Does Vitamin C Influence Neurodegenerative Diseases and Psychiatric Disorders?



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
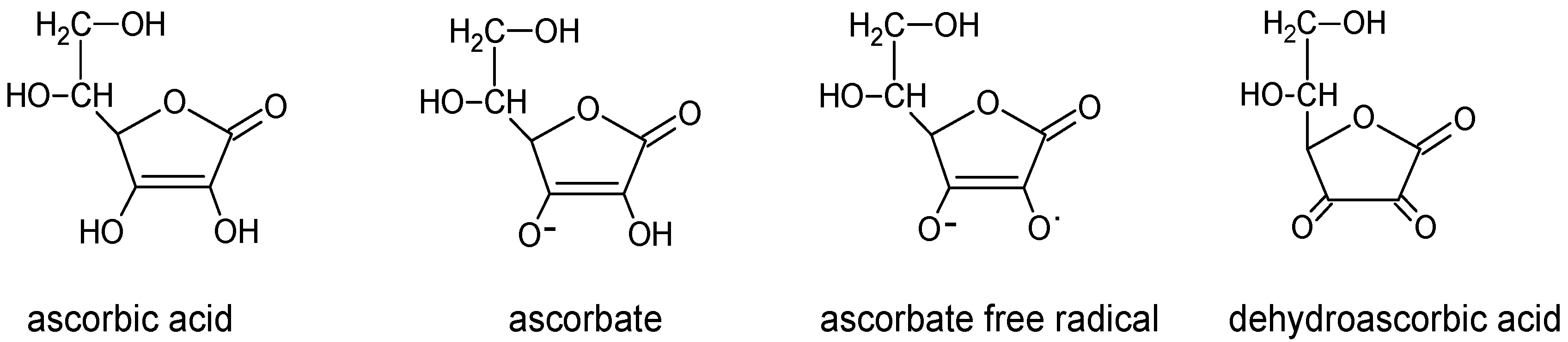
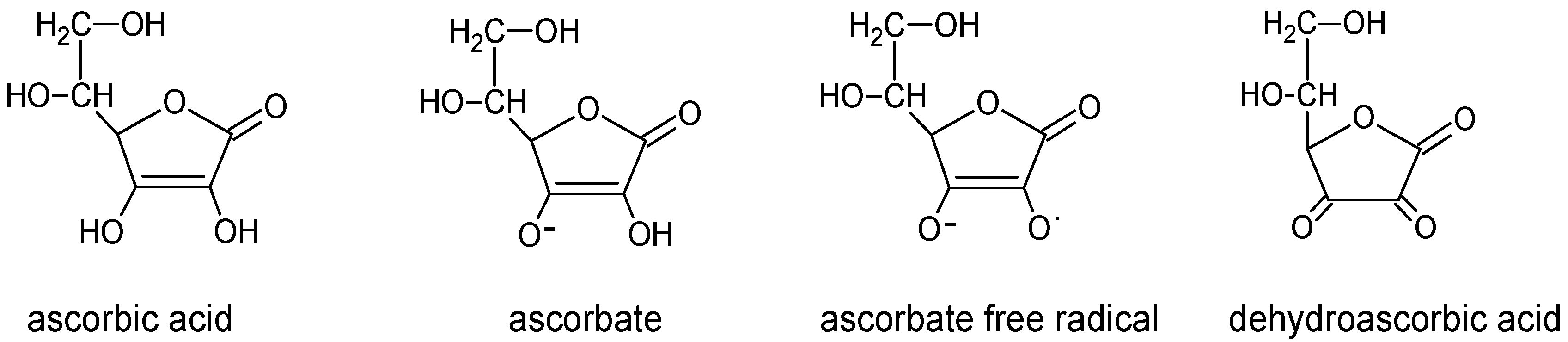
3. Vitamin C Transport Systems and Distribution in the Brain
4. Vitamin C Function in Central Nervous System
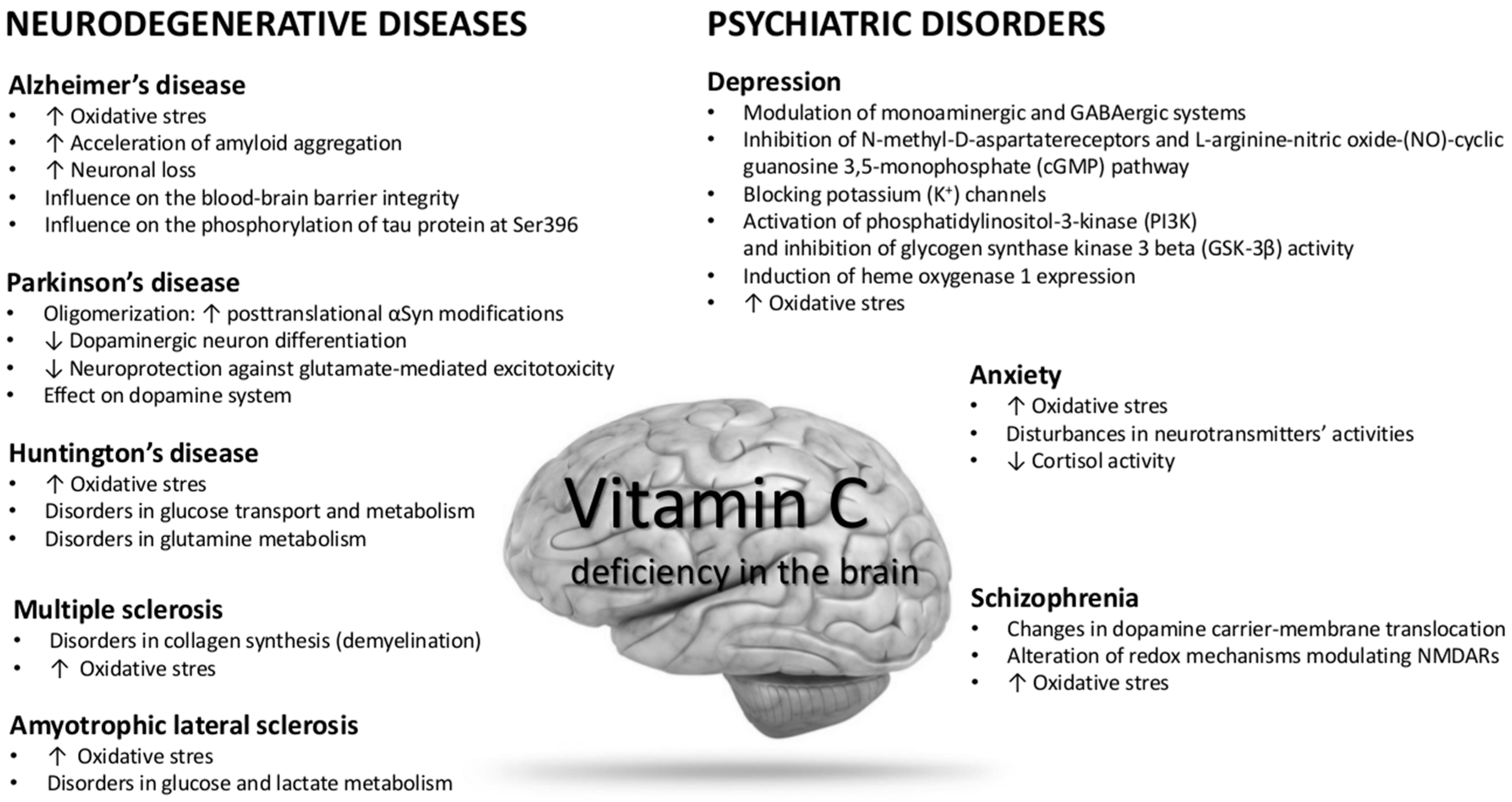
5. Role of Vitamin C in Neurodegenerative Diseases
5.1. Alzheimer’s Disease
5.2. Parkinson’s Disease
5.3. Huntington’s Disease
5.4. Multiple Sclerosis
5.5. Amyotrophic Lateral Sclerosis
6. Role of Vitamin C in Psychiatric Disorders
6.1. Depression
- modulation of monoaminergic systems [167] (e.g., Vit C was shown to activate the serotonin 1A (5-HT1A) receptor, this activation is a mechanism of action of many antidepressant, anxiolytic and antipsychotic drugs);
- modulation of GABAergic systems (via activation of GABAA receptors and a possible inhibition of GABAB receptors) [155];
- inhibition of N-methyl-d-aspartate (NMDA) receptors and l-arginine-nitric oxide (NO)-cyclic guanosine 3,5-monophosphate (cGMP) pathway—the blockade of NMDA receptor is associated with reduced levels of NO and cGMP, whereas reduction of NO levels within the hippocampus was shown to induce antidepressant-like effects [119];
- blocking potassium (K+) channels—Vit C administration was shown to produce an antidepressant-like effect in the tail suspension test via K+ channel inhibition [119]; as K+ channels were reported to belong to the physiological targets of NO and cGMP in the brain, their inhibition plays a significant role in the treatment of depression;
6.2. Anxiety
6.3. Schizophrenia
7. Conclusions
Conflicts of Interest
References
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Stevens, J.F. Vitamins C and E: Beneficial effects from a mechanistic perspective. Free Radic. Biol. Med. 2011, 51, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Said, H.M. Intestinal absorption of water-soluble vitamins in health and disease. Biochem. J. 2011, 437, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.M. Vitamin C requirements in parenteral nutrition. Gastroenterology 2009, 137, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Hart, A.; Cota, A.; Makhdom, A.; Harvey, E.J. The Role of Vitamin C in Orthopedic Trauma and Bone Health. Am. J. Orthop. 2015, 44, 306–311. [Google Scholar] [PubMed]
- Waly, M.I.; Al-Attabi, Z.; Guizani, N. Low Nourishment of Vitamin C Induces Glutathione Depletion and Oxidative Stress in Healthy Young Adults. Prev. Nutr. Food Sci. 2015, 20, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, C.F.; Bunge, M.B.; Bunge, R.P.; Wood, P.M. Differentiation of axon-related Schwann cells in vitro. I. Ascorbic acid regulates basal lamina assembly and myelin formation. J. Cell. Biol. 1987, 105, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Sawicka-Glazer, E.; Czuczwar, S.J. Vitamin C: A new auxiliary treatment of epilepsy? Pharmacol. Rep. 2014, 66, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Warner, T.A.; Kang, J.Q.; Kennard, J.K.; Harrison, F.E. Low brain ascorbic acid increases susceptibility to seizures in mouse models of decreased brain ascorbic acid transport and Alzheimer’s disease. Epilepsy Res. 2015, 110, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Vogt, L.; Schjoldager, J.G.; Jeannet, N.; Hasselholt, S.; Paidi, M.D.; Christen, S.; Lykkesfeldt, J. Maternal vitamin C deficiency during pregnancy persistently impairs hippocampal neurogenesis in offspring of guinea pigs. PLoS ONE 2012, 7, e48488. [Google Scholar] [CrossRef] [PubMed]
- Olajide, O.J.; Yawson, E.O.; Gbadamosi, I.T.; Arogundade, T.T.; Lambe, E.; Obasi, K.; Lawal, I.T.; Ibrahim, A.; Ogunrinola, K.Y. Ascorbic acid ameliorates behavioural deficits and neuropathological alterations in rat model of Alzheimer’s disease. Environ. Toxicol. Pharmacol. 2017, 50, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Ghosh, T.; Gupta, P.; Ghosh, R.; Kabir, S.N.; Roy, A. Dual Role of Vitamin C on the Neuroinflammation Mediated Neurodegeneration and Memory Impairments in Colchicine Induced Rat Model of Alzheimer Disease. J. Mol. Neurosci. 2016, 60, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Nualart, F.; Mack, L.; García, A.; Cisternas, P.; Bongarzone, E.R.; Heitzer, M.; Jara, N.; Martínez, F.; Ferrada, L.; Espinoza, F.; et al. Vitamin C Transporters, Recycling and the Bystander Effect in the Nervous System: SVCT2 versus Gluts. J. Stem Cell Res. Ther. 2014, 4, 209. [Google Scholar] [CrossRef] [PubMed]
- Corpe, C.P.; Tu, H.; Eck, P.; Wang, J.; Faulhaber-Walter, R.; Schnermann, J.; Margolis, S.; Padayatty, S.; Sun, H.; Wang, Y.; et al. Vitamin C transporter Slc23a1 links renal reabsorption, vitamin C tissue accumulation and perinatal survival in mice. J. Clin. Investig. 2010, 120, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na1-dependent l-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.N.; Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency affect cognitive development and function? Nutrients 2014, 6, 3818–3846. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, K.; Nakamura, G.; Akanuma, S.; Tomi, M.; Tachikawa, M. Dehydroascorbic acid uptake and intracellular ascorbic acid accumulation in cultured Müller glial cells (TR-MUL). Neurochem. Int. 2008, 52, 1351–1357. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.H.; Qu, Z.; May, J.M. Ascorbic Acid Transport in Brain Microvascular Pericytes. Biochem. Biophys. Res. Commun. 2015, 458, 262–267. [Google Scholar] [CrossRef] [PubMed]
- May, J.M. Vitamin C transport and its role in the central nervous system. Subcell. Biochem. 2012, 56, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.; Caprile, T.; Astuya, A.; Millán, C.; Reinicke, K.; Vera, J.C.; Vásquez, O.; Aguayo, L.G.; Nualart, F. High-affinity sodium-vitamin C co-transporters (SVCT) expression in embryonic mouse neurons. J. Neurochem. 2001, 78, 815–823. [Google Scholar] [CrossRef] [PubMed]
- García-Krauss, A.; Ferrada, L.; Astuya, A.; Salazar, K.; Cisternas, P.; Martínez, F.; Ramírez, E.; Nualart, F. Dehydroascorbic Acid Promotes Cell Death in Neurons Under Oxidative Stress: A Protective Role for Astrocytes. Mol. Neurobiol. 2016, 53, 5847–5863. [Google Scholar] [CrossRef]
- Covarrubias-Pinto, A.; Acuña, A.I.; Beltrán, F.A.; Torres-Díaz, L.; Castro, M.A. Old Things New View: Ascorbic Acid Protects the Brain in Neurodegenerative Disorders. Int. J. Mol. Sci. 2015, 16, 28194–28217. [Google Scholar]
- Mefford, I.N.; Oke, A.F.; Adams, R.N. Regional distribution of ascorbate in human brain. Brain Res. 1981, 212, 223–226. [Google Scholar] [CrossRef]
- Milby, K.; Oke, A.; Adams, R.N. Detailed mapping of ascorbate distribution in rat brain. Neurosci. Lett. 1982, 28, 169–174. [Google Scholar] [CrossRef]
- Rice, M.E.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1998, 82, 1213–1223. [Google Scholar] [CrossRef]
- Harrison, F.E.; May, J.M. Vitamin C function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Miele, M.; Boutelle, M.G.; Fillenz, M. The physiologically induced release of ascorbate in rat brain is dependent on impulse traffic, calcium influx and glutamate uptake. Neuroscience 1994, 62, 87–91. [Google Scholar] [CrossRef]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Olsen, C.L.; Bunge, R.P. Requisites for growth and myelination of urodele sensory neurons in tissue culture. J. Exp. Zool. 1986, 238, 373–384. [Google Scholar] [CrossRef] [PubMed]
- May, J.M.; Qu, Z.C. Ascorbic acid prevents oxidant-induced increases in endothelial permeability. Biofactors 2011, 37, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.M.; Chen, Y.J. Nitrosation-modulating effect of ascorbate in a model dynamic system of coexisting nitric oxide and superoxide. Free Radic. Res. 2010, 44, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.S.; Xu, A.; Vita, J.A.; Keaney, J.F., Jr. Ascorbate prevents the interaction of superoxide and nitric oxide only at very high physiological concentrations. Circ. Res. 1998, 83, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Mock, J.T.; Chaudhari, K.; Sidhu, A.; Sumien, N. The influence of vitamins E and C and exercise on brain aging. Exp. Gerontol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Majewska, M.D.; Bell, J.A.; London, E.D. Regulation of the NMDA receptor by redox phenomena: Inhibitory role of ascorbate. Brain Res. 1990, 537, 328–332. [Google Scholar] [CrossRef]
- Rebec, G.V.; Pierce, R.C. A vitamin as neuromodulator: Ascorbate release into the extracellular fluid of the brain regulates dopaminergic and glutamatergic transmission. Prog. Neurobiol. 1994, 43, 537–565. [Google Scholar] [CrossRef]
- Serra, P.A.; Esposito, G.; Delogu, M.R.; Migheli, R.; Rocchitta, G.; Grella, G.; Miele, E.; Miele, M.; Desole, M.S. Analysis of 3-morpholinosydnonimine and sodium nitroprusside effects on dopamine release in the striatum of freely moving rats: Role of nitric oxide, iron and ascorbic acid. Br. J. Pharmacol. 2000, 131, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.D.; Bauer, P.A. Ascorbate modulates 5-[3H]hydroxytryptamine binding to central 5-HT3 sites in bovine frontal cortex. J. Neurochem. 1988, 50, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Méndez, R.; Rivas-Arancibia, S. Vitamin C in Health and Disease: Its Role in the Metabolism of Cells and Redox State in the Brain. Front. Physiol. 2015, 23, 397. [Google Scholar] [CrossRef] [PubMed]
- Seitz, G.; Gebhardt, S.; Beck, J.F.; Böhm, W.; Lode, H.N.; Niethammer, D.; Bruchelt, G. Ascorbic acid stimulates DOPA synthesis and tyrosine hydroxylase gene expression in the human neuroblastoma cell line SK-N-SH. Neurosci. Lett. 1998, 244, 33–36. [Google Scholar] [CrossRef]
- Calero, C.I.; Vickers, E.; Cid, G.M.; Aguayo, L.G.; von Gersdorff, H.; Calvo, D.J. Allosteric modulation of retinal GABA receptors by ascorbic acid. J. Neurosci. 2011, 31, 9672–9682. [Google Scholar] [CrossRef] [PubMed]
- Majewska, M.D.; Bell, J.A. Ascorbic acid protects neurons from injury induced by glutamate and NMDA. Neuroreport 1990, 1, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.F.; Yazulla, S. Modulation of voltage-dependent k+ currents (Ik(v)) in retinal bipolar cells by ascorbate is mediated by dopamine d1 receptors. Vis. Neurosci. 1999, 16, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Joksovic, P.M.; Su, P.; Kang, H.W.; Van Deusen, A.; Baumgart, J.P.; David, L.S.; Snutch, T.P.; Barrett, P.Q.; Lee, J.H.; et al. Molecular mechanisms of subtype-specific inhibition of neuronal t-type calcium channels by ascorbate. J. Neurosci. 2007, 27, 12577–12583. [Google Scholar] [CrossRef] [PubMed]
- Kara, Y.; Doguc, D.K.; Kulac, E.; Gultekin, F. Acetylsalicylic acid and ascorbic acid combination improves cognition; via antioxidant effect or increased expression of NMDARs and nAChRs? Environ. Toxicol. Pharmacol. 2014, 37, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, M.I.; Rebec, G.V. Extracellular ascorbate modulates glutamate dynamics: Role of behavioral activation. BMC Neurosci. 2007, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, L.C.; Morris, P.E., Jr.; Spollen, J.J.; Ashe, S.C. Stereospecific effects of ascorbic acid and analogues on D1 and D2 agonist binding. Life Sci. 1992, 51, 921–930. [Google Scholar] [CrossRef]
- Castro, M.A.; Angulo, C.; Brauchi, S.; Nualart, F.; Concha, I.I. Ascorbic acid participates in a general mechanism for concerted glucose transport inhibition and lactate transport stimulation. Pflugers Arch. 2008, 457, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Beltrán, F.A.; Acuña, A.I.; Miro, M.P.; Anulo, C.; Concha, I.I.; Castro, M.A. Ascorbic acid-dependent GLUT3 inhibition is a critical step for switching neuronal metabolism. J. Cell. Physiol. 2011, 226, 3286–3294. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, S.; Gispert, S.; Cheng, J.; Wang, Y.H.; Chen, A.; Hoogstraten-Miller, S.; Miller, G.F.; Kwon, O.; Levine, M.; Guttentag, S.H.; et al. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat. Med. 2002, 8, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Yoon, G.H.; Kim, H.O.; Kim, M.O. Vitamin C neuroprotection against dose-dependent glutamate-induced neurodegeneration in the postnatal brain. Neurochem. Res. 2015, 40, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.C. The Genetics of Alzheimer’s Disease. Scientifica (Cairo) 2012, 2012, 46210. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Giovanello, K.S.; Saykin, A.J.; Xie, F.; Kong, D.; Wang, Y.; Yang, L.; Ibrahim, J.G.; Doraiswamy, P.M.; Zhu, H. Single-nucleotide polymorphisms are associated with cognitive decline at Alzheimer’s disease conversion within mild cognitive impairment patients. Alzheimers Dement. 2017, 8, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.; Henkel, A.W.; Henkel, M.K.; Redzic, Z.B.; Kornhuber, J.; Wiltfang, J. Pre-aggregated Aβ1 42 peptide increases tau aggregation and hyperphosphorylation after short-term application. Mol. Cell. Biochem. 2011, 349, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Marszałek, M. Alzheimer’s disease against peptides products of enzymatic cleavage of APP protein. Forming and variety of fibrillating peptides—Some aspects. Postepy Hig. Med. Dosw. 2016, 70, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Nagababu, E.; UsatyuK, P.V.; Enika, D.; Natarajan, V.; Rifkind, J.M. Vascular Endothelial Barrier Dysfunction Mediated by Amyloid-β Proteins. J. Alzheimers Dis. 2009, 17, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cui, J.; Fang, C.; Liu, M.; Min, G.; Li, L. S-Adenosylmethionine Attenuates Oxidative Stress and Neuroinflammation Induced by Amyloid-β Through Modulation of Glutathione Metabolism. J. Alzheimers Dis. 2017, 58, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G.; Lu, P.H.; Mintzd, J. Human brain myelination and amyloid beta deposition in Alzheimer’s disease. Alzheimers Dement. 2007, 3, 122–125. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: Implications for therapy. Acta Neuropathol. 2013, 126, 479. [Google Scholar] [CrossRef] [PubMed]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1792–1800. [Google Scholar] [CrossRef] [PubMed]
- Málaga-Trillo, E.; Ochs, K. Uncontrolled SFK-mediated protein trafficking in prion and Alzheimer’s disease. Prion 2016, 10, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Dixit, S.; Bernardo, A.; Walker, J.M.; Kennard, J.A.; Kim, G.Y.; Kessler, E.S.; Harrison, F.E. Vitamin C deficiency in the brain impairs cognition, increases amyloid accumulation and deposition, and oxidative stress in APP/PSEN1 and normally aging mice. ACS Chem. Neurosci. 2015, 6, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.S.; Lamb, J.; May, J.M.; Harrison, F.E. Behavioral and monoamine changes following severe vitamin C deficiency. J. Neurochem. 2013, 124, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Mukherjee, A.; Swarnakar, S.; Das, N. Nanocapsulated Ascorbic Acid in Combating Cerebral Ischemia Reperfusion—Induced Oxidative Injury in Rat Brain. Curr. Alzheimer. Res. 2016, 13, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Kennard, J.A.; Harrison, F.E. Intravenous ascorbate improves spatial memory in middle-aged APP/PSEN1 and wild type mice. Behav. Brain Res. 2014, 264, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Hosseini, A.H.; McDonald, M.P.; May, J.M. Vitamin C reduces spatial learning deficits in middle-aged and very old APP/PSEN1 transgenic and wild-type mice. Pharmacol. Biochem. Behav. 2009, 93, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Murata, N.; Ozawa, Y.; Kinoshita, N.; Irie, K.; Shirasawa, T.; Shimizu, T. Vitamin C restores behavioral deficits and amyloid-β oligomerization without affecting plaque formation in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 26, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.; Hackett, M.; Takechi, R. Antioxidants and Dementia Risk: Consideration through a Cerebrovascular Perspective. Nutrients 2016, 8, 828. [Google Scholar] [CrossRef] [PubMed]
- Kook, S.Y.; Lee, K.M.; Kim, Y.; Cha, M.Y.; Kang, S.; Baik, S.H.; Lee, H.; Park, R.; Mook-Jung, I. High-dose of vitamin C supplementation reduces amyloid plaque burden and ameliorates pathological changes in the brain of 5XFAD mice. Cell Death Dis. 2014, 5, 1083. [Google Scholar] [CrossRef] [PubMed]
- Allahtavakoli, M.; Amin, F.; Esmaeeli-Nadimi, A.; Shamsizadeh, A.; Kazemi-Arababadi, M.; Kennedy, D. Ascorbic Acid Reduces the Adverse Effects of Delayed Administration of Tissue Plasminogen Activator in a Rat Stroke Model. Basic Clin. Pharmacol. Toxicol. 2015, 117, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Park, J.; Kim, J.H.; Choi, J.Y.; Kim, J.Y.; Lee, K.M.; Lee, J.E. Dehydroascorbic Acid Attenuates Ischemic Brain Edema and Neurotoxicity in Cerebral Ischemia: An in vivo Study. Exp. Neurobiol. 2015, 24, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.L.; Huang, Y.H.; Shen, Y.C.; Huang, H.C.; Liu, pH. Ascorbic acid prevents blood-brain barrier disruption and sensory deficit caused by sustained compression of primary somatosensory cortex. J. Cereb. Blood Flow. Metab. 2010, 30, 1121–1136. [Google Scholar] [CrossRef] [PubMed]
- Polidori, M.C.; Mattioli, P.; Aldred, S.; Cecchetti, R.; Stahl, W.; Griffiths, H.; Senin, U.; Sies, H.; Mecocci, P. Plasma antioxidant status, immunoglobulin g oxidation and lipid peroxidation in demented patients: Relevance to Alzheimer disease and vascular dementia. Dement. Geriatr. Cogn. Disord. 2004, 18, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Schippling, S.; Kontush, A.; Arlt, S.; Buhmann, C.; Sturenburg, H.J.; Mann, U.; Griffiths, H.; Senin, U.; Sies, H.; Mecocci, P. Increased lipoprotein oxidation in Alzheimer’s disease. Free Radic. Biol. Med. 2000, 28, 351–360. [Google Scholar] [CrossRef]
- Lopes da Silva, S.; Vellas, B.; Elemans, S.; Luchsinger, J.; Kamphuis, P.; Yaffe, K.; Sijben, J.; Groenendijk, M.; Stijnen, T. Plasma nutrient status of patients with Alzheimer’s disease: Systematic review and meta-analysis. Alzheimers Dement. 2014, 10, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Arlt, S.; Müller-Thomsen, T.; Beisiegel, U.; Kontush, A. Effect of one-year vitamin C- and E-supplementation on cerebrospinal fluid oxidation parameters and clinical course in Alzheimer’s disease. Neurochem. Res. 2012, 37, 2706–2714. [Google Scholar] [CrossRef] [PubMed]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Alzheimer’s Disease Cooperative Study. Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Kantham, S.; Rao, V.M.; Palanivelu, M.K.; Pham, H.L.; Shaw, P.N.; McGeary, R.P.; Ross, B.P. Metal chelation, radical scavenging and inhibition of Aβ42 fibrillation by food constituents in relation to Alzheimer’s disease. Food Chem. 2016, 199, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Berti, V.; Murray, J.; Davies, M.; Spector, N.; Tsui, W.H.; Li, Y.; Williams, S.; Pirraglia, E.; Vallabhajosula, S.; McHugh, P.; et al. Nutrient patterns and brain biomarkers of Alzheimer’s disease in cognitively normal individuals. J. Nutr. Health Aging 2015, 19, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, S.; Man, Y.; Li, N.; Zhou, Y. Effects of vitamins E and C combined with β-carotene on cognitive function in the elderly. Exp. Ther. Med. 2015, 9, 1489–1493. [Google Scholar] [CrossRef] [PubMed]
- Zandi, P.P.; Anthony, J.C.; Khachaturian, A.S.; Stone, S.V.; Gustafson, D.; Tschanz, J.T.; Norton, M.C.; Welsh-Bohmer, K.A.; Breitner, J.C. Cache County Study Group. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: The Cache County Study. Arch. Neurol. 2004, 61, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Polidori, M.C.; Ruggiero, C.; Croce, M.F.; Raichi, T.; Mangialasche, F.; Cecchetti, R.; Pelini, L.; Paolacci, L.; Ercolani, S.; Mecocci, P. Association of increased carotid intima-media thickness and lower plasma levels of vitamin C and vitamin E in old age subjects: Implications for Alzheimer’s disease. J. Neural Transm. 2015, 122, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Liu, H.; Wang, X.; Chen, S.G.; Siedlak, S.L.; Kondo, E.; Choi, R.; Takeda, A.; Castellani, R.J.; Perry, G.; et al. Ectopic localization of FOXO3a protein in Lewy bodies in Lewy body dementia and Parkinson’s disease. Mol. Neurodegener. 2009, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health. Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.Y.; Giasson, B.L.; Bonini, N.M. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef] [PubMed]
- Elkon, H.; Melamed, E.; Offen, D. Oxidative stress, induced by 6-hydroxydopamine, reduces proteasome activities in PC12 cells: Implications for the pathogenesis of Parkinson’s disease. J. Mol. Neurosci. 2004, 24, 387–400. [Google Scholar] [CrossRef]
- Belluzzi, E.; Bisaglia, M.; Lazzarini, E.; Tabares, L.C.; Beltramini, M.; Bubacco, L. Human SOD2 modification by dopamine quinones affects enzymatic activity by promoting its aggregation: Possible implications for Parkinson’s disease. PLoS ONE 2012, 7, e38026. [Google Scholar] [CrossRef] [PubMed]
- Rokad, D.; Ghaisas, S.; Harischandra, D.S.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Role of neurotoxicants and traumatic brain injury in α-synuclein protein misfolding and aggregation. Brain Res. Bull. 2016. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Duda, J.E.; Xie, S.X.; Lee, E.B.; Van Deerlin, V.M.; Lopez, O.L.; Kofler, J.K.; et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet. Neurol. 2017, 16, 55–65. [Google Scholar] [CrossRef]
- Rieder, C.R.; Williams, A.C.; Ramsden, D.B. Selegiline increases heme oxygenase-1 expression and the cytotoxicity produced by dopaminetreatment of neuroblastoma SK-N-SH cells. Braz. J. Med. Biol. Res. 2004, 37, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Brundin, P. Parkinson’s disease and alpha synuclein: Is Parkinson’s disease a prion-like disorder? Mov. Disord. 2013, 28, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Seidel, K.; Bouzrou, M.; Heidemann, N.; Krüger, R.; Schöls, L.; den Dunnen, W.F.A.; Korf, H.W.; Rüb, U. Involvement of the cerebellum in Parkinson disease and dementia with Lewy bodies. Ann. Neurol. 2017. [CrossRef] [PubMed]
- Armstrong, R.A. Evidence from spatial pattern analysis for the anatomical spread of α-synuclein pathology in Parkinson’s disease dementia. Folia Neuropathol. 2017, 55, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Scheffold, A.; Holtman, I.R.; Dieni, S.; Brouwer, N.; Katz, S.F.; Jebaraj, B.M.; Kahle, P.J.; Hengerer, B.; Lechel, A.; Stilgenbauer, S.; et al. Telomere shortening leads to an acceleration of synucleinopathy and impaired microglia response in a genetic mouse model. Acta Neuropathol. Commun. 2016, 4, 87. [Google Scholar] [CrossRef] [PubMed]
- He, X.B.; Kim, M.; Kim, S.Y.; Yi, S.H.; Rhee, Y.H.; Kim, T.; Lee, E.H.; Park, C.H.; Dixit, S.; Harrison, F.E.; et al. Vitamin C facilitates dopamine neuron differentiation in fetal midbrain through TET1- and JMJD3-dependent epigenetic control manner. Stem Cells 2015, 33, 1320–1332. [Google Scholar] [CrossRef] [PubMed]
- Camarena, V.; Wang, G. The epigenetic role of vitamin C in health and disease. Cell. Mol. Life Sci. 2016, 73, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
- Minor, E.A.; Court, B.L.; Young, J.I.; Wang, G. Ascorbate Induces Ten-Eleven Translocation (Tet) Methylcytosine Dioxygenase-mediated Generation of 5-Hydroxymethylcytosine. J. Biol. Chem. 2013, 288, 13669–13674. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Schlachetzki, J.C.; Helling, S.; Bussmann, J.C.; Berlinghof, M.; Schaffer, T.E.; Marcus, K.; Winkler, J.; Klucken, J.; Becker, C.M. Oxidative stress-induced posttranslational modifications of α-synuclein: Specific modification of α-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol. Cell. Neurosci. 2013, 54, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Jinsmaa, Y.; Sullivan, P.; Sharabi, Y.; Goldstein, D.S. DOPAL is transmissible to and oligomerizes alpha-synuclein in human glial cells. Auton. Neurosci. 2016, 194, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jyoti, S.; Naz, F.; Shakya, B.; Rahul, A.M.; Siddique, Y.H. Effect of l-ascorbic Acid on the climbing ability and protein levels in the brain of Drosophila model of Parkinson’s disease. J. Neurosci. 2012, 122, 704–709. [Google Scholar] [CrossRef]
- Ballaz, S.; Morales, I.; Rodríguez, M.; Obeso, J.A. Ascorbate prevents cell death from prolonged exposure to glutamate in an in vitro model of human dopaminergic neurons. J. Neurosci. Res. 2013, 91, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Ezumi, M.; Takada-Takatori, Y.; Akaike, A.; Kume, T. Endogenous dopamine is involved in the herbicide paraquat-induced dopaminergic cell death. Toxicol. Sci. 2014, 139, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, M.S.; Schumacher-Schuh, A.; Cardoso, A.M.; Bochi, G.V.; Baldissarelli, J.; Kegler, A.; Santana, D.; Chaves, C.M.; Schetinger, M.R.; Moresco, R.N.; et al. Iron and Oxidative Stress in Parkinson’s Disease: An Observational Study of Injury Biomarkers. PLoS ONE 2016, 11, e0146129. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, M.J.; Carroll, D.W.; Brown, T.M. Ascorbate- and zinc-responsive parkinsonism. Ann. Pharmacother. 2014, 48, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Ide, K.; Yamada, H.; Umegaki, K.; Mizuno, K.; Kawakami, N.; Hagiwara, Y.; Matsumoto, M.; Yoshida, H.; Kim, K.; Shiosaki, E.; et al. Lymphocyte vitamin C levels as potential biomarker for progression of Parkinson’s disease. Nutrition 2015, 31, 406–408. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.C.; Gao, X.; Kim, I.Y.; Rimm, E.B.; Wang, M.; Weisskopf, M.G.; Schwarzschild, M.A.; Ascherio, A. Intake of antioxidant vitamins and risk of Parkinson’s disease. Mov. Disord. 2016, 31. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.; Healey, C.S.; McDougal-Chukwumah, L.D.; Brown, T.M. Old disease, new look? A first report of parkinsonism due to scurvy, and of refeeding-induced worsening of scurvy. Psychosomatics 2013, 54, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, H.; Hamamoto, M.; Ueda, M.; Nito, C.; Yamaguchi, H.; Katayama, Y. The effect of ascorbic acid on the pharmacokinetics of levodopa in elderly patients with Parkinson disease. Clin. Neuropharmacol. 2004, 27, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Mariam, I.; Ali, S.; Rehman, A. Ikram-Ul-Haq. l-Ascorbate, a strong inducer of l-dopa (3,4-dihydroxy-l-phenylalanine) production from pre-grown mycelia of Aspergillus oryzae NRRL-1560. Biotechnol. Appl. Biochem. 2010, 55, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Peña-Sánchez, M.; Riverón-Forment, G.; Zaldívar-Vaillant, T.; Soto-Lavastida, A.; Borrero-Sánchez, J.; Lara-Fernández, G.; Esteban-Hernández, E.M.; Hernández-Díaz, Z.; González-Quevedo, A.; Fernández-Almirall, I.; et al. Association of status redox with demographic, clinical and imaging parameters in patients with Huntington’s disease. Clin. Biochem. 2015, 48, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Sari, Y. Huntington’s Disease: From Mutant Huntingtin Protein to Neurotrophic Factor Therapy. Int. J. Biomed. Sci. 2011, 7, 89–100. [Google Scholar] [PubMed]
- Paulsen, J.S. Cognitive Impairment in Huntington Disease: Diagnosis and Treatment. Curr. Neurol. Neurosci. Rep. 2011, 11, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Long, J.D.; Paulsen, J.S.; Marder, K.; Zhang, Y.; Kim, J.I.; Mills, J.A. Researchers of the PREDICT-HD Huntington’s Study Group. Tracking motor impairments in the progression of Huntington’s disease. Mov. Disord. 2014, 29, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P., Jr. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Postert, T.; Lack, B.; Kuhn, W.; Jergas, M.; Andrich, J.; Braun, B.; Przuntek, H.; Sprengelmeyer, R.; Agelink, M.; Büttner, T. Basal ganglia alterations and brain atrophy in Huntington’s disease depicted by transcranial realtime sonography. J. Neurol. Neurosurg. Psychiatry 1999, 67, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Acuña, A.I.; Esparza, M.; Kramm, C.; Beltrán, F.A.; Parra, A.V.; Cepeda, C.; Toro, C.A.; Vidal, R.L.; Hetz, C.; Concha, I.I.; et al. A failure in energy metabolism and antioxidant uptake precede symptoms of Huntington’s disease in mice. Nat. Commun. 2013, 4, 2917. [Google Scholar] [CrossRef] [PubMed]
- Weydt, P.; Pineda, V.V.; Torrence, A.E.; Libby, R.T.; Satterfield, T.F.; Lazarowski, E.R.; Gilbert, M.L.; Morton, G.J.; Bammler, T.K.; Strand, A.D.; et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell. Metab. 2006, 4, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Tereshchenko, A.; McHugh, M.; Lee, J.K.; Gonzalez-Alegre, P.; Crane, K.; Dawson, J.; Nopoulos, P. Abnormal Weight and Body Mass Index in Children with Juvenile Huntington’s Disease. J. Huntingtons. Dis. 2015, 4, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. Huntington’s disease: Underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 2013, 38, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.S.; Chen, X.; Liu, J.; Bezprozvanny, I. Dopaminergic Signaling and Striatal Neurodegeneration in Huntington’s Disease. J. Neurosci. 2007, 27, 7899–7910. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias-Pinto, A.; Moll, P.; Solís-Maldonado, M.; Acuña, A.I.; Riveros, A.; Miró, M.P.; Papic, E.; Beltrán, F.A.; Cepeda, C.; Concha, I.I.; et al. Beyond the redox imbalance: Oxidative stress contributes to an impaired GLUT3 modulation in Huntington’s disease. Free Radic. Biol. Med. 2015, 89, 1085–1096. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; Dorner, J.L.; Bunner, K.D.; Gaither, T.W.; Klein, E.L.; Barton, S.J.; Rebec, G.V. Up-regulation of GLT1 reverses the deficit in cortically evoked striatal ascorbate efflux in the R6/2 mouse model of Huntington’s disease. J. Neurochem. 2012, 121, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Dorner, J.L.; Miller, B.R.; Klein, E.L.; Murphy-Nakhnikian, A.; Andrews, R.L.; Barton, S.J.; Rebec, G.V. Corticostriatal dysfunction underlies diminished striatal ascorbate release in the R6/2 mouse model of Huntington’s disease. Brain Res. 2009, 1290, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Rebec, G.V. Dysregulation of corticostriatal ascorbate release and glutamate uptake in transgenic models of Huntington’s disease. Antioxid. Redox Signal. 2013, 19, 2115–2128. [Google Scholar] [CrossRef] [PubMed]
- Rebec, G.V.; Conroy, S.K.; Barton, S.J. Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: Variations with behavioral state and repeated ascorbate treatment. Neuroscience 2006, 137, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Rebec, G.V.; Barton, S.J.; Marseilles, A.M.; Collins, K. Ascorbate treatment attenuates the Huntington behavioral phenotype in mice. Neuroreport 2003, 14, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Hadžović-Džuvo, A.; Lepara, O.; Valjevac, A.; Avdagić, N.; Hasić, S.; Kiseljaković, E.; Ibragić, S.; Alajbegović, A. Serum total antioxidant capacity in patients with multiple sclerosis. Bosn. J. Basic Med. Sci. 2011, 11, 33–36. [Google Scholar] [PubMed]
- Rottlaender, A.; Kuerten, S. Stepchild or Prodigy? Neuroprotection in Multiple Sclerosis (MS) Research. Int. J. Mol. Sci. 2015, 16, 14850–14865. [Google Scholar] [CrossRef] [PubMed]
- Morandi, E.; Tarlinton, R.E.; Gran, B. Multiple Sclerosis between Genetics and Infections: Human Endogenous Retroviruses in Monocytes and Macrophages. Front. Immunol. 2015, 6, 647. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Talamanca, A.A.; Castello, G.; Cordero, M.D.; d’Ischia, M.; Gadaleta, M.N.; Pallardó, F.V.; Petrović, S.; Tiano, L.; Zatterale, A. Oxidative stress and mitochondrial dysfunction across broad-ranging pathologies: Toward mitochondria-targeted clinical strategies. Oxid. Med. Cell. Longev. 2014, 2014, 541230. [Google Scholar] [CrossRef] [PubMed]
- Polachini, C.R.; Spanevello, R.M.; Zanini, D.; Baldissarelli, J.; Pereira, L.B.; Schetinger, M.R.; da Cruz, I.B.; Assmann, C.E.; Bagatini, M.D.; Morsch, V.M. Evaluation of Delta Aminolevulinic Dehydratase Activity, Oxidative Stress Biomarkers, and Vitamin D Levels in Patients with Multiple Sclerosis. Neurotox. Res. 2016, 29, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Tavazzi, B.; Batocchi, A.P.; Amorini, A.M.; Nociti, V.; D’Urso, S.; Longo, S.; Gullotta, S.; Picardi, M.; Lazzarino, G. Serum metabolic profile in multiple sclerosis patients. Epub Mult. Scler. Int. 2011, 2011, 167156. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Balabanov, R. Molecular mechanisms of oligodendrocyte injury in multiple sclerosis and experimental autoimmune encephalomyelitis. Int. J. Mol. Sci. 2012, 13, 10647–10659. [Google Scholar] [CrossRef] [PubMed]
- Besler, H.T.; Comoğlu, S.; Okçu, Z. Serum levels of antioxidant vitamins and lipid peroxidation in multiple sclerosis. Nutr. Neurosci. 2002, 5, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Hejazi, E.; Amani, R.; SharafodinZadeh, N.; Cheraghian, B. Comparison of Antioxidant Status and Vitamin D Levels between Multiple Sclerosis Patients and Healthy Matched Subjects. Mult. Scler. Int. 2014, 2014, 539854. [Google Scholar] [CrossRef] [PubMed]
- Odinak, M.M.; Bisaga, G.N.; Zarubina, I.V. New Approaches to Antioxidant Therapy in Multiple Sclerosis. Available online: http://www.ncbi.nlm.nih.gov/pubmed/12418396 (accessed on 26 June 2017).
- Babri, S.; Mehrvash, F.; Mohaddes, G.; Hatami, H.; Mirzaie, F. Effect of intrahippocampal administration of vitamin C and progesterone on learning in a model of multiple sclerosis in rats. Adv. Pharm. Bull. 2015, 5, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Matic, I.; Strobbe, D.; Frison, M.; Campanella, M. Controlled and Impaired Mitochondrial Quality in Neurons: Molecular Physiology and Prospective Pharmacology. Pharmacol. Res. 2015, 99, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Orrell, R.W.; Lane, R.J.; Ross, M. A systematic review of antioxidant treatment for amyotrophic lateral sclerosis/motor neuron disease. Amyotroph. Lateral Scler. 2008, 9, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Corcia, P.; Moreau, C.; Veau, S.; Fournier, C.; Vourc’h, P.; Emond, P.; Gordon, P.; Pradat, P.F.; Praline, J.; et al. 1H-NMR-based metabolomic profiling of CSF in early amyotrophic lateral sclerosis. PLoS ONE 2010, 5, e13223. [Google Scholar] [CrossRef]
- Nagano, S.; Fujii, Y.; Yamamoto, T.; Taniyama, M.; Fukada, K.; Yanagihara, T.; Sakoda, S. The efficacy of trientine or ascorbate alone compared to that of the combined treatment with these two agents in familial amyotrophic lateral sclerosis model mice. Exp. Neurol. 2003, 179, 176–180. [Google Scholar] [CrossRef]
- Netzahualcoyotzi, C.; Tapia, R. Degeneration of spinal motor neurons by chronic AMPA-induced excitotoxicity in vivo and protection by energy substrates. Acta Neuropathol. Commun. 2015, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.C.; O’Reilly, É.J.; Fondell, E.; Falcone, G.J.; McCullough, M.L.; Park, Y.; Kolonel, L.N.; Ascherio, A. Intakes of vitamin C and carotenoids and risk of amyotrophic lateral sclerosis: Pooled results from 5 cohort studies. Ann. Neurol. 2013, 73, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kihira, T.; Kobashi, G.; Washio, M.; Sasaki, S.; Yokoyama, T.; Miyake, Y.; Sakamoto, N.; Inaba, Y.; Nagai, M. Fruit and vegetable intake and risk of amyotrophic lateral sclerosis in Japan. Neuroepidemiology 2009, 32, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Spasojević, I.; Stević, Z.; Nikolić-Kokić, A.; Jones, D.R.; Blagojević, D.; Spasić, M.B. Different roles of radical scavengers—Ascorbate and urate in the cerebrospinal fluid of amyotrophic lateral sclerosis patients. Redox Rep. 2010, 15, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, N. Electrochemical detection of natural DNA damage induced by ferritin/ascorbic acid/H2O2 system and amplification of DNA damage by endonuclease Fpg. Biosens. Bioelectron. 2009, 25, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Bembnowska, M.; Jośko, J. Depressive behaviours among adolescents as a Public Health problem. Zdr. Publ. 2011, 121, 4260430. [Google Scholar]
- Lopresti, A.L. A review of nutrient treatments for paediatric depression. J. Affect. Disord. 2015, 181, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Moretti, M.; Budni, J.; Ribeiro, C.M.; Rieger, D.; Leal, R.B.; Rodrigues, A.L. Subchronic administration of ascorbic acid elicits antidepressant-like effect and modulates cell survival signaling pathways in mice. J. Nutr. Biochem. 2016, 38, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Luscher, B.; Shen, Q.; Sahir, N. The GABAergic Deficit Hypothesis of Major Depressive Disorder. Mol. Psychiatry 2011, 16, 383–406. [Google Scholar] [CrossRef] [PubMed]
- Rosa, P.B.; Neis, V.B.; Ribeiro, C.M.; Moretti, M.; Rodrigues, A.L.S. Antidepressant-like effects of ascorbic acid and ketamine involve modulation of GABAA and GABAB receptors. Pharmacol. Rep. 2016, 68, 996–1001. [Google Scholar] [CrossRef]
- Capuron, L.; Pagnoni, G.; Drake, D.F.; Woolwine, B.J.; Spivey, J.R.; Crowe, R.J.; Votaw, J.R.; Goodman, M.M.; Miller, A.H. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch. Gen. Psychiatry 2012, 69, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Bigdeli, T.B.; Ripke, S.; Peterson, R.E.; Trzaskowski, M.; Bacanu, S.A.; Abdellaoui, A.; Andlauer, T.F.; Beekman, A.T.; Berger, K.; Blackwood, D.H.; et al. Genetic effects influencing risk for major depressive disorder in China and Europe. Transl. Psychiatry 2017, 7, e1074. [Google Scholar] [CrossRef] [PubMed]
- Altshuler, L.L.; Abulseoud, O.A.; Foland-Ross, L.; Bartzokis, G.; Chang, S.; Mintz, J.; Hellemann, G.; Vinters, H.V. Amygdala astrocyte reduction in subjects with major depressive disorder but not bipolar disorder. Bipolar Disord. 2010, 12, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.A.; O’Neill, K.; Milner, J.; Mahajan, G.J.; Lawrence, T.J.; May, W.T.; Miguel-Hidalgo, J.; Rajkowska, G.; Stockmeiera, C.A. Density of GFAP-immunoreactive astrocytes is decreased in left hippocampi in major depressive disorder. Neuroscience 2016, 316, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Rajkowska, G.; Hughes, J.; Stockmeier, C.A.; Javier Miguel-Hidalgo, J.; Maciag, D. Coverage of blood vessels by astrocytic endfeet is reduced in major depressive disorder. Biol. Psychiatry 2013, 73, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Rubinow, M.J.; Mahajan, G.; May, W.; Overholser, J.C.; Jurjus, G.J.; Dieter, L.; Herbst, N.; Steffens, D.C.; Miguel-Hidalgo, J.J.; Rajkowska, G.; et al. Basolateral amygdala volume and cell numbers in major depressive disorder: A postmortem stereological study. Brain Struct. Funct. 2016, 21, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Wang, L.; Brew, B.J. Quinolinic acid selectively induces apoptosis of human astrocytes: Potential role in AIDS dementia complex. J. Neuroinflammation 2005, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Grant, R.; Adams, S.; Brew, B.J.; Guillemin, G.J. Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox. Res. 2009, 16, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Moretti, M.; Budni, J.; Ribeiro, C.M.; Rodrigues, A.L. Involvement of different types of potassium channels in the antidepressant-like effect of ascorbic acid in the mouse tail suspension test. Eur. J. Pharmacol. 2012, 687, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Moretti, M.; Budni, J.; Freitas, A.E.; Neis, V.B.; Ribeiro, C.M.; de Oliveira Balen, G.; Rieger, D.K.; Leal, R.B.; Rodrigues, A.L. TNF-α-induced depressive-like phenotype and p38(MAPK) activation are abolished by ascorbic acid treatment. Eur. Neuropsychopharmacol. 2015, 25, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Fei, J.; Chen, Y.; Ying, Y.L.; Ma, L.; Song, X.Q.; Wang, L.; Chen, E.Z.; Mao, E.Q. Pharmacological preconditioning with vitamin C attenuates intestinal injury via the induction of heme oxygenase-1 after hemorrhagic shock in rats. PLoS ONE 2014, 9, 99134. [Google Scholar] [CrossRef]
- Binfaré, R.W.; Rosa, A.O.; Lobato, K.R.; Santos, A.R.; Rodrigues, A.L.S. Ascorbic acid administration produces an antidepressant-like effect: Evidence for the involvement of monoaminergic neurotransmission. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Robaczewska, J.; Kędziora-Kornatowska, K.; Kucharski, R.; Nowak, M.; Muszalik, M.; Kornatowski, M.; Kędziora, J. Decreased expression of heme oxygenase is associated with depressive symptoms and may contribute to depressive and hypertensive comorbidity. Redox Rep. 2016, 21, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Gariballa, S. Poor vitamin C status is associated with increased depression symptoms following acute illness in older people. Int. J. Vitam. Nutr. Res. 2014, 84, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, A.; Verma, A.K.; Srivastava, M.; Srivastava, R. Oxidative stress and major depression. J. Clin. Diagn. Res. 2014, 8, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Agrawal, M.; Gautam, M.; Sharma, P.; Gautam, A.S.; Gautam, S. Role of antioxidants in generalised anxiety disorder and depression. Indian J. Psychiatry 2012, 54, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Prohan, M.; Amani, R.; Nematpour, S.; Jomehzadeh, N.; Haghighizadeh, M.H. Total antioxidant capacity of diet and serum, dietary antioxidant vitamins intake, and serum hs-CRP levels in relation to depression scales in university male students. Redox Rep. 2014, 19, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Choi, J.Y.; Lee, H.H.; Park, Y. Associations between Dietary Pattern and Depression in Korean Adolescent Girls. J. Pediatr. Adolesc. Gynecol. 2015, 28, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Rubio-López, N.; Morales-Suárez-Varela, M.; Pico, Y.; Livianos-Aldana, L.; Llopis-González, A. Nutrient Intake and Depression Symptoms in Spanish Children: The ANIVA Study. Int. J. Environ. Res. Public Health 2016, 13, 352. [Google Scholar] [CrossRef] [PubMed]
- Amr, M.; El-Mogy, A.; Shams, T.; Vieira, K.; Lakhan, S.E. Efficacy of vitamin C as an adjunct to fluoxetine therapy in pediatric major depressive disorder: A randomized, double-blind, placebo-controlled pilot study. Nutr. J. 2013, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Robitaille, L.; Eintracht, S.; Hoffer, L.J. Vitamin C provision improves mood in acutely hospitalized patients. Nutrition 2011, 27, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, X.J.; Robitaille, L.; Eintracht, S.; MacNamara, E.; Hoffer, L.J. Effects of vitamin C and vitamin D administration on mood and distress in acutely hospitalizedpatients. Am. J. Clin. Nutr. 2013, 98, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Khajehnasiri, F.; Akhondzadeh, S.; Mortazavi, S.B.; Allameh, A.; Sotoudeh, G.; Khavanin, A.; Zamanian, Z. Are Supplementation of Omega-3 and Ascorbic Acid Effective in Reducing Oxidative Stress and Depression among Depressed Shift Workers? Int. J. Vitam. Nutr. Res. 2016, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fritz, H.; Flower, G.; Weeks, L.; Cooley, K.; Callachan, M.; McGowan, J.; Skidmore, B.; Kirchner, L.; Seely, D. Intravenous Vitamin C and Cancer: A Systematic Review. Integr. Cancer Ther. 2014, 13, 280–300. [Google Scholar] [CrossRef] [PubMed]
- Grillon, C.H. Models and mechanisms of anxiety: Evidence from startle studies. Psychopharmacology 2008, 199, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.N.; Lowther, C.L.; van Nobelen, M. Prolonged treatment with vitamins C and E separately and together decreases anxiety-related open-field behavior and acoustic startle in hooded rats. Pharmacol. Biochem. Behav. 2011, 97, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Kori, R.S.; Aladakatti, R.H.; Desai, S.D.; Das, K.K. Effect of Drug Alprazolam on Restrained Stress Induced Alteration of Serum Cortisol and Antioxidant Vitamins (Vitamin C and E) in Male Albino Rats. J. Clin. Diagn. Res. 2016, 10, AF07–AF09. [Google Scholar] [CrossRef] [PubMed]
- Boufleur, N.; Antoniazzi, C.T.; Pase, C.S.; Benvegnú, D.M.; Dias, V.T.; Segat, H.J.; Roversi, K.; Roversi, K.; Nora, M.D.; Koakoskia, G.; et al. Neonatal handling prevents anxiety-like symptoms in rats exposed to chronic mild stress: Behavioral and oxidative parameters. Stress 2013, 16, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, M.; Kondob, Y.; Isakaa, A.; Ishigamib, A.; Suzukia, E. Vitamin C impacts anxiety-like behavior and stress-induced anorexia relative to social environment in SMP30/GNL knockout mice. Nutr. Res. 2016, 36, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Hughes, N.R.; Hancock, J.N.; Thompson, R.M. Anxiolysis and recognition memory enhancement with long-term supplemental ascorbic acid (vitamin C) in normal rats: Possible dose dependency and sex differences. Ann. Neurosci. Psychol. 2015, 2, 2. Available online: http://www.vipoa.org/neuropsychol (accessed on 26 June 2017).
- Choi, J.Y.; dela Peña, I.C.; Yoon, S.Y.; Woo, T.E.; Choi, Y.J.; Shin, C.Y.; Ryu, J.H.; Lee, Y.S.; Yu, G.Y.; Cheong, J.H. Is the anti-stress effect of vitamin C related to adrenal gland function in rat? Food Sci. Biotechnol. 2011, 20, 429–435. [Google Scholar] [CrossRef]
- Puty, B.; Maximino, C.; Brasil, A.; da Silva, W.L.; Gouveia, A., Jr.; Oliveira, K.R.; Batista Ede, J.; Crespo-Lopez, M.E.; Rocha, F.A.; Herculano, A.M. Ascorbic acid protects against anxiogenic-like effect induced by methylmercury in zebrafish: Action on the serotonergic system. Zebrafish 2014, 11, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Angrini, M.A.; Leslie, J.C. Vitamin C attenuates the physiological and behavioural changes induced by long-term exposure to noise. Behav. Pharmacol. 2012, 23, 119–125. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, I.J.; de Souza, V.V.; Motta, V.; Da-Silva, S.L. Effects of Oral Vitamin C Supplementation on Anxiety in Students: A Double-Blind, Randomized, Placebo-Controlled Trial. Pak. J. Biol. Sci. 2015, 18, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Mazloom, Z.; Ekramzadeh, M.; Hejazi, N. Efficacy of supplementary vitamins C and E on anxiety, depression and stress in type 2 diabetic patients: A randomized, single-blind, placebo-controlled trial. Pak. J. Biol. Sci. 2013, 16, 1597–1600. [Google Scholar] [PubMed]
- McCabe, D.; Lisy, K.; Lockwood, C.; Colbeck, M. The impact of essential fatty acid, B vitamins, vitamin C, magnesium and zinc supplementation on stress levels in women: A systematic review. JBI Database Syst. Rev. Implement. Rep. 2017, 15, 402–453. [Google Scholar] [CrossRef]
- Arroll, M.A.; Wilder, L.; Neil, J. Nutritional interventions for the adjunctive treatment of schizophrenia: A brief review. Nutr. J. 2014, 13, 91. [Google Scholar] [CrossRef] [PubMed]
- Sershen, H.; Hashim, A.; Dunlop, D.S.; Suckow, R.F.; Cooper, T.B.; Javitt, D.C. Modulating NMDA Receptor Function with D-Amino Acid Oxidase Inhibitors: Understanding Functional Activity in PCP-Treated Mouse Model. Neurochem. Res. 2016, 41, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C. Twenty-five years of glutamate in schizophrenia: Are we there yet? Schizophr. Bull. 2012, 38, 911–913. [Google Scholar] [CrossRef] [PubMed]
- Wabaidur, S.M.; Alothman, Z.A.; Naushad, M. Determination of dopamine in pharmaceutical formulation using enhanced luminescence from europium complex. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 93, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Morera-Fumero, A.L.; Díaz-Mesa, E.; Abreu-Gonzalez, P.; Fernandez-Lopez, L.; Cejas-Mendez, M.D. Low levels of serum total antioxidant capacity and presence at admission and absence at discharge of a day/night change as a marker of acute paranoid schizophrenia relapse. Psychiatry Res. 2017, 249, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Hoffer, L.J. Vitamin Therapy in Schizophrenia. Isr. J. Psychiatry Relat. Sci. 2008, 45, 3–10. [Google Scholar] [PubMed]
- Sarandol, A.; Kirli, S.; Akkaya, C.; Altin, A.; Demirci, M.; Sarandol, E. Oxidative-antioxidative systems and their relation with serum S100 B levels in patients with schizophrenia: Effects of short term antipsychotic treatment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; McKinney, S.B.; Ross, B.M.; Wahle, K.W.; Boyle, S.P. Biomarkers of oxidative stress in schizophrenic and control subjects. Prostaglandins Leukot. Essent. Fatty Acids 2007, 76, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Konarzewska, B.; Stefańska, E.; Wendołowicz, A.; Cwalina, U.; Golonko, A.; Małus, A.; Kowzan, U.; Szulc, A.; Rudzki, L.; Ostrowska, L. Visceral obesity in normal-weight patients suffering from chronic schizophrenia. BMC Psychiatry 2014, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, P.V.; Dean, O.; Andreazza, A.C.; Berk, M.; Kapczinski, F. Antioxidant treatments for schizophrenia. Cochrane Database Syst. Rev. 2016, 2, CD008919. [Google Scholar] [CrossRef] [PubMed]
- Bentsen, H.; Osnes, K.; Refsum, H.; Solberg, D.K.; Bøhmer, T. A randomized placebo-controlled trial of an omega-3 fatty acid and vitamins E + C in schizophrenia. Transl. Psychiatry 2013, 3, e335. [Google Scholar] [CrossRef] [PubMed]
- Sivrioglu, E.Y.; Kirli, S.; Sipahioglu, D.; Gursoy, B.; Sarandöl, E. The impact of omega-3 fatty acids, vitamins E and C supplementation on treatment outcome and side effects in schizophrenia patients treated with haloperidol: An open-label pilot study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Heiser, P.; Sommer, O.; Schmidt, A.J.; Clement, H.W.; Hoinkes, A.; Hopt, U.T.; Schulz, E.; Krieg, J.C.; Dobschütz, E. Effects of antipsychotics and vitamin C on the formation of reactive oxygen species. J. Psychopharmacol. 2010, 24, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Kawazoe, T.; Park, H.K.; Iwana, S.; Tsuge, H.; Fukui, K. Human d-amino acid oxidase: An update and review. Chem. Rec. 2007, 7, 305–315. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kocot, J.; Luchowska-Kocot, D.; Kiełczykowska, M.; Musik, I.; Kurzepa, J. Does Vitamin C Influence Neurodegenerative Diseases and Psychiatric Disorders? Nutrients 2017, 9, 659. https://doi.org/10.3390/nu9070659
Kocot J, Luchowska-Kocot D, Kiełczykowska M, Musik I, Kurzepa J. Does Vitamin C Influence Neurodegenerative Diseases and Psychiatric Disorders? Nutrients. 2017; 9(7):659. https://doi.org/10.3390/nu9070659
Chicago/Turabian StyleKocot, Joanna, Dorota Luchowska-Kocot, Małgorzata Kiełczykowska, Irena Musik, and Jacek Kurzepa. 2017. "Does Vitamin C Influence Neurodegenerative Diseases and Psychiatric Disorders?" Nutrients 9, no. 7: 659. https://doi.org/10.3390/nu9070659
APA StyleKocot, J., Luchowska-Kocot, D., Kiełczykowska, M., Musik, I., & Kurzepa, J. (2017). Does Vitamin C Influence Neurodegenerative Diseases and Psychiatric Disorders? Nutrients, 9(7), 659. https://doi.org/10.3390/nu9070659