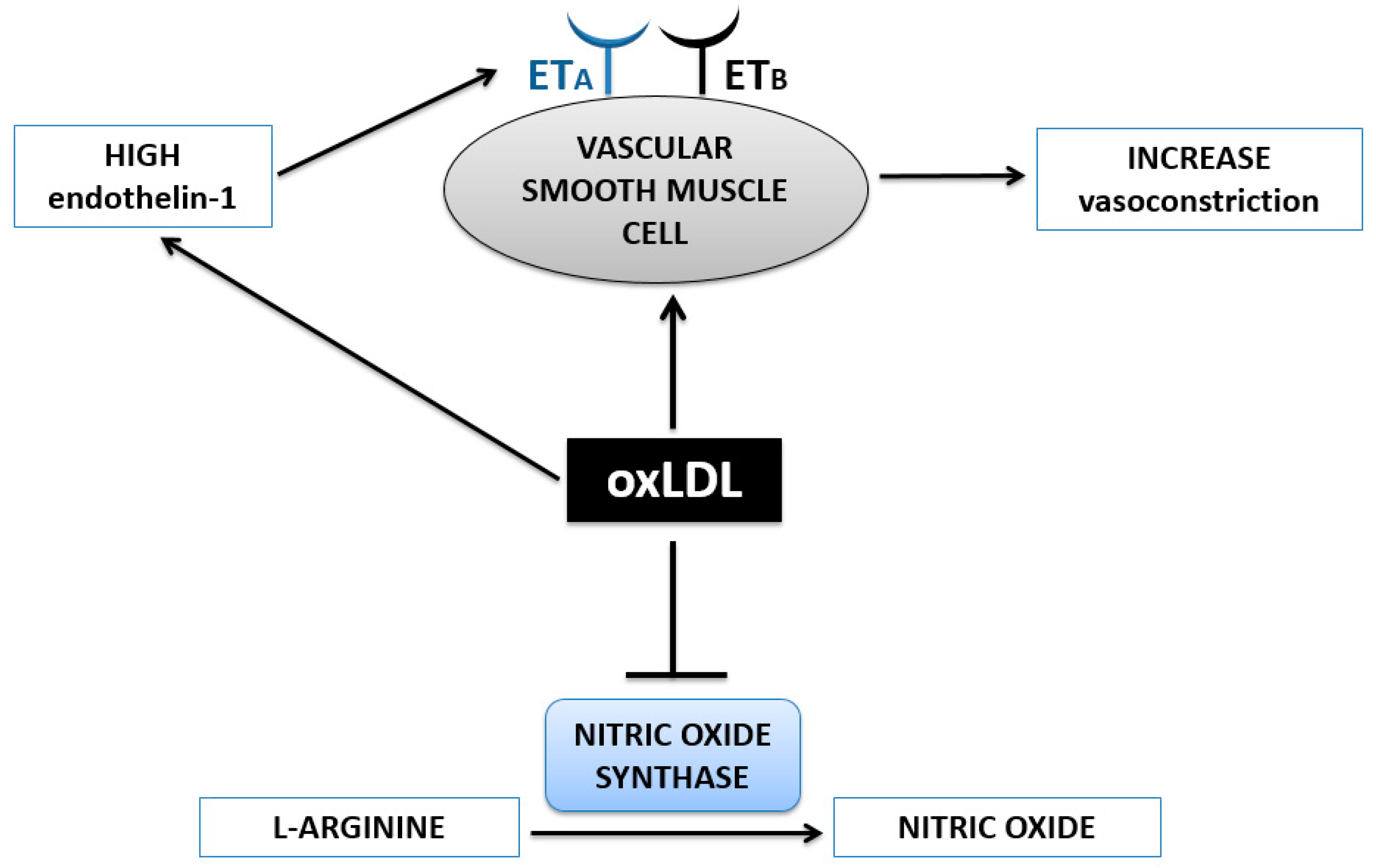
3. Hypertension
OxLDL and Vasoconstriction
4. Treatments
4.1. Dysbiosis Treatment
4.2. Prebiotics
4.3. Probiotics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kamada, N.; Seo, S.-U.; Chen, G.Y.; Núñez, G. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Trine, N.; Nicolas, P.; Florence, L.; Takuji, Y.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Bello, M.G.; Blaser, M.J.; Ley, R.E.; Knight, R. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology 2011, 140, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.; Scott, K.P.; Khan, S.; Martin, J.C.; Berry, S.H.; Stevenson, M.; Okpapi, A.; Munro, M.J.; Hold, G.L. First-Pass Meconium Samples from Healthy Term Vaginally-Delivered Neonates: An Analysis of the Microbiota. PLoS ONE 2015, 10, e0133320. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.; Tremaroli, V.; Nielsen, J.; Bäckhed, F. Assessing the human gut microbiota in metabolic diseases. Diabetes 2013, 62, 3341–3349. [Google Scholar]
- Watson, J.; Jones, R.C.; Cortes, C.; Gerber, S.I.; Golash, R.G.; Price, J.; Bancroft, E.; Mascola, L.; Gorwitz, R.J.; Jernigan, D.B.; et al. Community-associated methicillin-resistant Staphylococcus aureus infection among healthy newborns—Chicago and Los Angeles County, 2004. (Reprinted from MMWR 2006, 55, 329–332). JAMA 2006, 296, 36–38. [Google Scholar]
- Palmer, C.; Bik, E.M.; DiGiulio, D.B.; Relman, D.A.; Brown, P.O. Development of the Human Infant Intestinal Microbiota. PLoS Biol. 2007, 5, e177. [Google Scholar] [CrossRef] [PubMed]
- Koenig, J.E.; Spor, A.; Scalfone, N.; Fricker, A.D.; Stombaugh, J.; Knight, R.; Angenent, L.T.; Ley, R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108, 4578–4585. [Google Scholar] [CrossRef] [PubMed]
- Zivkovic, A.M.; German, J.B.; Lebrilla, C.B.; Mills, D.A. Human milk glycobiome and its impact on the infant gastrointestinal microbiota. Proc. Natl. Acad. Sci. USA 2011, 108, 4653–4658. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Laparra, J.M.; Sanz, Y. Interactions of gut microbiota with functional food components and nutraceuticals. Pharmacol. Res. 2010, 61, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.W.; de Souza, R.; Kendall, C.W.C.; Emam, A.; Jenkins, D.J.A. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, G.; Lange, K.; Havinga, R.; van Dijk, T.H.; Gerding, A.; van Eunen, K.; Müller, M.; Groen, A.K.; Hooiveld, G.J.; Bakker, B.M. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G900–G910. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T. The gut is still the biggest lymphoid organ in the body. Mucosal Immunol. 2008, 1, 246–247. [Google Scholar]
- Serino, M.; Blasco-Baque, V.; Nicolas, S.; Burcelin, R. Far from the eyes, close to the heart: Dysbiosis of gut microbiota and cardiovascular consequences. Curr. Cardiol. Rep. 2014, 16, 540. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26. [Google Scholar] [CrossRef] [PubMed]
- Al Khodor, S.; Reichert, B.; Shatat, I.F. The Microbiome and Blood Pressure: Can Microbes Regulate Our Blood Pressure? Front. Pediatr. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Dunne, J.L.; Triplett, E.W.; Gevers, D.; Xavier, R.; Insel, R.; Danska, J.; Atkinson, M.A. The intestinal microbiome in type 1 diabetes. Clin. Exp. Immunol. 2014, 177, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.J.; Prabhu, K.S.; Vijay-Kumar, M. The microbiome and obesity—An established risk for certain types of cancer. Cancer J. Sudbury Mass 2014, 20, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Gross, M. Does the gut microbiome hold clues to obesity and diabetes? Curr. Biol. 2013, 23, R359–R362. [Google Scholar] [CrossRef] [PubMed]
- Francescone, R.; Hou, V.; Grivennikov, S.I. Microbiome, inflammation, and cancer. Cancer J. Sudbury Mass 2014, 20, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, K.E.; Lynch, S.V. Microbiota in allergy and asthma and the emerging relationship with the gut microbiome. Cell Host Microbe 2015, 17, 592–602. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, C.; Tan, J.; Macia, L.; Mackay, C.R. The nutrition-gut microbiome—Physiology axis and allergic diseases. Immunol. Rev. 2017, 278, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Emoto, T.; Yamashita, T.; Sasaki, N.; Hirota, Y.; Hayashi, T.; So, A.; Kasahara, K.; Yodoi, K.; Matsumoto, T.; Mizoguchi, T. Analysis of Gut Microbiota in Coronary Artery Disease Patients: A Possible Link between Gut Microbiota and Coronary Artery Disease. J. Atheroscler. Thromb. 2016, 23, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.W. Gut microbiota and cardiometabolic outcomes: Influence of dietary patterns and their associated components. Am. J. Clin. Nutr. 2014, 100, 369S–377S. [Google Scholar] [CrossRef] [PubMed]
- Gui, T.; Shimokado, A.; Sun, Y.; Akasaka, T.; Muragaki, Y. Diverse roles of macrophages in atherosclerosis: From inflammatory biology to biomarker discovery. Mediat. Inflamm. 2012, 2012, 693083. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Hansson, G.K.; Hansson, G.K. Mechanisms of disease: Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Status Report on Noncommunicable Diseases 2014; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Brown, J.M.; Hazen, S.L. The gut microbial endocrine organ: bacterially derived signals driving cardiometabolic diseases. Annu. Rev. Med. 2015, 66, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Manco, M.; Putignani, L.; Bottazzo, G.F. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr. Rev. 2010, 31, 817–844. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Robertson, A.-K.L.; Söderberg-Nauclér, C. Inflammation and atherosclerosis. Annu. Rev. Pathol. 2006, 1, 297–329. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-H.; Fu, Y.-C.; Zhang, D.-W.; Yin, K.; Tang, C.-K. Foam cells in atherosclerosis. Clin. Chim. Acta Int. J. Clin. Chem. 2013, 424, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Shashkin, P.; Dragulev, B.; Ley, K. Macrophage differentiation to foam cells. Curr. Pharm. Des. 2005, 11, 3061–3072. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Keaney, J.F. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Annema, W.; Tietge, U.J. Regulation of reverse cholesterol transport—A comprehensive appraisal of available animal studies. Nutr. Metab. 2012, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Spady, D.K. Reverse cholesterol transport and atherosclerosis regression. Circulation 1999, 100, 576–578. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, R.; Mu, H.; Wang, X.; Yao, Q.; Chen, C. Reverse cholesterol transport and cholesterol efflux in atherosclerosis. QJM Mon. J. Assoc. Physicians 2005, 98, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R. An overview of reverse cholesterol transport. Eur. Heart J. 1998, 19, A31–A35. [Google Scholar] [CrossRef]
- Cuchel, M.; Rader, D.J. Macrophage reverse cholesterol transport: Key to the regression of atherosclerosis? Circulation 2006, 113, 2548–2555. [Google Scholar] [CrossRef] [PubMed]
- Lo Sasso, G.; Murzilli, S.; Salvatore, L.; D’Errico, I.; Petruzzelli, M.; Conca, P.; Jiang, Z.Y.; Calabresi, L.; Parini, P.; Moschetta, A. Intestinal specific LXR activation stimulates reverse cholesterol transport and protects from atherosclerosis. Cell Metab. 2010, 12, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Castrillo, A.; Joseph, S.B.; Vaidya, S.A.; Haberland, M.; Fogelman, A.M.; Cheng, G.; Tontonoz, P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol. Cell 2003, 12, 805–816. [Google Scholar] [CrossRef]
- Harris, K.; Kassis, A.; Major, G.; Chou, C.J. Is the gut microbiota a new factor contributing to obesity and its metabolic disorders? J. Obes. 2012, 2012, 879151. [Google Scholar] [PubMed]
- Neves, A.L.; Coelho, J.; Couto, L.; Leite-Moreira, A.; Roncon-Albuquerque, R. Metabolic endotoxemia: A molecular link between obesity and cardiovascular risk. J. Mol. Endocrinol. 2013, 51, R51–R64. [Google Scholar] [CrossRef] [PubMed]
- Barton, G.M.; Kagan, J.C. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat. Rev. Immunol. 2009, 9, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Guzzo, C.; Ayer, A.; Basta, S.; Banfield, B.W.; Gee, K. IL-27 enhances LPS-induced proinflammatory cytokine production via upregulation of TLR4 expression and signaling in human monocytes. J. Immunol. 2012, 188, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.-H.; Huang, X.-Q.; Tan, H.-M. Vascular fibrosis in atherosclerosis. Cardiovasc. Pathol. 2013, 22, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Shirwany, N.A.; Zou, M. Arterial stiffness: A brief review. Acta Pharmacol. Sin. 2010, 31, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Webb, N.R. Getting to the core of atherosclerosis. Nat. Med. 2008, 14, 1015–1016. [Google Scholar] [CrossRef] [PubMed]
- Van der Wal, A.C.; Becker, A.E. Atherosclerotic plaque rupture—Pathologic basis of plaque stability and instability. Cardiovasc. Res. 1999, 41, 334–344. [Google Scholar] [CrossRef]
- Jones, M.L.; Martoni, C.J.; Ganopolsky, J.G.; Labbé, A.; Prakash, S. The human microbiome and bile acid metabolism: Dysbiosis, dysmetabolism, disease and intervention. Expert Opin. Biol. Ther. 2014, 14, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Martoni, C.J.; Parent, M.; Prakash, S. Cholesterol-lowering efficacy of a microencapsulated bile salt hydrolase—Active Lactobacillus reuteri NCIMB 30242 yoghurt formulation in hypercholesterolaemic adults. Br. J. Nutr. 2012, 107, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.I.A.; Gibson, G.R. Effects of consumption of probiotics and prebiotics on serum lipid levels in humans. Crit. Rev. Biochem. Mol. Biol. 2002, 37, 259–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; DuGar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Menzel, T.; Lührs, H.; Zirlik, S.; Schauber, J.; Kudlich, T.; Gerke, T.; Gostner, A.; Neumann, M.; Melcher, R.; Scheppach, W. Butyrate inhibits leukocyte adhesion to endothelial cells via modulation of VCAM-1. Inflamm. Bowel Dis. 2004, 10, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Segain, J.; de la Bletiere, D.R.; Bourreille, A.; Leray, V.; Gervois, N.; Rosales, C.; Ferrier, L.; Bonnet, C.; Blottiere, H.; Galmiche, J. Butyrate inhibits inflammatory responses through NFκB inhibition: Implications for Crohn’s disease. Gut 2000, 47, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.; Brar, M.; Kirjavainen, P.; Chen, Y.; Peng, J.; Li, D.; Leung, F.C.; El Nezami, H. High fat diet induced atherosclerosis is accompanied with low colonic bacterial diversity and altered abundances that correlates with plaque size, plasma A-FABP and cholesterol: a pilot study of high fat diet and its intervention with Lactobacillus rhamnosus GG (LGG) or telmisartan in ApoE−/− mice. BMC Microbiol. 2016, 16. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed]
- Savoia, C.; Schiffrin, E.L. Inflammation in hypertension. Curr. Opin. Nephrol. Hypertens. 2006, 15, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, T.F.; Barton, M. Biology of the endothelium. Clin. Cardiol. 1997, 20, II-3-10. [Google Scholar] [PubMed]
- Subah Packer, C. Estrogen protection, oxidized LDL, endothelial dysfunction and vasorelaxation in cardiovascular disease: New insights into a complex issue. Cardiovasc. Res. 2007, 73, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.M. Secondary endothelial dysfunction: Hypertension and heart failure. J. Mol. Cell. Cardiol. 1999, 31, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Peluso, I.; Morabito, G.; Urban, L.; Ioannone, F.; Serafini, M. Oxidative stress in atherosclerosis development: the central role of LDL and oxidative burst. Endocr. Metab. Immune Disord. Drug Targets 2012, 12, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.X.; Zhou, B.; Chen, Z.; Ren, Q.; Lu, S.H.; Sawamura, T.; Han, Z.C. Oxidized low density lipoprotein impairs endothelial progenitor cells by regulation of endothelial nitric oxide synthase. J. Lipid Res. 2006, 47, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.; Lüscher, T.F. Release of endothelin from the porcine aorta. Inhibition by endothelium-derived nitric oxide. J. Clin. Investig. 1990, 85, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Yeoh, B.S.; Vijay-Kumar, M. Gut microbiome as a novel cardiovascular therapeutic target. Curr. Opin. Pharmacol. 2016, 27, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Roberfroid, M. Prebiotics: the concept revisited. J. Nutr. 2007, 137, 830S–837S. [Google Scholar] [PubMed]
- Delzenne, N.M.; Neyrinck, A.M.; Bäckhed, F.; Cani, P.D. Targeting gut microbiota in obesity: Effects of prebiotics and probiotics. Nat. Rev. Endocrinol. 2011, 7, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Koutsos, A.; Tuohy, K.M.; Lovegrove, J.A. Apples and Cardiovascular Health—Is the Gut Microbiota a Core Consideration? Nutrients 2015, 7, 3959–3998. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, K.; Ohashi, Y.; Kawasumi, K.; Terada, A.; Fujisawa, T. Effect of apple intake on fecal microbiota and metabolites in humans. Anaerobe 2010, 16, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Pietro Femia, A.; Luceri, C.; Bianchini, F.; Salvadori, M.; Salvianti, F.; Pinzani, P.; Dolara, P.; Calorini, L.; Caderni, G. Marie Ménard apples with high polyphenol content and a low-fat diet reduce 1,2-dimethylhydrazine-induced colon carcinogenesis in rats: Effects on inflammation and apoptosis. Mol. Nutr. Food Res. 2012, 56, 1353–1357. [Google Scholar] [CrossRef] [PubMed]
- Aprikian, O.; Duclos, V.; Guyot, S.; Besson, C.; Manach, C.; Bernalier, A.; Morand, C.; Rémésy, C.; Demigné, C. Apple pectin and a polyphenol-rich apple concentrate are more effective together than separately on cecal fermentations and plasma lipids in rats. J. Nutr. 2003, 133, 1860–1865. [Google Scholar] [PubMed]
- Watzl, B.; Girrbach, S.; Roller, M. Inulin, oligofructose and immunomodulation. Br. J. Nutr. 2005, 93, S49–S55. [Google Scholar] [CrossRef] [PubMed]
- Pokusaeva, K.; Fitzgerald, G.F.; van Sinderen, D. Carbohydrate metabolism in Bifidobacteria. Genes Nutr. 2011, 6, 285–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xiao, G.; Yao, Y.; Guo, S.; Lu, K.; Sheng, Z. The role of Bifidobacteria in gut barrier function after thermal injury in rats. J. Trauma 2006, 61, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.E. Probiotics: Definition, sources, selection, and uses. Clin. Infect. Dis. 2008, 46, S58–S61, discussion S144–S151. [Google Scholar] [CrossRef] [PubMed]
- Kailasapathy, K.; Chin, J. Survival and therapeutic potential of probiotic organisms with reference to Lactobacillus acidophilus and Bifidobacterium spp. Immunol. Cell Biol. 2000, 78, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, M.C.; Lajo, T.; Carrión, J.M.; Cuñé, J. Cholesterol-lowering efficacy of Lactobacillus plantarum CECT 7527, 7528 and 7529 in hypercholesterolaemic adults. Br. J. Nutr. 2013, 109, 1866–1872. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Hill, C.; Gahan, C.G.M. Bile Salt Hydrolase Activity in Probiotics. Appl. Environ. Microbiol. 2006, 72, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Liu, X.M.; Zhang, Q.X.; Shen, Z.; Tian, F.W.; Zhang, H.; Sun, Z.H.; Zhang, H.P.; Chen, W. Influence of consumption of probiotics on the plasma lipid profile: A meta-analysis of randomised controlled trials. Nutr. Metab. Cardiovasc. Dis. NMCD 2011, 21, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Ishimwe, N.; Daliri, E.B.; Lee, B.H.; Fang, F.; Du, G. The perspective on cholesterol-lowering mechanisms of probiotics. Mol. Nutr. Food Res. 2015, 59, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Martoni, C.J.; Prakash, S. Cholesterol lowering and inhibition of sterol absorption by Lactobacillus reuteri NCIMB 30242: A randomized controlled trial. Eur. J. Clin. Nutr. 2012, 66, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Tuohy, K.M.; Fava, F.; Viola, R. ‘The way to a man’s heart is through his gut microbiota’—Dietary pro- and prebiotics for the management of cardiovascular risk. Proc. Nutr. Soc. 2014, 73, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.; Rivière, A.; Moens, F.; Van den Abbeele, P.; Geirnaert, A.; Rogelj, I.; Leroy, F.; De Vuyst, L. Inulin-type fructan fermentation by Bifidobacteria depends on the strain rather than the species and region in the human intestine. Appl. Microbiol. Biotechnol. 2016, 100, 4097–4107. [Google Scholar] [CrossRef] [PubMed]
- Dewulf, E.M.; Cani, P.D.; Claus, S.P.; Fuentes, S.; Puylaert, P.G.B.; Neyrinck, A.M.; Bindels, L.B.; de Vos, W.M.; Gibson, G.R.; Thissen, J. Insight into the prebiotic concept: Lessons from an exploratory, double blind intervention study with inulin-type fructans in obese women. Gut 2013, 62, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.P.; Martin, J.C.; Duncan, S.H.; Flint, H.J. Prebiotic stimulation of human colonic butyrate-producing bacteria and Bifidobacteria, in vitro. FEMS Microbiol. Ecol. 2014, 87, 30–40. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment: | Prebiotics | Probiotics |
|---|---|---|
| Definition: | Dietary constituents that fertilize and promote healthy gut microbiota composition | Beneficial live microorganisms that can colonize the human gut to develop or restore healthy gut microbiota composition |
| Examples and Effects: | Plant polyphenols Fruits and vegetables (e.g., apples): reduce inflammation [76] and total cholesterol levels [74,77], promote bifidobacteria growth [75] Dietary fructans Foods high in inulin and/or oligofructose: promote bifidobacteria growth [90], restore butyrate-producing bacterial populations [91,92] | Lactobaccillus strains L. reuteri (microencapsulated in yogurt): reduce LDL-cholesterol, serum total cholesterol, and non-HDL cholesterol [88] L. plantarum (capsules): reduce serum total cholesterol [84] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lau, K.; Srivatsav, V.; Rizwan, A.; Nashed, A.; Liu, R.; Shen, R.; Akhtar, M. Bridging the Gap between Gut Microbial Dysbiosis and Cardiovascular Diseases. Nutrients 2017, 9, 859. https://doi.org/10.3390/nu9080859
Lau K, Srivatsav V, Rizwan A, Nashed A, Liu R, Shen R, Akhtar M. Bridging the Gap between Gut Microbial Dysbiosis and Cardiovascular Diseases. Nutrients. 2017; 9(8):859. https://doi.org/10.3390/nu9080859
Chicago/Turabian StyleLau, Kimberley, Varun Srivatsav, Ayesha Rizwan, Andrew Nashed, Rui Liu, Rui Shen, and Mahmood Akhtar. 2017. "Bridging the Gap between Gut Microbial Dysbiosis and Cardiovascular Diseases" Nutrients 9, no. 8: 859. https://doi.org/10.3390/nu9080859
APA StyleLau, K., Srivatsav, V., Rizwan, A., Nashed, A., Liu, R., Shen, R., & Akhtar, M. (2017). Bridging the Gap between Gut Microbial Dysbiosis and Cardiovascular Diseases. Nutrients, 9(8), 859. https://doi.org/10.3390/nu9080859