Vitamin C Deficiency Reduces Muscarinic Receptor Coronary Artery Vasoconstriction and Plasma Tetrahydrobiopterin Concentration in Guinea Pigs



Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Study
2.2. Euthanasia
2.3. Wire Myography and Tissue Preparation
2.4. Biochemical Analysis
2.5. Data and Statistical Analysis
2.6. Materials
3. Results
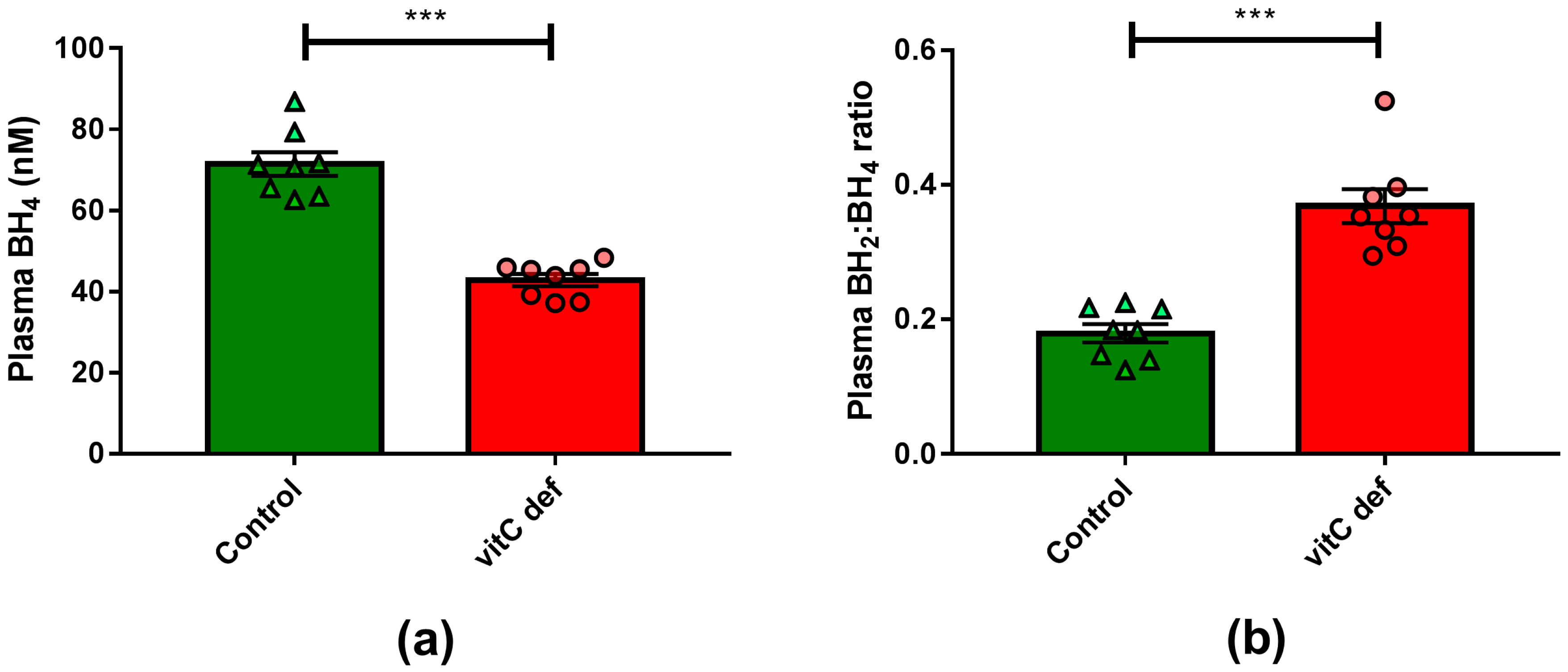
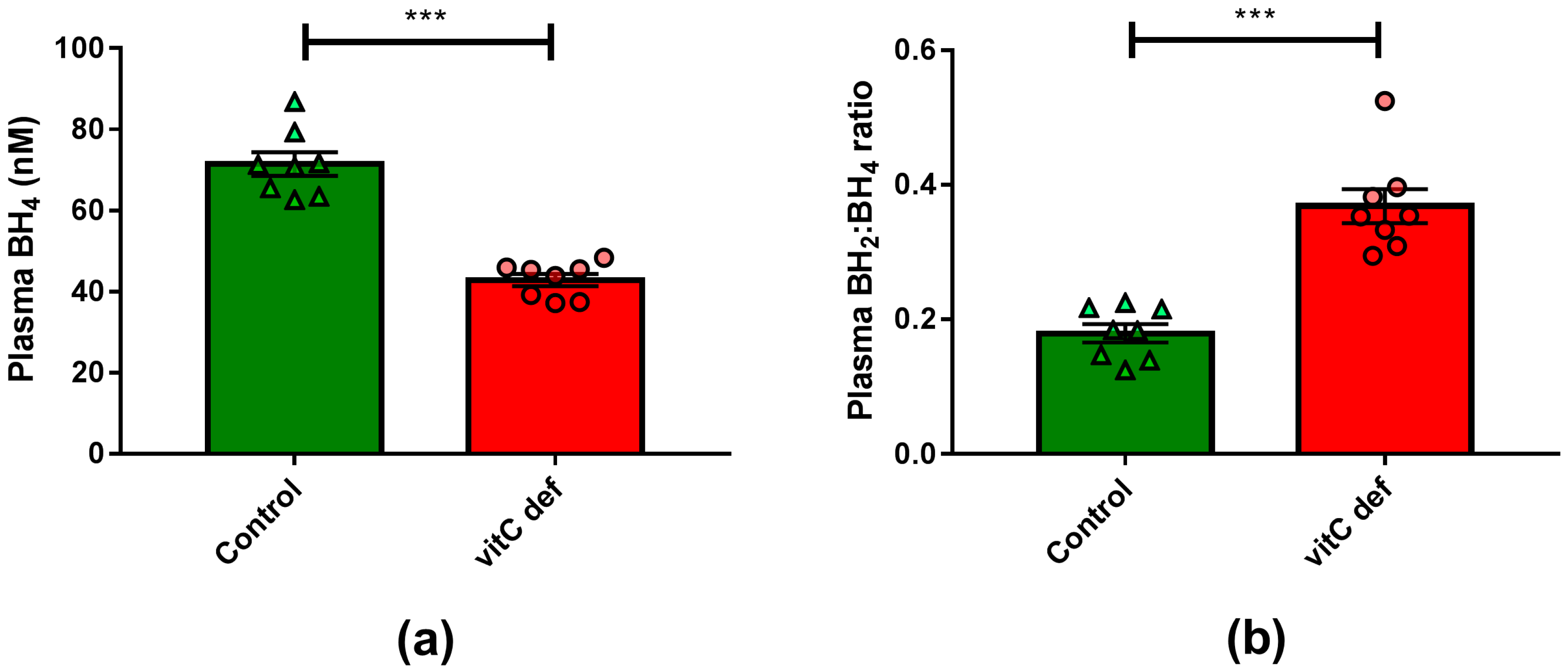
3.1. Effects of Vitamin C Deficiency on Weight Gain of Animals, Plasma Ascorbate Concentration and Plasma BH4 Concentration
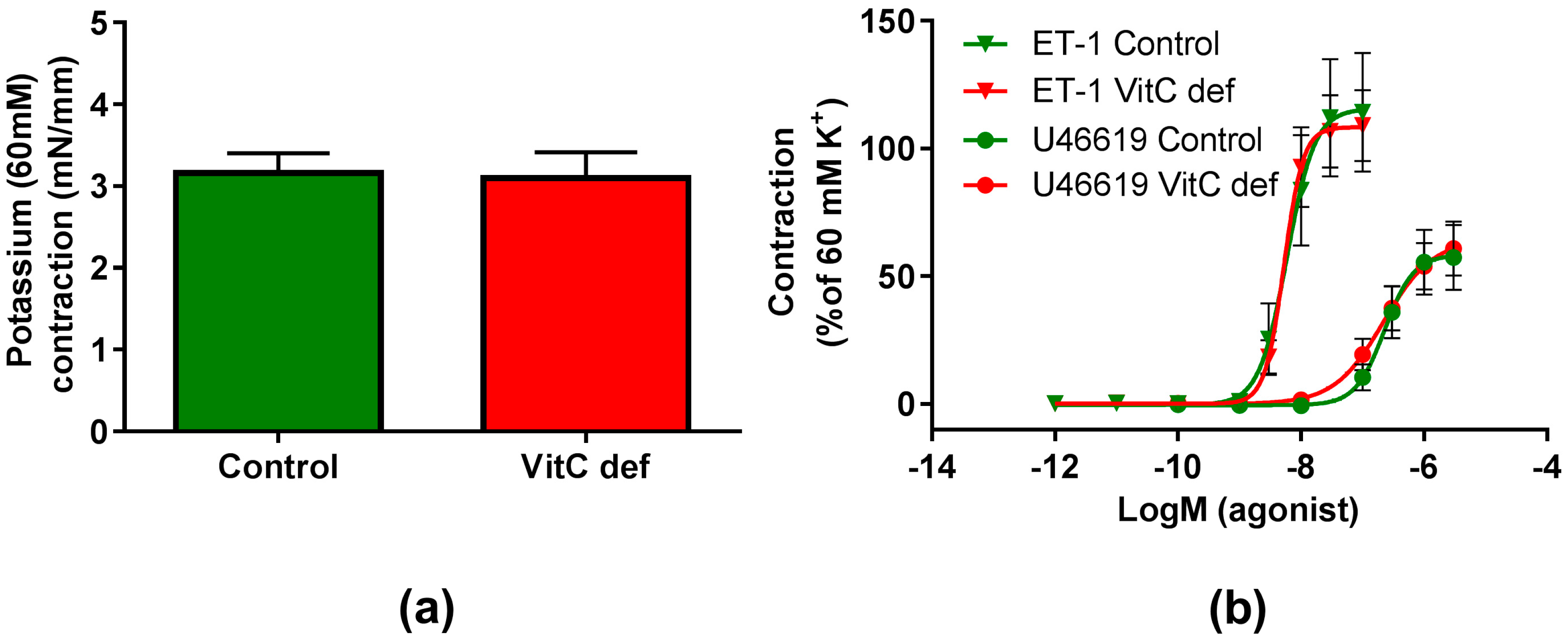
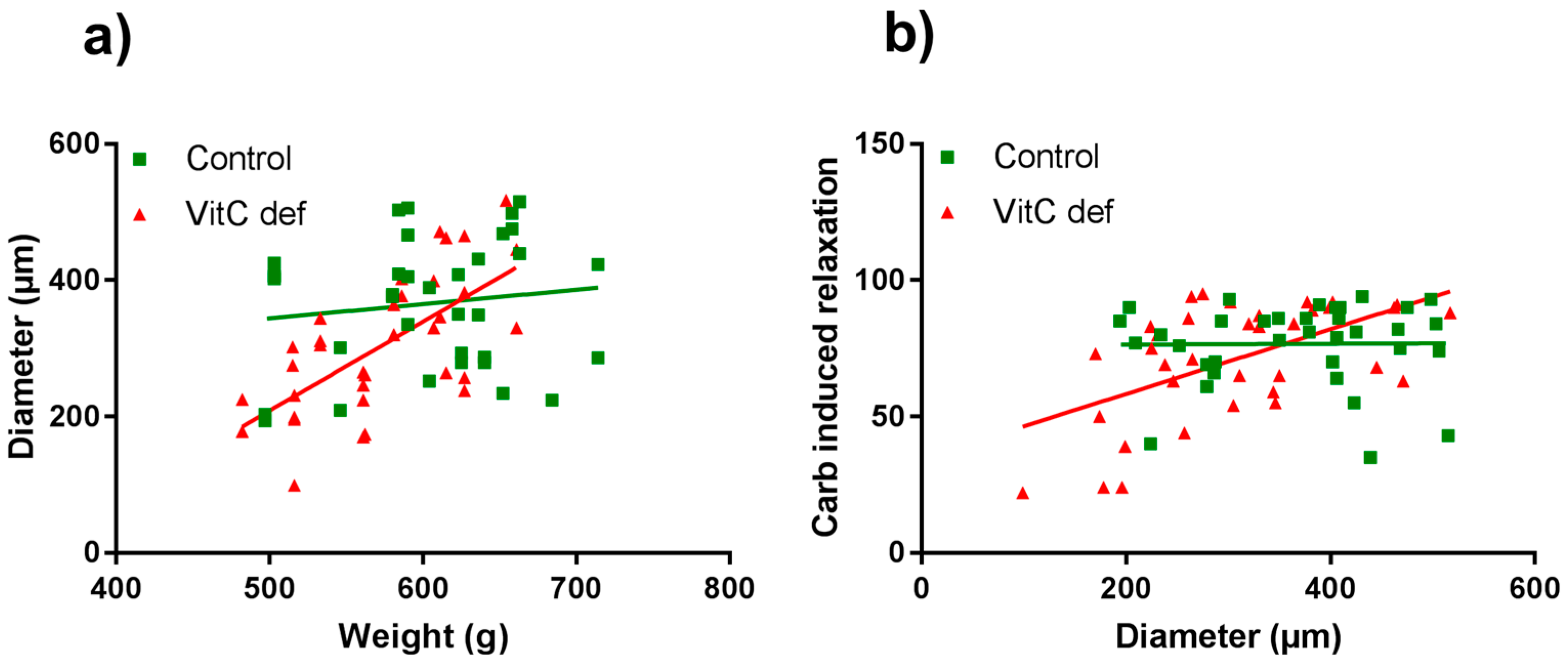
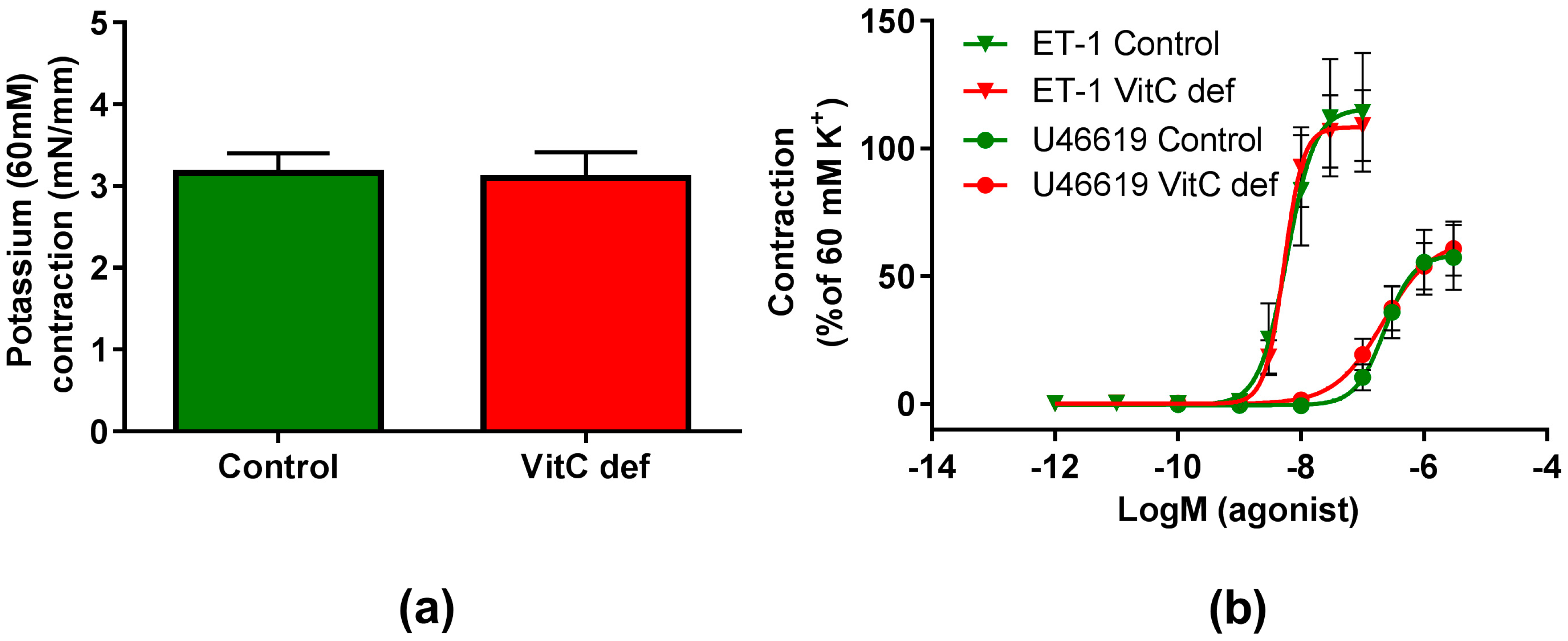
3.2. Contractile Reactivity
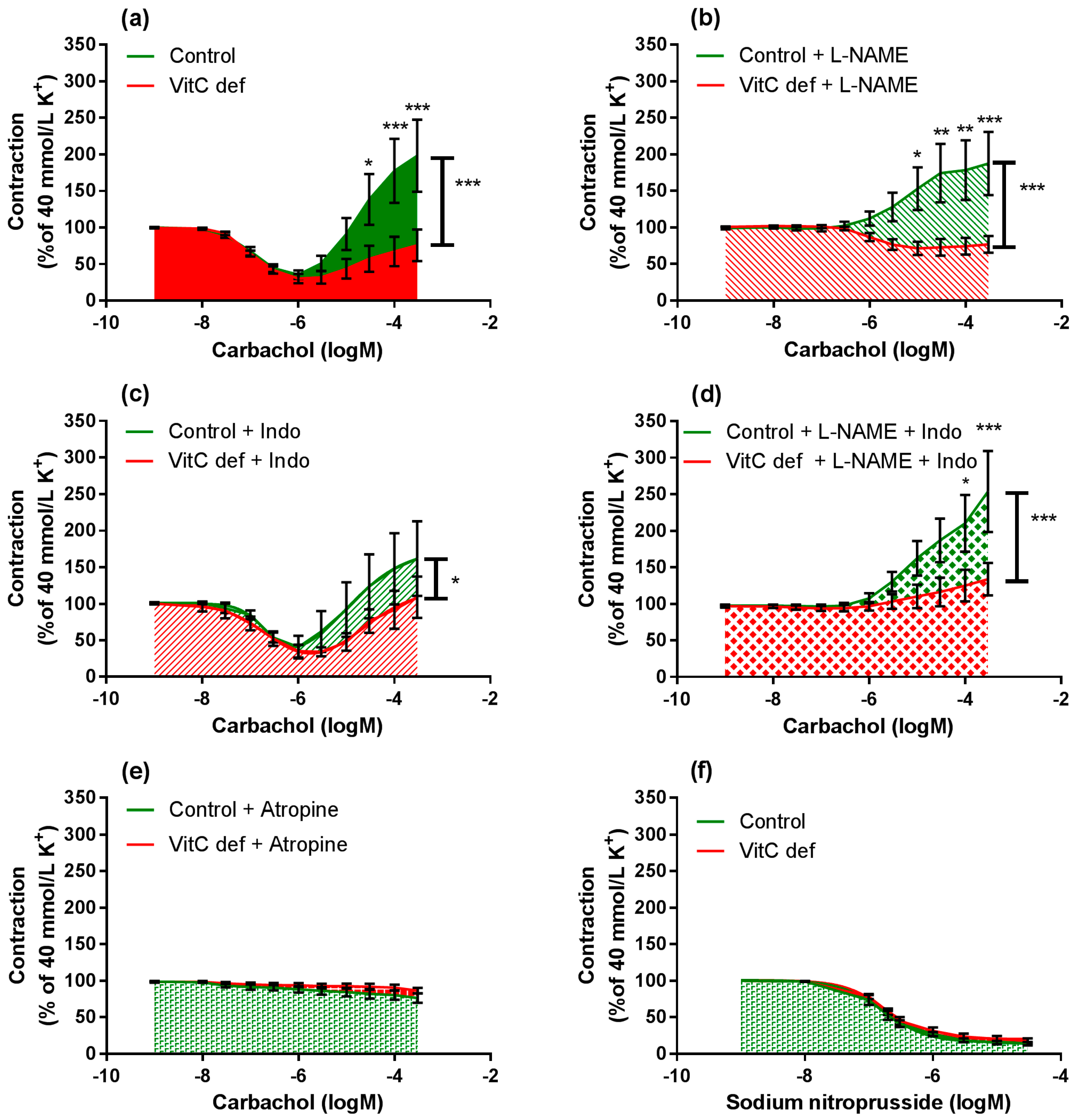
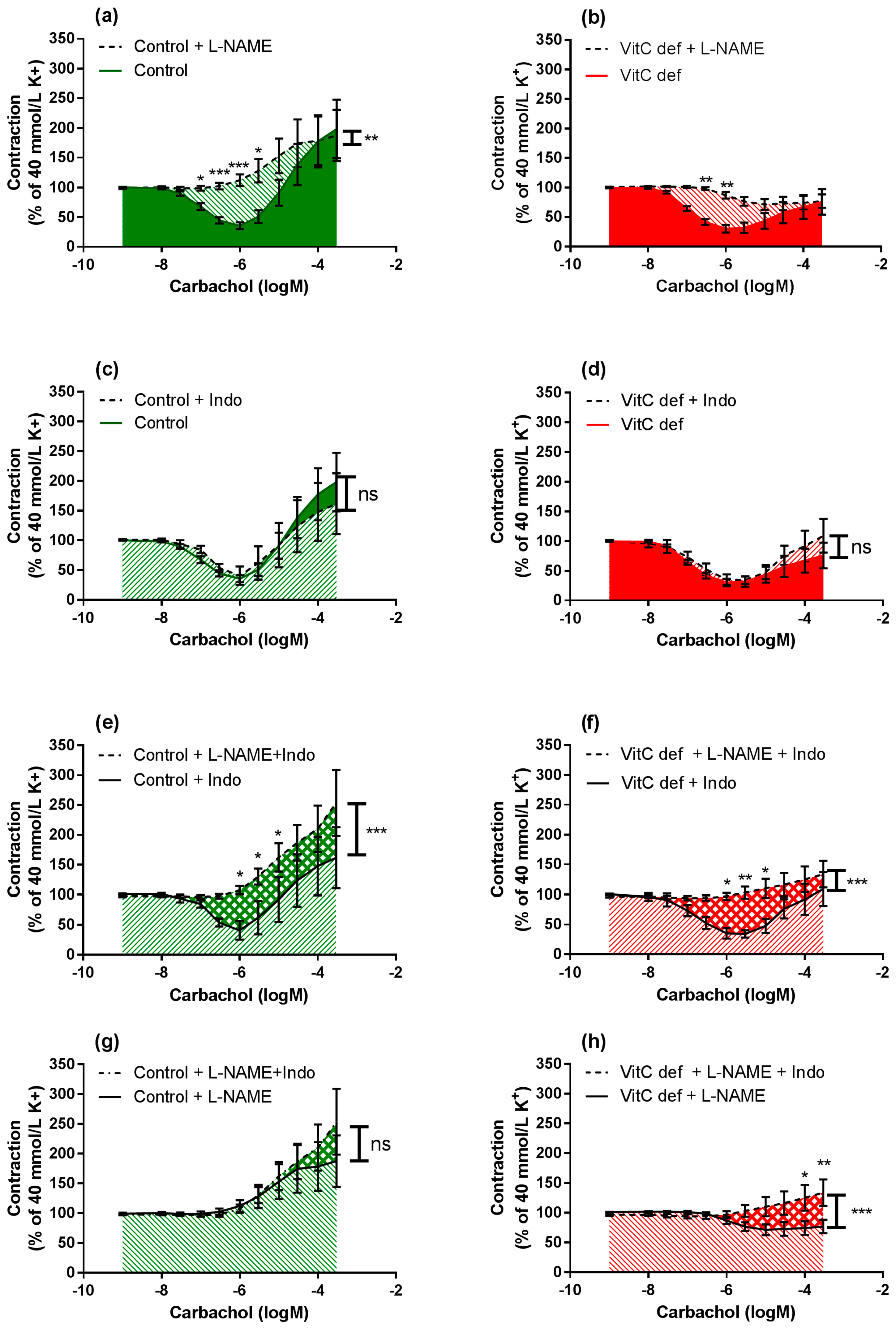
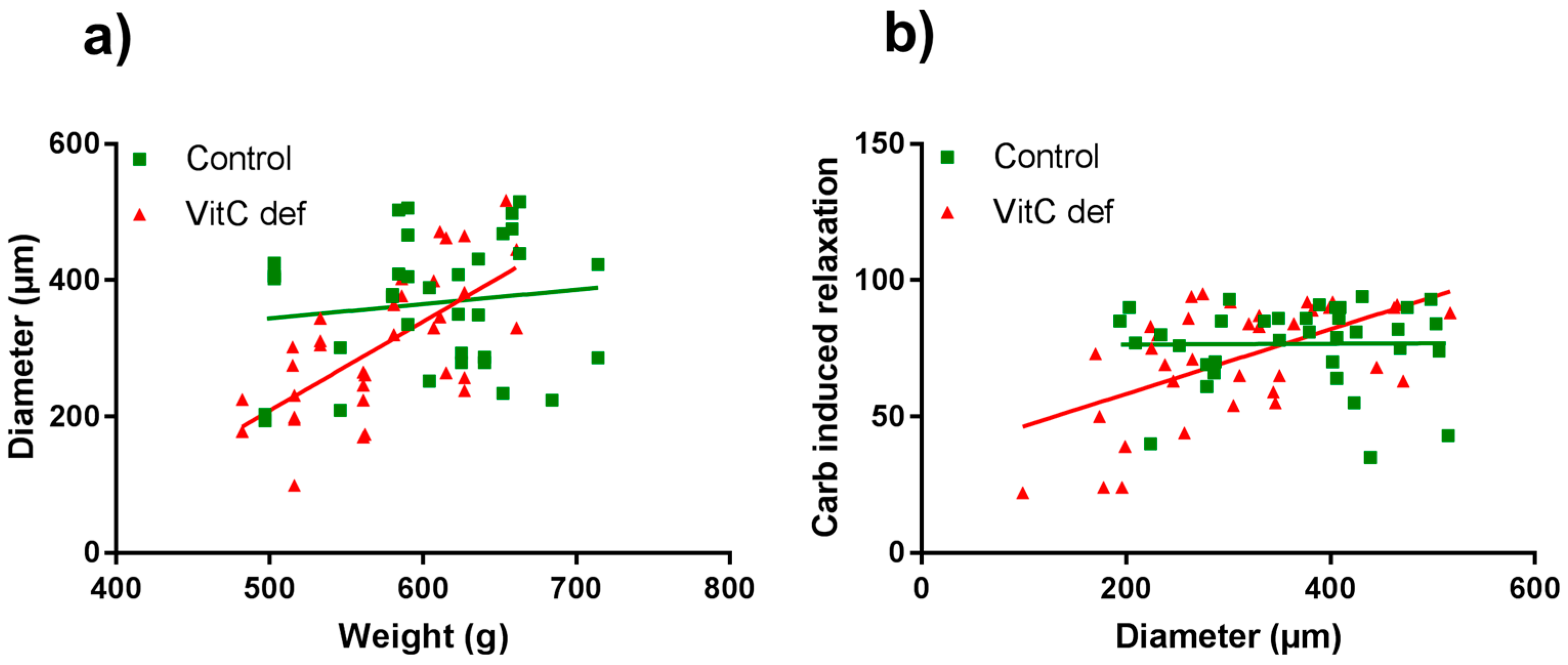
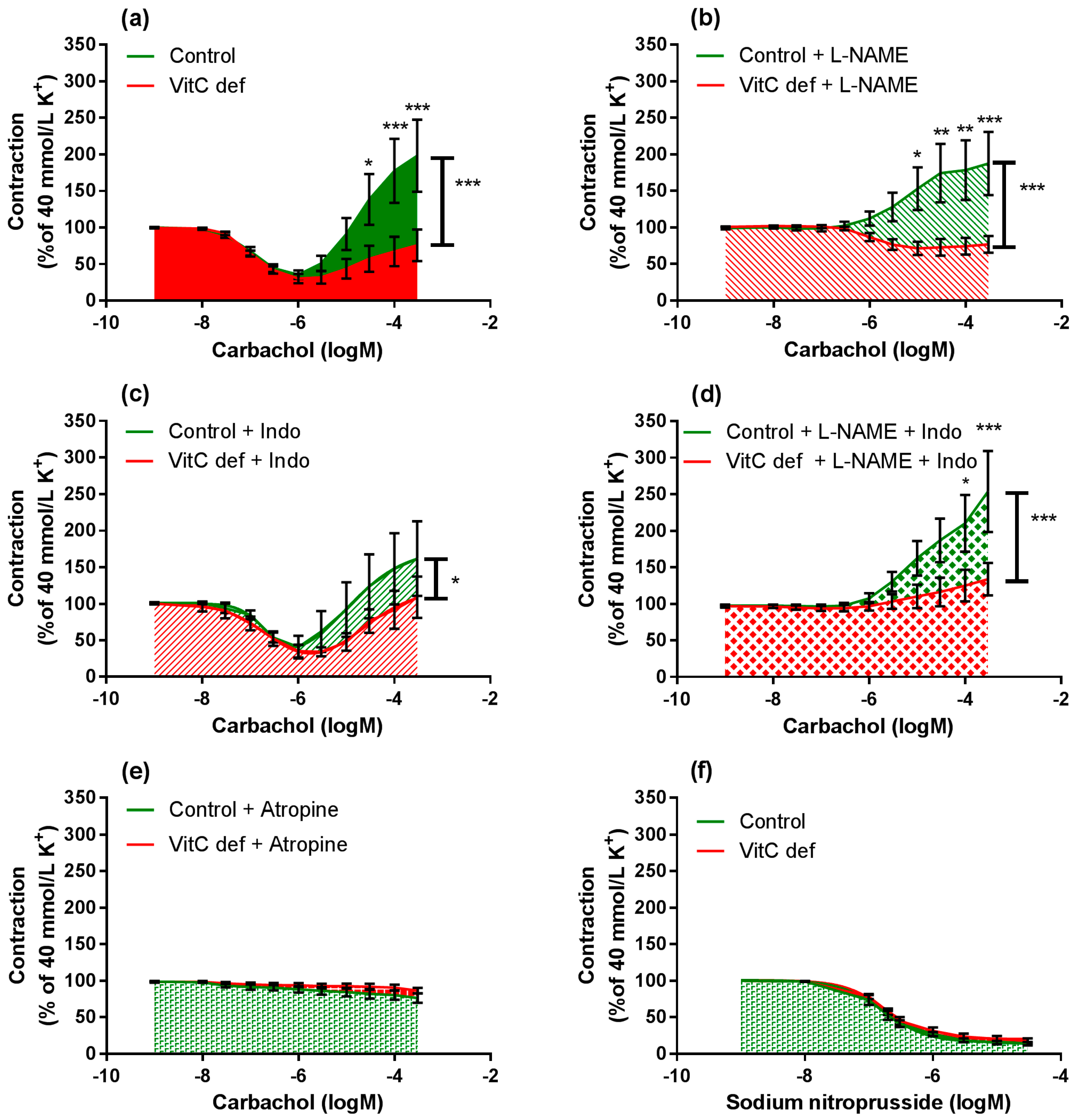
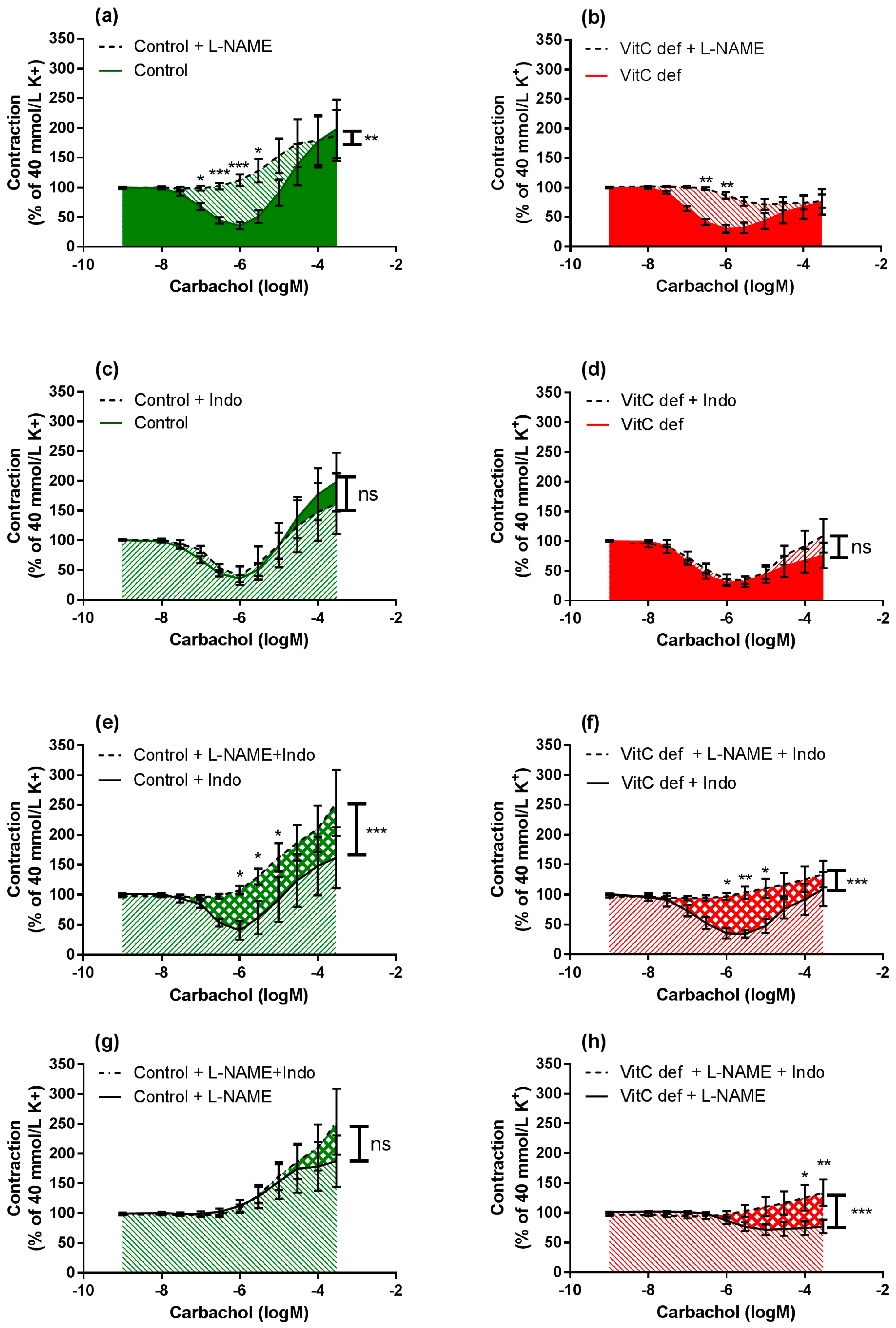
3.3. Vascular Responses to Carbachol
3.4. Relaxing Responses to SNP
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Myint, P.K.; Luben, R.N.; Wareham, N.J.; Khaw, K.-T. Association Between Plasma Vitamin C Concentrations and Blood Pressure in the European Prospective Investigation Into Cancer-Norfolk Population-Based Study. Hypertension 2011, 58, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Myint, P.K.; Luben, R.N.; Welch, A.A.; Bingham, S.A.; Wareham, N.J.; Khaw, K.T. Plasma vitamin C concentrations predict risk of incident stroke over 10 years in 20,649 participants of the European Prospective Investigation into Cancer Norfolk prospective population study. Am. J. Clin. Nutr. 2008, 87, 64–69. [Google Scholar] [PubMed]
- NyyssÖnen, K.; Parviainen, M.T.; Salonen, R.; Tuomilehto, J.; Salonen, J.T. Vitamin C deficiency and risk of myocardial infarction: Prospective population study of men from eastern Finland. BMJ 1997, 314, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Pfister, R.; Michels, G.; Bragelmann, J.; Sharp, S.J.; Luben, R.; Wareham, N.J.; Khaw, K.T. Plasma vitamin C and risk of hospitalisation with diagnosis of atrial fibrillation in men and women in EPIC-Norfolk prospective study. Int. J. Cardiol. 2014, 177, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Pfister, R.; Sharp, S.J.; Luben, R.; Wareham, N.J.; Khaw, K.T. Plasma vitamin C predicts incident heart failure in men and women in European Prospective Investigation into Cancer and Nutrition-Norfolk prospective study. Am. Heart J. 2011, 162, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Wannamethee, S.G.; Bruckdorfer, K.R.; Shaper, A.G.; Papacosta, O.; Lennon, L.; Whincup, P.H. Plasma Vitamin C, but Not Vitamin E, Is Associated With Reduced Risk of Heart Failure in Older Men. Circ. Heart Fail. 2013, 6, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, R.L.; Carroll, M.D.; Ford, E.S.; Lacher, D.A. Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am. J. Clin. Nutr. 2009, 90, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Heller, R.; Unbehaun, A.; Schellenberg, B.; Mayer, B.; Werner-Felmayer, G.; Werner, E.R. L-ascorbic acid potentiates endothelial nitric oxide synthesis via a chemical stabilization of tetrahydrobiopterin. J. Biol. Chem. 2001, 276, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, A.; Lykkesfeldt, J. Does vitamin C enhance nitric oxide bioavailability in a tetrahydrobiopterin-dependent manner? In vitro, in vivo and clinical studies. Nitric Oxide Biol. Chem. Off. J. Nitric Oxide Soc. 2014, 36, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.A.; Milstien, S.; Katusic, Z.S. Effect of vitamin C on the availability of tetrahydrobiopterin in human endothelial cells. J. Cardiovasc. Pharmacol. 2001, 37, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Tejero, J.; Stuehr, D. Tetrahydrobiopterin in nitric oxide synthase. IUBMB Life 2013, 65, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Vivar, J.; Martasek, P.; Whitsett, J.; Joseph, J.; Kalyanaraman, B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: An EPR spin trapping study. Biochem. J. 2002, 362, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, A.; Hasselholt, S.; Tveden-Nyborg, P.; Lykkesfeldt, J. Guinea pig ascorbate status predicts tetrahydrobiopterin plasma concentration and oxidation ratio in vivo. Nutr. Res. 2013, 33, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Touyz, R.M.; Park, J.B.; Schiffrin, E.L. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR. Hypertension 2001, 38, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Griendling, K.K. Redox signaling, vascular function, and hypertension. Antioxid. Redox Signal. 2008, 10, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H. Peroxynitrite and protein tyrosine nitration of prostacyclin synthase. Prostaglandins Lipid Mediat. 2007, 82, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Li, H.; He, C.; Lin, M.; Lyons, T.J.; Xie, Z. Tyrosine nitration of prostacyclin synthase is associated with enhanced retinal cell apoptosis in diabetes. Am. J. Pathol. 2011, 179, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Frei, B.; Stocker, R.; England, L.; Ames, B.N. Ascorbate: The most effective antioxidant in human blood plasma. Adv. Exp. Med. Biol. 1990, 264, 155–163. [Google Scholar] [PubMed]
- De Jongh, R.; Haenen, G.R.; van Koeveringe, G.A.; Dambros, M.; De Mey, J.G.; van Kerrebroeck, P.E. Oxidative stress reduces the muscarinic receptor function in the urinary bladder. Neurourol. Urodyn. 2007, 26, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Sawiris, P.; Chanaud, N.; Enwonwu, C.O. Impaired inositol trisphosphate generation in carbachol-stimulated submandibular gland acinar cells from ascorbate deficient guinea pigs. J. Nutr. Biochem. 1995, 6, 557–563. [Google Scholar] [CrossRef]
- Sawiris, P.G.; Enwonwu, C.O. Ascorbate deficiency impairs the muscarinic-cholinergic and ss-adrenergic receptor signaling systems in the guinea pig submandibular salivary gland. J. Nutr. 2000, 130, 2876–2882. [Google Scholar] [PubMed]
- Gulbenkian, S.; Edvinsson, L.; Saetrum Opgaard, O.; Valenca, A.; Wharton, J.; Polak, J.M. Neuropeptide Y modulates the action of vasodilator agents in guinea-pig epicardial coronary arteries. Regul. Pept. 1992, 40, 351–362. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Ascorbate and dehydroascorbic acid as reliable biomarkers of oxidative stress: Analytical reproducibility and long-term stability of plasma samples subjected to acidic deproteinization. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2513–2516. [Google Scholar] [CrossRef] [PubMed]
- DMT Normalization Guide. Available online: https://www.dmt.dk/uploads/6/5/6/8/65689239/dmt_normalization_guide.pdf (accessed on 22 June 2017).
- Brunner, F.; Kuhberger, E.; Groschner, K.; Poch, G.; Kukovetz, W.R. Characterization of muscarinic receptors mediating endothelium-dependent relaxation of bovine coronary artery. Eur. J. Pharmacol. 1991, 200, 25–33. [Google Scholar] [CrossRef]
- Ren, L.M.; Nakane, T.; Chiba, S. Muscarinic receptor subtypes mediating vasodilation and vasoconstriction in isolated, perfused simian coronary arteries. J. Cardiovasc. Pharmacol. 1993, 22, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Lamping, K.G.; Wess, J.; Cui, Y.; Nuno, D.W.; Faraci, F.M. Muscarinic (M) receptors in coronary circulation: Gene-targeted mice define the role of M2 and M3 receptors in response to acetylcholine. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Brunner, F.; Kuhberger, E.; Schloos, J.; Kukovetz, W.R. Characterization of muscarinic receptors of bovine coronary artery by functional and radioligand binding studies. Eur. J. Pharmacol. 1991, 196, 247–255. [Google Scholar] [CrossRef]
- Duckles, S.P.; Garcia-Villalon, A.L. Characterization of vascular muscarinic receptors: Rabbit ear artery and bovine coronary artery. J. Pharmacol. Exp. Ther. 1990, 253, 608–613. [Google Scholar] [PubMed]
- Entzeroth, M.; Doods, H.N.; Mayer, N. Characterization of porcine coronary muscarinic receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1990, 341, 432–438. [Google Scholar] [CrossRef]
- Van Charldorp, K.J.; van Zwieten, P.A. Comparison of the muscarinic receptors in the coronary artery, cerebral artery and atrium of the pig. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1989, 339, 403–408. [Google Scholar] [CrossRef]
- Findlay, G.M. The Effects of an Unbalanced Diet in the Production of Guinea-pig Scurvy. Biochem. J. 1921, 15, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Karaki, H.; Urakawa, N.; Kutsky, P. Potassium-induced contraction in smooth muscle. Nihon Heikatsukin Gakkai Zasshi 1984, 20, 427–444. [Google Scholar] [CrossRef] [PubMed]
- Mombouli, J.V.; Illiano, S.; Nagao, T.; Scott-Burden, T.; Vanhoutte, P.M. Potentiation of endothelium-dependent relaxations to bradykinin by angiotensin I converting enzyme inhibitors in canine coronary artery involves both endothelium-derived relaxing and hyperpolarizing factors. Circ. Res. 1992, 71, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Wada, K.; Kamisaki, Y.; Miyagawa, I. Effect of ischemia-reperfusion on contractile function of rat urinary bladder: Possible role of nitric oxide. Life Sci. 1998, 62, PL149–PL156. [Google Scholar] [CrossRef]
- Hisadome, Y.; Saito, M.; Kono, T.; Satoh, I.; Kinoshita, Y.; Satoh, K. Beneficial effect of preconditioning on ischemia-reperfusion injury in the rat bladder in vivo. Life Sci. 2007, 81, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Kots, A.Y.; Martin, E.; Sharina, I.G.; Murad, F. A short history of cGMP, guanylyl cyclases, and cGMP-dependent protein kinases. Handb. Exp. Pharmacol. 2009, 191, 1–14. [Google Scholar]
- Priviero, F.B.; Zemse, S.M.; Teixeira, C.E.; Webb, R.C. Oxidative stress impairs vasorelaxation induced by the soluble guanylyl cyclase activator BAY 41-2272 in spontaneously hypertensive rats. Am. J. Hypertens. 2009, 22, 493–499. [Google Scholar] [CrossRef] [PubMed]
- De Beer, V.J.; Taverne, Y.J.; Kuster, D.W.; Najafi, A.; Duncker, D.J.; Merkus, D. Prostanoids suppress the coronary vasoconstrictor influence of endothelin after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1080–H1089. [Google Scholar] [CrossRef] [PubMed]
- Hori, M.; Nishida, K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc. Res. 2009, 81, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Edlund, A.; Sollevi, A.; Wennmalm, A. The role of adenosine and prostacyclin in coronary flow regulation in healthy man. Acta Physiol. Scand. 1989, 135, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.Z.; Bache, R.J. Effect of indomethacin on coronary blood flow during graded treadmill exercise in the dog. Am. J. Physiol. 1984, 247, H452–H458. [Google Scholar] [PubMed]
- Lykkesfeldt, J.; Poulsen, H.E. Is vitamin C supplementation beneficial? Lessons learned from randomised controlled trials. Br. J. Nutr. 2010, 103, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Frei, B.; Birlouez-Aragon, I.; Lykkesfeldt, J. Authors’ perspective: What is the optimum intake of vitamin C in humans? Crit. Rev. Food Sci. Nutr. 2012, 52, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Frikke-Schmidt, H.; Lykkesfeldt, J. Role of marginal vitamin C deficiency in atherogenesis: In vivo models and clinical studies. Basic Clin. Pharmacol. Toxicol. 2009, 104, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency increase lifestyle-associated vascular disease progression? Evidence based on experimental and clinical studies. Antioxid. Redox Signal. 2013, 19, 2084–2104. [Google Scholar] [CrossRef] [PubMed]
- Dart, A.M.; Du, X.J.; Kingwell, B.A. Gender, sex hormones and autonomic nervous control of the cardiovascular system. Cardiovasc. Res. 2002, 53, 678–687. [Google Scholar] [CrossRef]
- Niihashi, M.; Esumi, M.; Kusumi, Y.; Sato, Y.; Sakurai, I. Expression of muscarinic receptor genes in the human coronary artery. Angiology 2000, 51, 295–300. [Google Scholar] [PubMed]
- Ludmer, P.L.; Selwyn, A.P.; Shook, T.L.; Wayne, R.R.; Mudge, G.H.; Alexander, R.W.; Ganz, P. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N. Engl. J. Med. 1986, 315, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Yasue, H.; Horio, Y.; Nakamura, N.; Fujii, H.; Imoto, N.; Sonoda, R.; Kugiyama, K.; Obata, K.; Morikami, Y.; Kimura, T. Induction of coronary artery spasm by acetylcholine in patients with variant angina: Possible role of the parasympathetic nervous system in the pathogenesis of coronary artery spasm. Circulation 1986, 74, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.P.; Mosfer, S.I.; Lang, D.; Donaldson, F.; Lewis, M.J. Vitamin C and quinapril abrogate LVH and endothelial dysfunction in aortic-banded guinea pigs. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1704–H1710. [Google Scholar] [PubMed]
- LeBlanc, A.J.; Hoying, J.B. Adaptation of the Coronary Microcirculation in Aging. Microcirculation 2016, 23, 157–167. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Controls | N | VitC Deficient | |
|---|---|---|---|---|
| Weight (g) | 16 | 625 ± 14 | 16 | 602 ± 16 |
| Ascorbate concentration, (µM) | 16 | 60.2 ± 5.1 | 16 | 2.3 ± 0.1 **** |
| Ascorbate total, (µM) | 16 | 61.5 ± 5.3 | 16 | 2.3 ± 0.1 **** |
| % DHA | 16 | 1.9 ± 0.5 | 16 | 2.2 ± 1.0 |
| N, n | Controls | N, n | VitC Deficient | |
|---|---|---|---|---|
| Diameter | 16, 35 | 366 ± 16 | 16, 35 | 312 ± 12 * |
| Potassium, tension | 16, 35 | 3.2 ± 0.2 | 16, 35 | 3.1 ± 0.3 |
| Endothelin-1 | 12, 17 | Emax 116 ± 16 | 12, 17 | Emax 108 ± 8 |
| pEC50 8.2 ± 0.1 | pEC50 8.3 ± 0.1 | |||
| Sarafotoxin 6c | 8, 20 | Emax NC | 8, 20 | Emax 3 ± 1.6 |
| pEC50 NC | pEC50 8.5 ± 0.7 | |||
| U46619 | 16, 35 | Emax 58 ± 9 | 16, 35 | Emax 64 ± 13 |
| pEC50 6.6 ± 0.1 | pEC50 6.7 ± 0.2 |
| Controls | VitC Deficient | |||||
|---|---|---|---|---|---|---|
| N, n | Pearson r | p Values | N, n | Pearson r | p Values | |
| Diameter vs. weight | 16, 35 | 0.182 | 0.500 | 16, 35 | 0.729 | 0.001 ** |
| Diameter vs. carb induced dilatation | 16, 35 | 0.010 | 0.954 | 16, 35 | 0.555 | 0.0005 *** |
| Diameter vs. carb induced contraction | 16, 35 | −0.174 | 0.518 | 16, 35 | −0.286 | 0.284 |
| BH4 vs. carb induced dilatation | 8, 15 | 0.403 | 0.105 | 8, 15 | 0.057 | 0.840 |
| BH4 vs. carb induced contraction | 8, 15 | −0.372 | 0.172 | 8, 15 | 0.232 | 0.406 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skovsted, G.F.; Tveden-Nyborg, P.; Lindblad, M.M.; Hansen, S.N.; Lykkesfeldt, J. Vitamin C Deficiency Reduces Muscarinic Receptor Coronary Artery Vasoconstriction and Plasma Tetrahydrobiopterin Concentration in Guinea Pigs. Nutrients 2017, 9, 691. https://doi.org/10.3390/nu9070691
Skovsted GF, Tveden-Nyborg P, Lindblad MM, Hansen SN, Lykkesfeldt J. Vitamin C Deficiency Reduces Muscarinic Receptor Coronary Artery Vasoconstriction and Plasma Tetrahydrobiopterin Concentration in Guinea Pigs. Nutrients. 2017; 9(7):691. https://doi.org/10.3390/nu9070691
Chicago/Turabian StyleSkovsted, Gry Freja, Pernille Tveden-Nyborg, Maiken Marie Lindblad, Stine Normann Hansen, and Jens Lykkesfeldt. 2017. "Vitamin C Deficiency Reduces Muscarinic Receptor Coronary Artery Vasoconstriction and Plasma Tetrahydrobiopterin Concentration in Guinea Pigs" Nutrients 9, no. 7: 691. https://doi.org/10.3390/nu9070691
APA StyleSkovsted, G. F., Tveden-Nyborg, P., Lindblad, M. M., Hansen, S. N., & Lykkesfeldt, J. (2017). Vitamin C Deficiency Reduces Muscarinic Receptor Coronary Artery Vasoconstriction and Plasma Tetrahydrobiopterin Concentration in Guinea Pigs. Nutrients, 9(7), 691. https://doi.org/10.3390/nu9070691