Chronic Fructose Ingestion as a Major Health Concern: Is a Sedentary Lifestyle Making It Worse? A Review

{kind=link}
Abstract
:1. Introduction
2. Fructose Intake, Absorption and Metabolism
2.1. Fructose Intake
2.2. Fructose Absorption
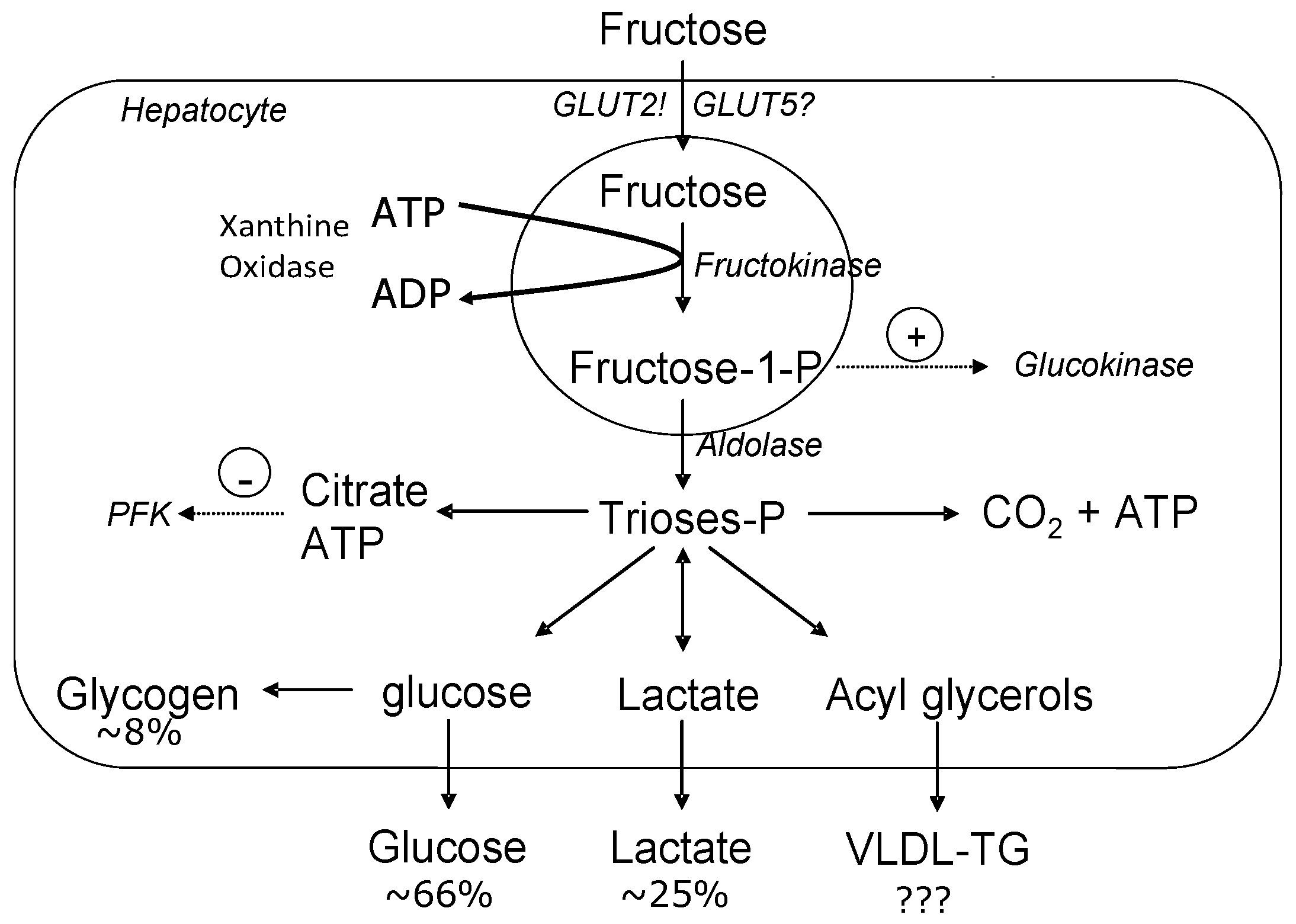
2.3. Fructose Metabolism
3. Fructose-Induced Lipogenesis
4. Fructose and Postprandial Lipemia
5. Fructose and Insulin Resistance
6. Fructose and Inflammation
7. Physical Inactivity and Fructose Consumption
7.1. Physical Inactivity
7.2. Physical Activity and Inflammation
7.3. Fructose Ingestion and Physical Activity
8. Conclusions
Conflicts of Interest
References
- U.S. Department of Health and Human Services. National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). Overweight and Obesity Statistics. Updated October 2012. Available online: https://www.niddk.nih.gov/health-information/health-statistics/overweight-obesity (accessed on 4 April 2017).
- Elliott, S.S.; Keim, N.L.; Stern, J.S.; Teff, K.; Havel, P.J. Fructose, weight gain, and the insulin resistance syndrome. Am. J. Clin. Nutr. 2002, 76, 911–922. [Google Scholar] [PubMed]
- Mayes, P.A. Intermediary metabolism of fructose. Am. J. Clin. Nutr. 1993, 58, 754S–765S. [Google Scholar] [PubMed]
- Miller, A.; Adeli, K. Dietary fructose and the metabolic syndrome. Curr. Opin. Gastroenterol. 2008, 24, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.; Hu, F. Fructose and cardiometabolic health. What the Evidence from sugar-sweetened Beverages Tells Us. J. Am. Coll. Cardiol. 2015, 66, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed]
- Douard, V.; Ferraris, R. Regulation of the fructose transporter GLUT5 in health and disease. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E227–E237. [Google Scholar] [CrossRef] [PubMed]
- Kneepkens, C.M.; Vonk, R.J.; Fernandes, J. Incomplete intestinal absorption of fructose. Arch. Dis Child. 1984, 59, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, K.; Riby, J.E.; Fujisawa, T.; Kretchmer, N. Absorption of fructose by isolated small intestine of rats is via a specific saturable carrier in the absence of glucose and by the disaccharidase-related transport system in the presence of glucose. J. Nutr. 1995, 125, 2156–2164. [Google Scholar] [PubMed]
- Truswell, A.S.; Seach, J.M.; Thorburn, A.W. Incomplete absorption of pure fructose in healthy subjects and the facilitating effect of glucose. Am. J. Clin. Nutr. 1988, 48, 1424–1430. [Google Scholar] [PubMed]
- Haidari, M.; Leung, N.; Mahbub, F.; Uffelman, K.D.; Kohen-Avramoglu, R.; Lewis, G.F.; Adeli, K. Fasting and postprandial overproduction of intestinally derived lipoproteins in an animal model of insulin resistance. Evidence that chronic fructose feeding in the hamster is accompanied by enhanced intestinal de novo lipogenesis and ApoB48-containing lipoprotein overproduction. J. Biol. Chem. 2002, 277, 31646–31655. [Google Scholar] [PubMed]
- Havel, P.J. Dietary fructose: Implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr. Rev. 2005, 63, 133–157. [Google Scholar] [CrossRef] [PubMed]
- Le, K.A.; Tappy, L. Metabolic effects of fructose. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Carmona, A.; Freedland, R.A. Comparison among the lipogenic potential of various substrates in rat hepatocytes: The differential effects of fructose-containing diets on hepatic lipogenesis. J. Nutr. 1989, 119, 1304–1310. [Google Scholar] [PubMed]
- Lê, K.A.; Faeh, D.; Stettler, R.; Ith, M.; Kreis, R.; Vermathen, P.; Boesch, C.; Ravussin, E.; Tappy, L. A 4-wk high-fructose diet alters lipid metabolism without affecting insulin sensitivity or ectopic lipids in healthy humans. Am. J. Clin. Nutr. 2006, 84, 1374–1379. [Google Scholar] [PubMed]
- Dai, S.; Todd, M.E.; Lee, S.; McNeill, J.H. Fructose loading induces cardiovascular and metabolic changes in non-diabetic and diabetic rats. Can. J. Physiol. Pharmacol. 1994, 72, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Rizkalla, S.W.; Lerer-Metzger, M.; Boillot, J. A fructose-rich diet decreases insulin-stimulated glucose incorporation into lipids but not glucose transport in adipocytes of normal and diabetic rats. J. Nutr. 1995, 125, 164–171. [Google Scholar] [PubMed]
- Swarbrick, M.M.; Stanhope, K.L.; Elliott, S.S.; Graham, J.L.; Krauss, R.M.; Christiansen, M.P.; Griffen, S.C.; Keim, N.L.; Havel, P.J. Consumption of fructose-sweetened beverages for 10 weeks increases postprandial triacylglycerol and apolipoprotein-B concentrations in overweight and obese women. Br. J. Nutr. 2008, 100, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Parks, E.J.; Skokan, L.E.; Timlin, M.T.; Dingfelder, C.S. Dietary sugars stimulate fatty acid synthesis in adults. J. Nutr. 2008, 138, 1039–1046. [Google Scholar] [PubMed]
- Faeh, D.; Minehira, K.; Schwarz, J.M.; Periasamy, R.; Park, S.; Tappy, L. Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes 2005, 54, 1907–1913. [Google Scholar] [CrossRef] [PubMed]
- Swanson, J.E.; Laine, D.C.; Thomas, W.; Bantle, J.P. Metabolic effects of dietary fructose in healthy subjects. Am. J. Clin. Nutr. 1992, 55, 851–856. [Google Scholar] [PubMed]
- Bantle, J.P.; Raatz, S.K.; Thomas, W.; Georgopoulos, A. Effects of dietary fructose on plasma lipids in healthy subjects. Am. J. Clin. Nutr. 2000, 72, 1128–1134. [Google Scholar] [PubMed]
- Schwarz, J.; Noworolski, S.; Wen, M.; Dyachenko, A.; Prior, J.; Weinberg, M.; Herraiz, L.; Tai, V.; Bergeron, N.; Bersot, T.; et al. Effect of a High-Fructose Weight-Maintaining Diet on Lipogenesis and Liver Fat. J. Clin. Endocrinol. Metab. 2015, 100, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Sayed, A.; Binnert, C.; Le, K.A.; Bortolotti, M.; Schneiter, P.; Tappy, L. A high-fructose diet impairs basal and stress-mediated lipid metabolism in healthy male subjects. Br. J. Nutr. 2008, 100, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; Vasan, R.S. Triglycerides as vascular risk factors: New epidemiologic insights. Curr. Opin. Cardiol. 2009, 24, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Alipour, A.; Elte, J.W.; van Zaanen, H.C.; Rietveld, A.P.; Cabezas, M.C. Postprandial inflammation and endothelial dysfuction. Biochem. Soc. Trans. 2007, 35, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.F.; Fielding, B.A.; Frayn, K.N. Mechanisms for the acute effect of fructose on postprandial lipemia. Am. J. Clin. Nutr. 2007, 85, 1511–1520. [Google Scholar] [PubMed]
- Chang, Y.C.; Chuang, L.M. The role of oxidative stress in the pathogenesis of type 2 diabetes: From molecular mechanism to clinical implication. Am. J. Transl. Res. 2010, 2, 316–331. [Google Scholar] [PubMed]
- Rutledge, A.C.; Adeli, K. Fructose and the metabolic syndrome: Pathophysiology and molecular mechanisms. Nutr. Rev. 2007, 65, S13–S23. [Google Scholar] [CrossRef] [PubMed]
- Rebrin, K.; Steil, G.M.; Getty, L.; Bergman, R.N. Free fatty acid as a link in the regulation of hepatic glucose output by peripheral insulin. Diabetes 1995, 44, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- LDL Cholesterol. New measurements of risk. Mayo Clin. Health Lett. 2011, 29, 1–3.
- Pagliassotti, M.J.; Prach, P.A. Quantity of sucrose alters the tissue pattern and time course of insulin resistance in young rats. Am. J. Physiol. 1995, 269, R641–R646. [Google Scholar] [PubMed]
- Morino, K.; Petersen, K.F.; Shulman, G.I. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 2006, 55, S9–S15. [Google Scholar] [CrossRef] [PubMed]
- Delarue, J.; Magnan, C. Free fatty acids and insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Kahn, C.R. Insulin signaling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Cortright, R.N.; Azevedo, J.L., Jr.; Zhou, Q.; Sinha, M.; Pories, W.J.; Itani, S.I.; Dohm, G.L. Protein kinase C modulates insulin action in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E553–E562. [Google Scholar] [PubMed]
- Thorburn, A.W.; Storlien, L.H.; Jenkins, A.B.; Khouri, S.; Kraegen, E.W. Fructose-induced in vivo insulin resistance and elevated plasma triglyceride levels in rats. Am. J. Clin. Nutr. 1989, 49, 1155–1163. [Google Scholar] [PubMed]
- Yoo, S.; Ahn, H.; Park, Y.K. High Dietary Fructose Intake on Cardiovascular Disease Related Parameters in Growing Rats. Nutrients 2017, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Dhawan, T.; Young, S.; Yong, W.H.; Boros, L.G.; Heaney, A.P. Fructose impairs glucose-induced hepatic triglyceride synthesis. Lipids Health Dis. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Sunehag, A.L.; Toffolo, G.; Campioni, M.; Bier, D.M.; Haymond, M.W. Short-term high dietary fructose intake had no effects on insulin sensitivity and secretion or glucose and lipid metabolism in healthy, obese adolescents. J. Pediatr. Endocrinol. Metab. 2008, 21, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Dirlewanger, M.; Schneiter, P.; Jequier, E.; Tappy, L. Effects of fructose on hepatic glucose metabolism in humans. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E907–E911. [Google Scholar] [PubMed]
- Catena, C.; Giacchetti, G.; Novello, M.; Colussi, G.; Cavarape, A.; Sechi, L.A. Cellular mechanisms of insulin resistance in rats with fructose-induced hypertension. Am. J. Hypertens. 2003, 16, 973–978. [Google Scholar] [CrossRef]
- Ueno, M.; Bezerra, R.M.; Silva, M.S.; Tavares, D.Q.; Carvalho, C.R.; Saad, M.J. A high-fructose diet induces changes in pp185 phosphorylation in muscle and liver of rats. Braz. J. Med. Biol. Res. 2000, 33, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Dupas, J.; Goanvec, C.; Guernec, A.; Feray, A.; Goanvec, C.; Samson, N.; Bougaran, P.; Guerrero, F.; Mansourati, J. Metabolic Syndrome and Hypertension Resulting from Fructose Enriched Diet in Wistar Rats. Biomed. Res. Int. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Alipour, A.; van Oostrom, A.J.; Izraeljan, A.; Verseyden, C.; Collins, J.M.; Frayn, K.N.; Plokker, T.W.M.; Elte, J.W.F.; Cabezas, M.C. Leukocyte activation by triglyceride-rich lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Zeyda, M.; Stulnig, T.M. Obesity, inflammation, and insulin resistance—A mini-review. Gerontology 2009, 55, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar] [PubMed]
- Pradhan, A.D.; Ridker, P.M. Do atherosclerosis and type 2 diabetes share a common inflammatory basis? Eur. Heart J. 2002, 23, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Pickup, J.C.; Mattock, M.B.; Chusney, G.D.; Burt, D. NIDDM as a disease of the innate immune system: Association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 1997, 40, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Kanuri, G.; Spruss, A.; Wagnerberger, S.; Bischoff, S.C.; Bergheim, I. Role of tumor necrosis factor alpha (TNFalpha) in the onset of fructose-induced nonalcoholic fatty liver disease in mice. J. Nutr. Biochem. 2011, 22, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H. SREBP-1c and TFE3, energy transcription factors that regulate hepatic insulin signaling. J. Mol. Med. 2007, 85, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Lozada, L.G.; Mu, W.; Roncal, C.; Sautin, Y.Y.; Abdelmalek, M.; Reungjui, S.; Le, M.; Nakagawa, T.; Lan, H.Y.; Yu, X.; et al. Comparison of free fructose and glucose to sucrose in the ability to cause fatty liver. Eur. J. Nutr. 2010, 49, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Lakka, T.A.; Laaksonen, D.E.; Lakka, H.M.; Männikkö, N.I.K.O.; Niskanen, L.K.; Rauramaa, R.A.I.N.E.R.; Salonen, J.T. Sedentary lifestyle, poor cardiorespiratory fitness, and the metabolic syndrome. Med. Sci. Sports Exerc. 2003, 35, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Haskell, W.L.; Lee, I.M.; Pate, R.R.; Powell, K.E.; Blair, S.N.; Franklin, B.A.; Macera, C.A.; Heatg, G.W.; Thompson, P.D.; Bauman, A. Physical activity and public health: Updated recommendation for adults from the American College of Sports Medicine and the American Heart Association. Med. Sci. Sports Exerc. 2007, 39, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.T.; Hamilton, D.G.; Zderic, T.W. Role of low energy expenditure and sitting in obesity, metabolic syndrome, type 2 diabetes, and cardiovascular disease. Diabetes 2007, 56, 2655–2667. [Google Scholar] [CrossRef] [PubMed]
- Healy, G.N.; Dunstan, D.W.; Salmon, J.; Cerin, E.; Shaw, J.E.; Zimmet, P.Z.; Owen, N. Objectively measured light-intensity physical activity is independently associated with 2-h plasma glucose. Diabetes Care 2007, 30, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Bey, L.; Hamilton, M.T. Suppression of skeletal muscle lipoprotein lipase activity during physical inactivity: A molecular reason to maintain daily low-intensity activity. J. Physiol. 2003, 551, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.N.; Heady, J.A.; Raffle, P.A.; Roberts, C.G.; Parks, J.W. Coronary heart-disease and physical activity of work. Lancet 1953, 265, 1111–1120. [Google Scholar] [CrossRef]
- Hsia, J.; Wu, L.; Allen, C.; Oberman, A.; Lawson, W.E.; Torréns, J.; Safford, M.; Limacher, M.C. Physical activity and diabetes risk in postmenopausal women. Am. J. Prev. Med. 2005, 28, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Wijndaele, K.; Healy, G.N.; Dunstan, D.W.; Barnett, A.G.; Salmon, J.; Shaw, J.E.; Zimmet, P.Z.; Owen, N. Increased cardiometabolic risk is associated with increased TV viewing time. Med. Sci. Sports Exerc. 2010, 42, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Healy, G.N.; Dunstan, D.W.; Salmon, J.; Shaw, J.E.; Zimmet, P.Z.; Owen, N. Television time and continuous metabolic risk in physically active adults. Med. Sci. Sports Exerc. 2008, 40, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, P.A.; Healy, G.N.; Eakin, E.G.; Clark, B.K.; Dunstan, D.W.; Shaw, J.E.; Zimmet, P.Z.; Owen, N. Associations between television viewing time and overall sitting time with the metabolic syndrome in older men and women: The Australian diabetes, obesity and lifestyle study. J. Am. Geriatr. Soc. 2011, 59, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.W.; Morrato, E.H.; Ghushchyan, V.; Wyatt, H.R.; Hill, J.O. Obesity, inactivity, and the prevalence of diabetes and diabetes-related cardiovascular comorbidities in the U.S., 2000–2002. Diabetes Care 2005, 28, 1599–1603. [Google Scholar] [CrossRef] [PubMed]
- Zderic, T.W.; Hamilton, M.T. Physical inactivity amplifies the sensitivity of skeletal muscle to the lipid-induced downregulation of lipoprotein lipase activity. J. Appl. Physiol. 2006, 100, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.; Bihain, B.E.; Bengtsson-Olivecrona, G.; Deckelbaum, R.J.; Carpentier, Y.A.; Olivecrona, T. Fatty acid control of lipoprotein lipase: A link between energy metabolism and lipid transport. Proc. Natl. Acad. Sci. USA 1990, 87, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Slentz, C.A.; Houmard, J.A.; Johnson, J.L.; Bateman, L.A.; Tanner, C.J.; McCartney, J.S.; Duscha, B.D.; Kraus, W.E. Inactivity, exercise training and detraining, and plasma lipoproteins. STRRIDE: A randomized, controlled study of exercise intensity and amount. J. Appl. Physiol. 2007, 103, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Herd, S.L.; Hardman, A.E.; Boobis, L.H.; Cairns, C.J. The effect of 13 weeks of running training followed by 9 d of detraining on postprandial lipaemia. Br. J. Nutr. 1998, 80, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Rockl, K.S.; Witczak, C.A.; Goodyear, L.J. Signaling mechanisms in skeletal muscle: Acute responses and chronic adaptations to exercise. IUBMB Life 2008, 60, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Jessen, N.; Goodyear, L.J. Contraction signaling to glucose transport in skeletal muscle. J. Appl. Physiol. 2005, 99, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.; Pedersen, B.K. The role of exercise-induced myokines in muscle homeostasis and the defense against chronic diseases. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Vider, J.; Laaksonen, D.E.; Kilk, A.; Atalay, M.; Lehtmaa, J.; Zilmer, M.; Sen, C.K. Physical exercise induces activation of NF-kappaB in human peripheral blood lymphocytes. Antioxid. Redox Signal. 2001, 3, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Lakka, T.A.; Laaksonen, D.E. Physical activity in prevention and treatment of the metabolic syndrome. Appl. Physiol. Nutr. Metab. 2007, 32, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Steensberg, A.; Fischer, C.; Keller, C.; Keller, P.; Plomgaard, P.; Febbraio, M.; Saltin, B. Searching for the exercise factor: Is IL-6 a candidate? J. Muscle Res. Cell. Motil. 2003, 24, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an endocrine organ: Focus on muscle-derived interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef] [PubMed]
- Currell, K.; Jeukendrup, A.E. Superior endurance performance with ingestion of multiple transportable carbohydrates. Med. Sci. Sports Exerc. 2008, 40, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, A.J.; Fairchild, T.J.; Redmond, J.; Wang, L.; Keslacy, S.; Kanaley, J.A. Physical Activity Offsets the Negative Effects of a High Fructose Diet. Med. Sci. Sports Exerc. 2014, 46, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bidwell, A.J. Chronic Fructose Ingestion as a Major Health Concern: Is a Sedentary Lifestyle Making It Worse? A Review. Nutrients 2017, 9, 549. https://doi.org/10.3390/nu9060549
Bidwell AJ. Chronic Fructose Ingestion as a Major Health Concern: Is a Sedentary Lifestyle Making It Worse? A Review. Nutrients. 2017; 9(6):549. https://doi.org/10.3390/nu9060549
Chicago/Turabian StyleBidwell, Amy J. 2017. "Chronic Fructose Ingestion as a Major Health Concern: Is a Sedentary Lifestyle Making It Worse? A Review" Nutrients 9, no. 6: 549. https://doi.org/10.3390/nu9060549
APA StyleBidwell, A. J. (2017). Chronic Fructose Ingestion as a Major Health Concern: Is a Sedentary Lifestyle Making It Worse? A Review. Nutrients, 9(6), 549. https://doi.org/10.3390/nu9060549