Vitamin D Insufficiency Exacerbates Adipose Tissue Macrophage Infiltration and Decreases AMPK/SIRT1 Activity in Obese Rats


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Diets
2.2. Measurements of Serum Metabolic Parameters
2.3. Histological Analysis of Adipose Tissue and Cell Size Determination
2.4. Immunohistochemistry and Crown-Like Structure (CLS) Quantification
2.5. RNA Isolation, Reverse Transcription and Real-Time Quantitative Polymerase Chain Reaction (RT-PCR)
2.6. Cytokine Assay
2.7. SIRT1 Activity Assay
2.8. AMPK Activity Assay
2.9. Statistical Analysis
3. Results
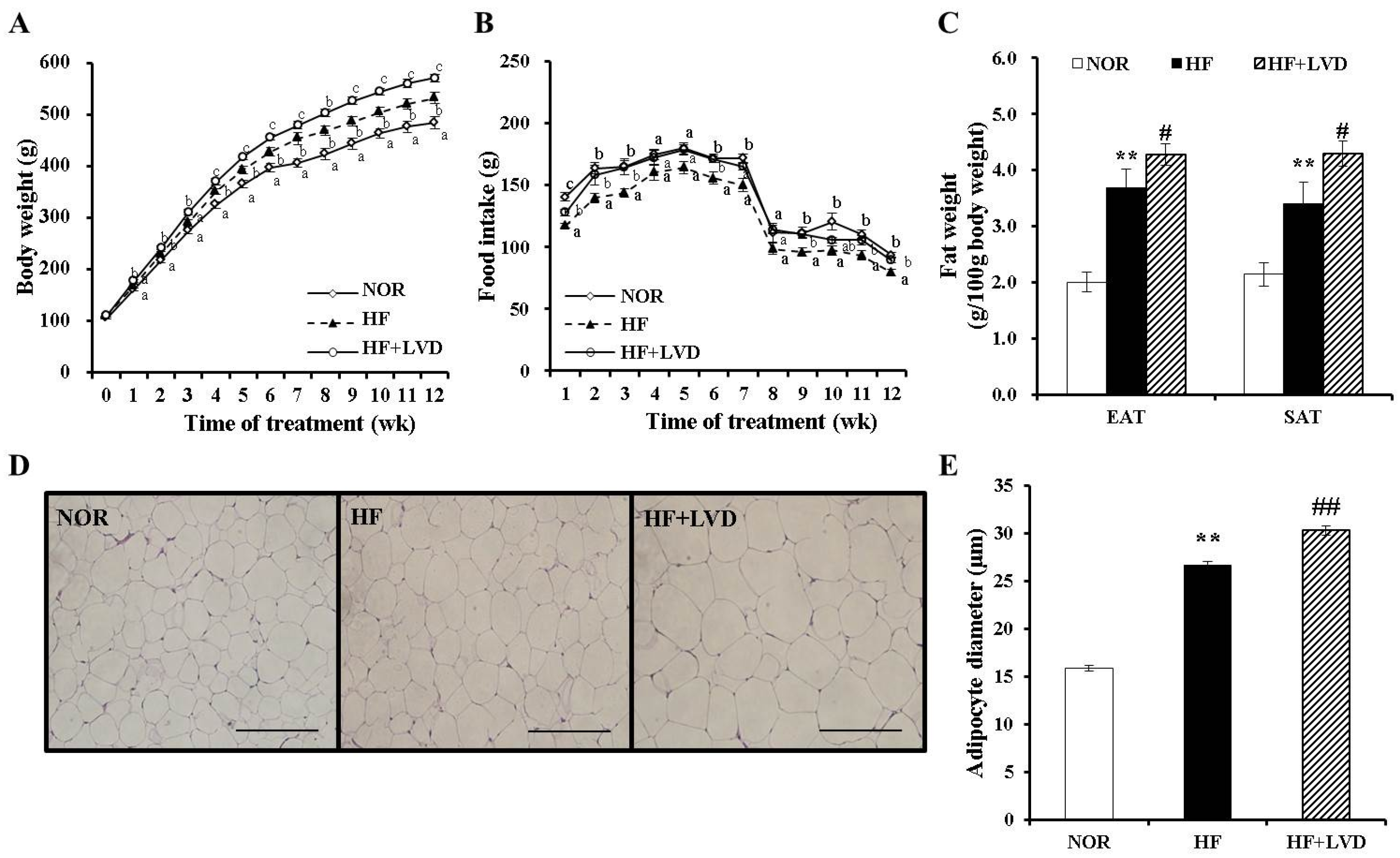
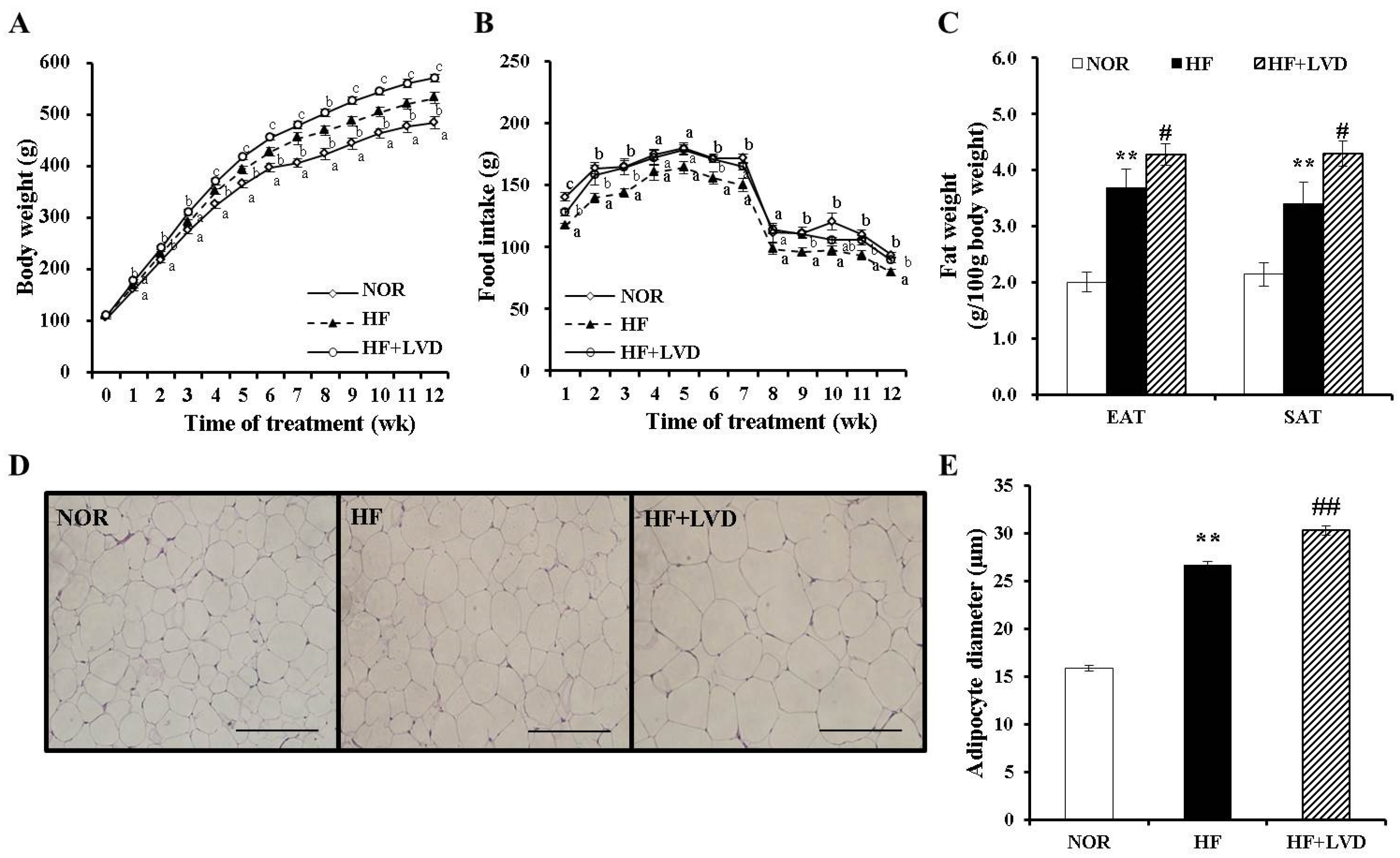
3.1. Dietary Vitamin D-Insufficient Diet Exacerbates High Fat Diet-Increased Body Weight Gain and Fat Deposition
3.2. Influence of a Vitamin D-Insufficient Diet on Serum Vitamin D and Metabolic Parameters
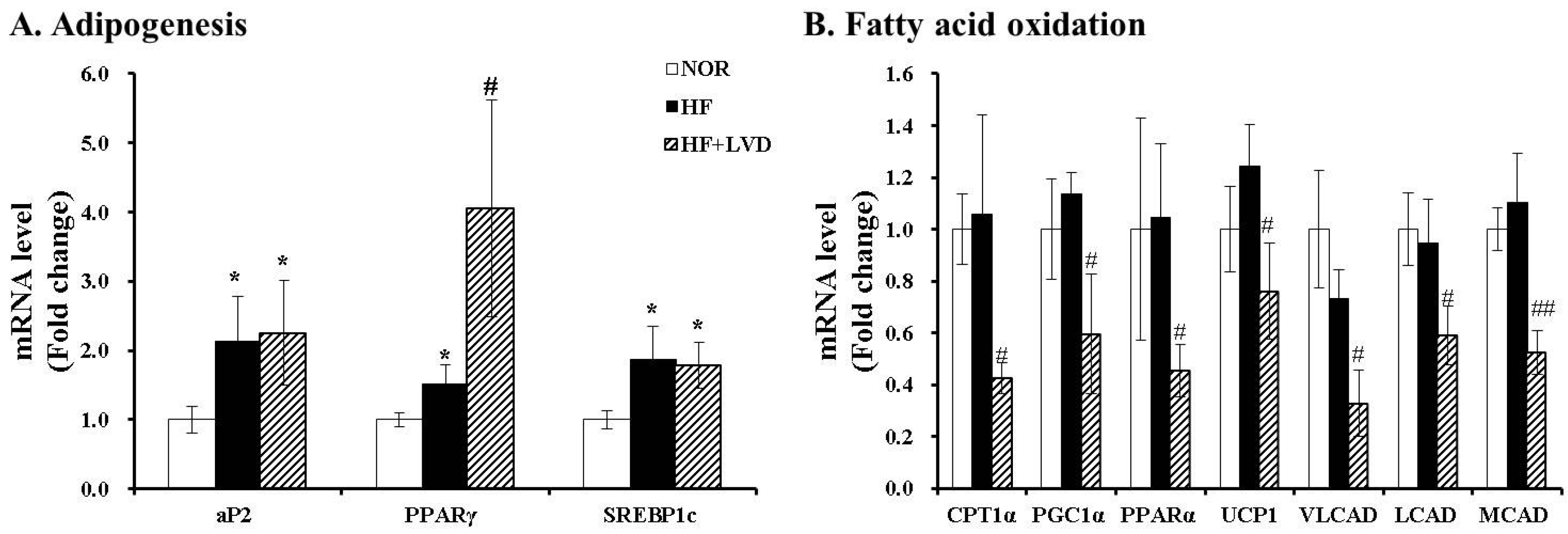
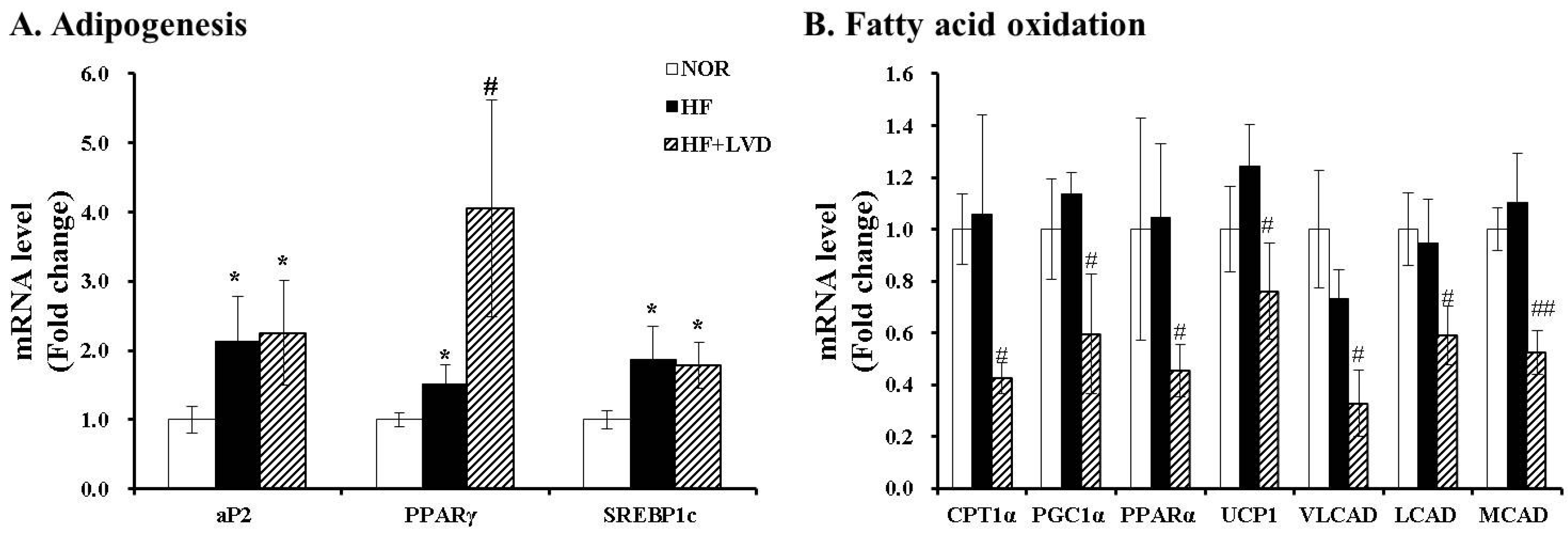
3.3. Influene of Vitamin D Insufficiency on Gene Expression invovled in Adipogensis and Fat Oxidation
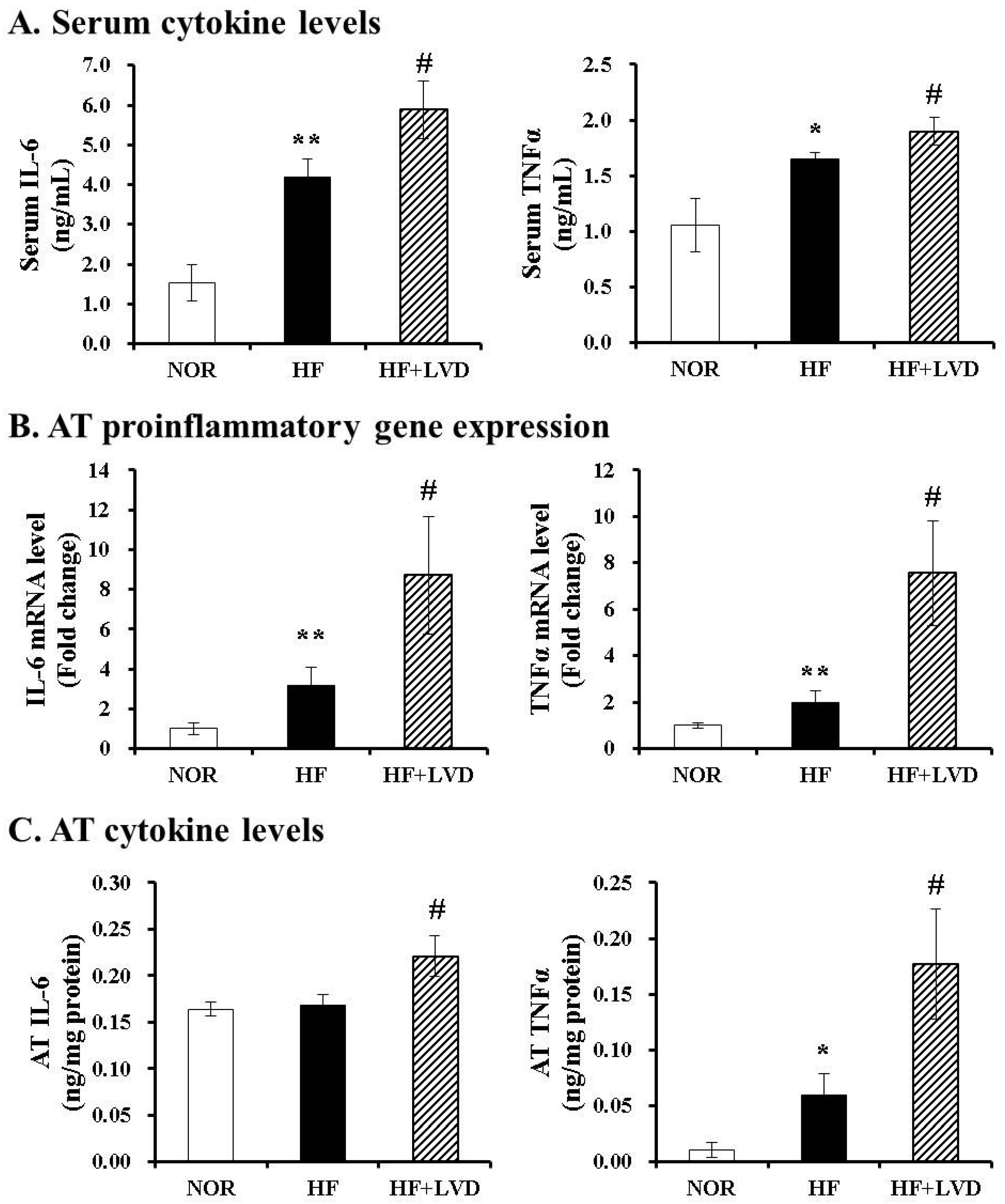
3.4. Vitamin D-Insufficient Diet Increases Inflammatory Cytokines in Serum and Adipose Tissues of Obese Rats
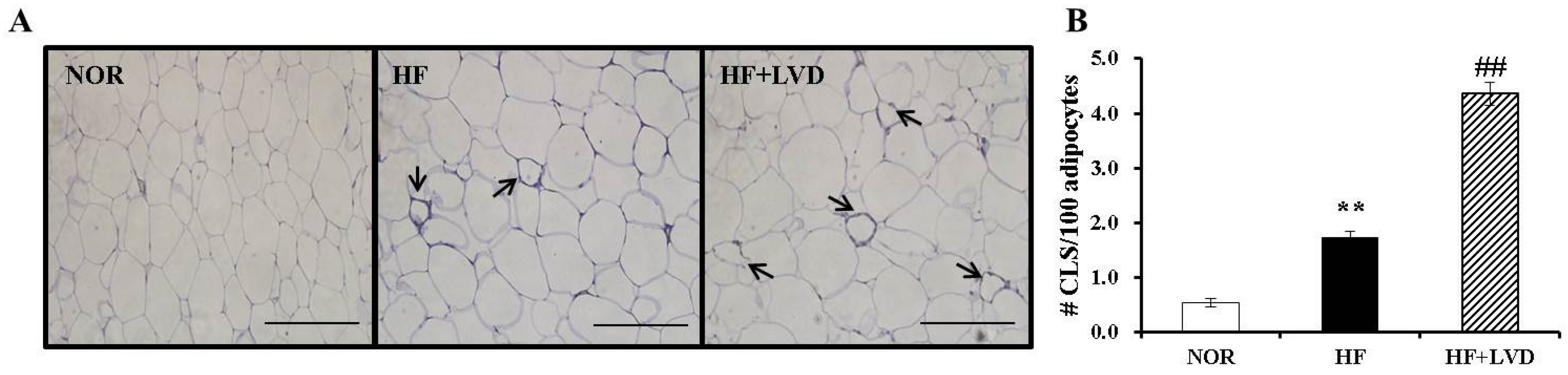
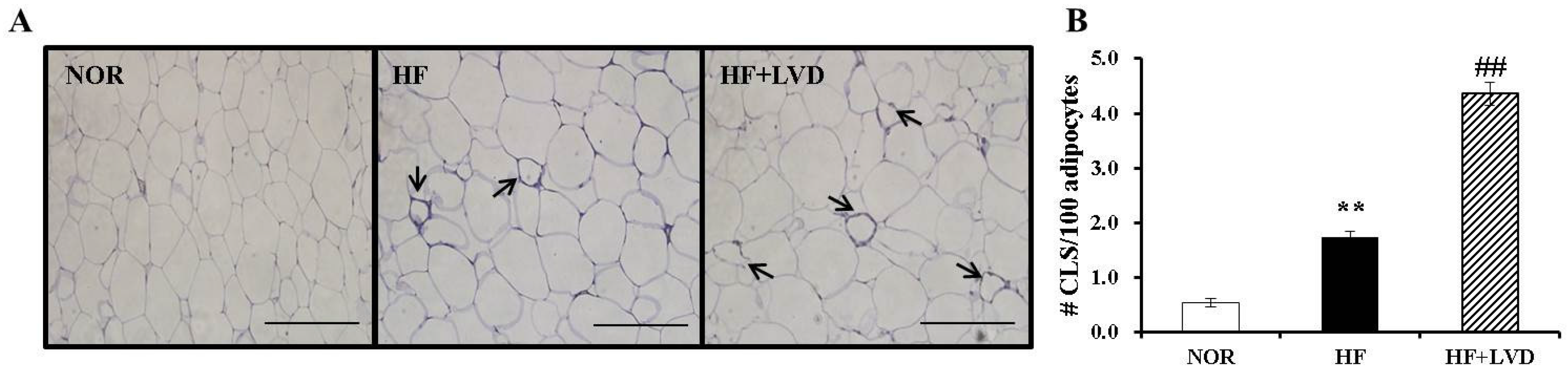
3.5. Vitamin D Insufficiency Significantly Increases Macrophage Infiltration in Obese Adipose Tissue
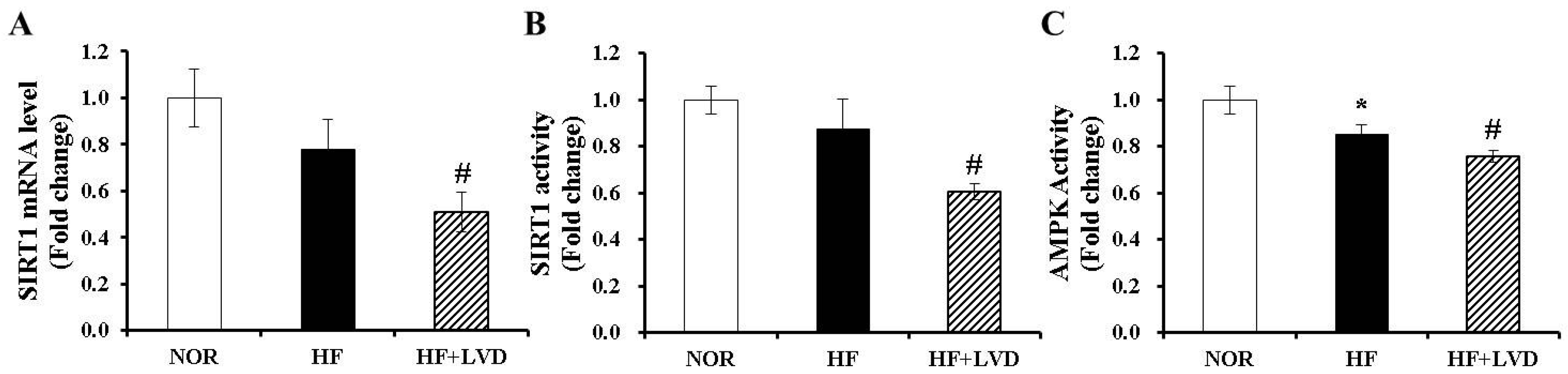
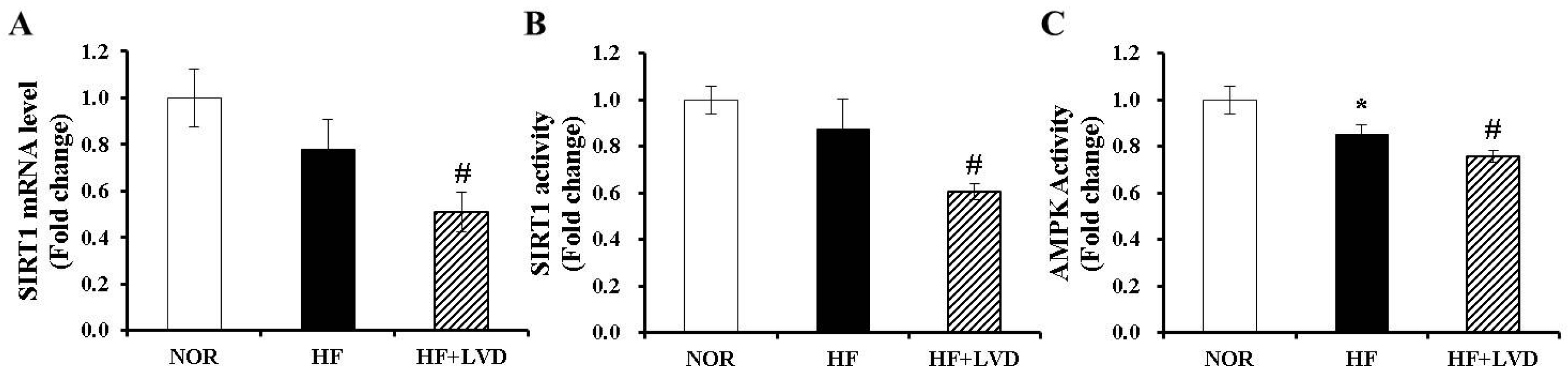
3.6. Vitamin D Insufficiency Decreases AMPK and SIRT1 Activity in Obese Adipose Tissue
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Arner, P.; Spalding, K.L. Fat cell turnover in humans. Biochem. Biophys. Res. Commun. 2010, 396, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Strissel, K.J.; Stancheva, Z.; Miyoshi, H.; Perfield, J.W., II; DeFuria, J.; Jick, Z.; Greenberg, A.S.; Obin, M.S. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 2007, 56, 2910–2918. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P.; Beattie, J.H. Physiological role of adipose tissue: White adipose tissue as an endocrine and secretory organ. Proc. Nutr. Soc. 2001, 60, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Eagon, J.C.; Trujillo, M.E.; Scherer, P.E.; Klein, S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 2007, 56, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between adipocyte size and adipokine expression and secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Antuna-Puente, B.; Feve, B.; Fellahi, S.; Bastard, J.P. Adipokines: The missing link between insulin resistance and obesity. Diabetes Metab. 2008, 34, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Goldner, W.S.; Stoner, J.A.; Thompson, J.; Taylor, K.; Larson, L.; Erickson, J.; McBride, C. Prevalence of vitamin D insufficiency and deficiency in morbidly obese patients: A comparison with non-obese controls. Obes. Surg. 2008, 18, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Molero, I.; Rojo-Martinez, G.; Morcillo, S.; Gutierrez, C.; Rubio, E.; Perez-Valero, V.; Esteva, I.; Ruiz de Adana, M.S.; Almaraz, M.C.; Colomo, N.; et al. Hypovitaminosis D and incidence of obesity: A prospective study. Eur. J. Clin. Nutr. 2013, 67, 680–682. [Google Scholar] [PubMed]
- Arunabh, S.; Pollack, S.; Yeh, J.; Aloia, J.F. Body fat content and 25-hydroxyvitamin D levels in healthy women. J. Clin. Endocrinol. Metab. 2003, 88, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Snijder, M.B.; van Dam, R.M.; Visser, M.; Deeg, D.J.; Dekker, J.M.; Bouter, L.M.; Seidell, J.C.; Lips, P. Adiposity in relation to vitamin D status and parathyroid hormone levels: A population-based study in older men and women. J. Clin. Endocrinol. Metab. 2005, 90, 4119–4123. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Massaro, J.M.; Fox, C.S.; Larson, M.G.; Keyes, M.J.; McCabe, E.L.; Robins, S.J.; O’Donnell, C.J.; Hoffmann, U.; Jacques, P.F.; et al. Adiposity, cardiometabolic risk, and vitamin D status: The framingham heart study. Diabetes 2010, 59, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Rajakumar, K.; de las Heras, J.; Chen, T.C.; Lee, S.; Holick, M.F.; Arslanian, S.A. Vitamin D status, adiposity, and lipids in black american and caucasian children. J. Clin. Endocrinol. Metab. 2011, 96, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Bellia, A.; Garcovich, C.; D’Adamo, M.; Lombardo, M.; Tesauro, M.; Donadel, G.; Gentileschi, P.; Lauro, D.; Federici, M.; Lauro, R.; et al. Serum 25-hydroxyvitamin D levels are inversely associated with systemic inflammation in severe obese subjects. Intern. Emerg. Med. 2013, 8, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rodriguez, E.; Aparicio, A.; Andres, P.; Ortega, R.M. Moderate vitamin D deficiency and inflammation related markers in overweight/obese schoolchildren. Int. J. Vitam. Nutr. Res. 2014, 84, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zemel, M.B. Calcium and 1,25-dihydroxyvitamin D3 regulation of adipokine expression. Obesity 2007, 15, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zemel, M.B. Calcitriol and calcium regulate cytokine production and adipocyte-macrophage cross-talk. J. Nutr. Biochem. 2008, 19, 392–399. [Google Scholar] [PubMed]
- Lorente-Cebrian, S.; Eriksson, A.; Dunlop, T.; Mejhert, N.; Dahlman, I.; Astrom, G.; Sjolin, E.; Wahlen, K.; Carlberg, C.; Laurencikiene, J.; et al. Differential effects of 1alpha,25-dihydroxycholecalciferol on MCP-1 and adiponectin production in human white adipocytes. Eur. J. Nutr. 2012, 51, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Marcotorchino, J.; Gouranton, E.; Romier, B.; Tourniaire, F.; Astier, J.; Malezet, C.; Amiot, M.J.; Landrier, J.F. Vitamin D reduces the inflammatory response and restores glucose uptake in adipocytes. Mol. Nutr. Food Res. 2012, 56, 1771–1782. [Google Scholar] [CrossRef] [PubMed]
- Mutt, S.J.; Karhu, T.; Lehtonen, S.; Lehenkari, P.; Carlberg, C.; Saarnio, J.; Sebert, S.; Hypponen, E.; Jarvelin, M.R.; Herzig, K.H. Inhibition of cytokine secretion from adipocytes by 1,25-dihydroxyvitamin D(3) via the NF-κB pathway. FASEB J. 2012, 26, 4400–4407. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Trayhurn, P.; Bing, C. 1,25-dihydroxyvitamin D3 inhibits the cytokine-induced secretion of MCP-1 and reduces monocyte recruitment by human preadipocytes. Int. J. Obes. 2013, 37, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.E.; Szeto, F.L.; Zhang, W.; Ye, H.; Kong, J.; Zhang, Z.; Sun, X.J.; Li, Y.C. Involvement of the vitamin D receptor in energy metabolism: Regulation of uncoupling proteins. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E820–E828. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.E.; Kong, J.; Zhang, W.; Szeto, F.L.; Ye, H.; Deb, D.K.; Brady, M.J.; Li, Y.C. Targeted expression of human vitamin D receptor in adipocytes decreases energy expenditure and induces obesity in mice. J. Biol. Chem. 2011, 286, 33804–33810. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, C.J.; Bae, J.; Esposito, D.; Komarnytsky, S.; Hu, P.; Chen, J.; Zhao, L. 1,25-Dihydroxyvitamin D3/vitamin D receptor suppresses brown adipocyte differentiation and mitochondrial respiration. Eur. J. Nutr. 2015, 54, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.S.; O’Brien, E.L.; Bigornia, S.; Mott, M.; Cacicedo, J.M.; Xu, X.J.; Gokce, N.; Apovian, C.; Ruderman, N. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem. Biophys. Res. Commun. 2011, 404, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Gaidhu, M.P.; Anthony, N.M.; Patel, P.; Hawke, T.J.; Ceddia, R.B. Dysregulation of lipolysis and lipid metabolism in visceral and subcutaneous adipocytes by high-fat diet: Role of ATGL, HSL, and AMPK. Am. J. Physiol. Cell. Physiol. 2010, 298, C961–C971. [Google Scholar] [CrossRef] [PubMed]
- Caton, P.W.; Kieswich, J.; Yaqoob, M.M.; Holness, M.J.; Sugden, M.C. Metformin opposes impaired AMPK and SIRT1 function and deleterious changes in core clock protein expression in white adipose tissue of genetically-obese db/db mice. Diabetes Obes. Metab. 2011, 13, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Rossmeisl, M.; Flachs, P.; Brauner, P.; Sponarova, J.; Matejkova, O.; Prazak, T.; Ruzickova, J.; Bardova, K.; Kuda, O.; Kopecky, J. Role of energy charge and AMP-activated protein kinase in adipocytes in the control of body fat stores. Int. J. Obes. Relat. Metab. Disord. 2004, 28 (Suppl. 4), S38–S44. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Kahn, B.B.; Shi, H.; Xue, B.Z. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J. Biol. Chem. 2010, 285, 19051–19059. [Google Scholar] [CrossRef] [PubMed]
- Galic, S.; Fullerton, M.D.; Schertzer, J.D.; Sikkema, S.; Marcinko, K.; Walkley, C.R.; Izon, D.; Honeyman, J.; Chen, Z.P.; van Denderen, B.J.; et al. Hematopoieti-educes mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J. Clin. Investig. 2011, 121, 4903–4915. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Chalkiadaki, A.; Guarente, L. High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell. Metab. 2012, 16, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Mayoral, R.; Osborn, O.; McNelis, J.; Johnson, A.M.; Oh da, Y.; Izquierdo, C.L.; Chung, H.; Li, P.; Traves, P.G.; Bandyopadhyay, G.; et al. Adipocyte SIRT1 knockout promotes PPARgamma activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol. Metab. 2015, 4, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. Sirt1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Kim, Y. Vitamin D decreases adipocyte lipid storage and increases NAD-SIRT1 pathway in 3T3-L1 adipocytes. Nutrition 2016, 32, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Nuclear Regulatory Commission (NRC). Nutrient Requirements of Laboratory Animals, 4th ed.; National Academy Press: Washington, DC, USA, 1995. [Google Scholar]
- Fleet, J.C.; Gliniak, C.; Zhang, Z.; Xue, Y.; Smith, K.B.; McCreedy, R.; Adedokun, S.A. Serum metabolite profiles and target tissue gene expression define the effect of cholecalciferol intake on calcium metabolism in rats and mice. J. Nutr. 2008, 138, 1114–1120. [Google Scholar] [PubMed]
- Park, C.Y.; Lee, W.H.; Fleet, J.C.; Allen, M.R.; McCabe, G.P.; Walsh, D.M.; Weaver, C.M. Calcium and vitamin D intake maintained from preovariectomy independently affect calcium metabolism and bone properties in sprague dawley rats. Osteoporos. Int. 2014, 25, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hollis, B.W. Circulating 25-hydroxyvitamin D levels indicative of vitamin D sufficiency: Implications for establishing a new effective dietary intake recommendation for vitamin D. J. Nutr. 2005, 135, 317–322. [Google Scholar] [PubMed]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M. Evaluation, treatment, and prevention of vitamin D deficiency: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef] [PubMed]
- Caron-Jobin, M.; Morisset, A.S.; Tremblay, A.; Huot, C.; Legare, D.; Tchernof, A. Elevated serum 25(OH)D concentrations, vitamin D, and calcium intakes are associated with reduced adipocyte size in women. Obesity 2011, 19, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Marcotorchino, J.; Tourniaire, F.; Astier, J.; Karkeni, E.; Canault, M.; Amiot, M.J.; Bendahan, D.; Bernard, M.; Martin, J.C.; Giannesini, B.; et al. Vitamin D protects against diet-induced obesity by enhancing fatty acid oxidation. J. Nutr. Biochem. 2014, 25, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, I.N.; Song, Q. High vitamin D and calcium intakes reduce diet-induced obesity in mice by increasing adipose tissue apoptosis. Mol. Nutr. Food Res. 2014, 58, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [PubMed]
- Vega, R.B.; Huss, J.M.; Kelly, D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 2000, 20, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, D.M.; Rinaldo, P.; Rhead, W.J.; Tian, L.; Millington, D.S.; Vockley, J.; Hamm, D.A.; Brix, A.E.; Lindsey, J.R.; Pinkert, C.A.; et al. Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc. Natl. Acad. Sci. USA 1998, 95, 15592–15597. [Google Scholar] [CrossRef] [PubMed]
- Goetzman, E.S. The regulation of acyl-CoA dehydrogenases in adipose tissue by rosiglitazone. Obesity 2009, 17, 196–198. [Google Scholar] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor gamma (PPAR gamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar] [PubMed]
- Lira, F.S.; Rosa, J.C.; Cunha, C.A.; Ribeiro, E.B.; do Nascimento, C.O.; Oyama, L.M.; Mota, J.F. Supplementing alpha-tocopherol (vitamin E) and vitamin D3 in high fat diet decrease IL-6 production in murine epididymal adipose tissue and 3T3-L1 adipocytes following lps stimulation. Lipids Health Dis. 2011, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Karkeni, E.; Marcotorchino, J.; Tourniaire, F.; Astier, J.; Peiretti, F.; Darmon, P.; Landrier, J.F. Vitamin D limits chemokine expression in adipocytes and macrophage migration in vitro and in male mice. Endocrinology 2015, 156, 1782–1793. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Elfers, C.T.; Figlewicz, D.P.; Melhorn, S.J.; Morton, G.J.; Hoofnagle, A.; Yeh, M.M.; Nelson, J.E.; Kowdley, K.V. Vitamin D deficiency in obese rats exacerbates nonalcoholic fatty liver disease and increases hepatic resistin and toll-like receptor activation. Hepatology 2012, 55, 1103–1111. [Google Scholar] [PubMed]
- Villena, J.A.; Viollet, B.; Andreelli, F.; Kahn, A.; Vaulont, S.; Sul, H.S. Induced adiposity and adipocyte hypertrophy in mice lacking the AMPK-activated protein kinase-alpha2 subunit. Diabetes 2004, 53, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.E.; Brocklehurst, K.J.; Marley, A.E.; Carey, F.; Carling, D.; Beri, R.K. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with aicar, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994, 353, 33–36. [Google Scholar] [CrossRef]
- Lihn, A.S.; Jessen, N.; Pedersen, S.B.; Lund, S.; Richelsen, B. AICAR stimulates adiponectin and inhibits cytokines in adipose tissue. Biochem. Biophys. Res. Commun. 2004, 316, 853–858. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NOR | HF | HF+LVD | |
|---|---|---|---|
| Casein (g) | 200 | 200 | 200 |
| l-Cystine (g) | 3 | 3 | 3 |
| Corn starch (g) | 550 | 72.8 | 72.8 |
| Maltodextrin (g) | 150 | 100 | 100 |
| Sucrose (g) | 0 | 172.8 | 172.8 |
| Cellulose (g) | 50 | 50 | 50 |
| Soybean oil (g) | 25 | 25 | 25 |
| Lard (g) | 20 | 177.5 | 177.5 |
| Total kcal | 4057 | 4057 | 4057 |
| Carbohydrate (% of energy) | 70 | 35 | 35 |
| Fat (% of energy) | 10 | 45 | 45 |
| Vitamin D (IU/kg diet) | 1000 | 1000 | 25 |
| Gene | GeneBank No. | Forward Sequence (5′-3′) | Reverse Sequence (5′-3′) | Product Size (bp) |
|---|---|---|---|---|
| aP2 | NM_053365 | TCACCCCAGATGACAGGAAA | CATGACACATTCCACCACCA | 140 |
| β-actin | NM_031144 | GTCGTACCACTGGCATTGTG | TCTCAGCTGTGGTGGTGAAG | 180 |
| CPT1α | NM_031559.2 | ATGACGGCTATGGTGTCTCC | GTGAGGCCAAACAAGGTGAT | 154 |
| IL-6 | NM_012585 | ATAGTCCTTCCTACCCCAAC | TGCCGAGTAGACCTCATAGT | 143 |
| LCAD | NM_012819.1 | CCTACAGCTGCATGAAACCA | GACGATCTGTCTTGCGATCA | 229 |
| MCAD | NM_016986.2 | TATGCCCTGGACAGGAAAAC | CCTTCGCAATAGAGGCAAAG | 172 |
| MCP-1 | NM_031530 | ACTCACCTGCTGCTACTCAT | CTACAGCTTCTTTGGGACAC | 101 |
| PGC1α | NM_031347.1 | ATGAGAAGCGGGAGTCTGAA | TGCATTCCTCAATTTCACCA | 159 |
| PPARα | NM_013196.1 | TACCTGTGAACACGATCTGA | GCTAGTCTTTCCTGCGAGTA | 136 |
| PPARγ | NM_001145366 | TGTGGGGATAAAGCATCAGC | CAAGGCACTTCTGAAACCGA | 175 |
| SIRT1 | XM_008774951.1 | AGGGAACCTCTGCCTCATCT | GAGGTGTTGGTGGCAACTCT | 199 |
| SREBP1c | AF286470 | AGGAGGCCATCTTGTTGCTT | GTTTTGACCCTTAGGGCAGC | 134 |
| TNFα | NM_012675 | CCCCTTTATCGTCTACTCCT | ACTACTTCAGCGTCTCGTGT | 139 |
| UCP1 | NM_012682.2 | GACTCGGATCCTGGAACGTC | GCATAGGAGCCCAGCATAGG | 151 |
| VLCAD | NM_012891.2 | GCATCTTGCTCTATGGCACA | ACTTTCCACAGGGGCTAGGT | 156 |
| NOR (n = 9) | HF (n = 7) | HF+LVD (n = 7) | |
|---|---|---|---|
| Body weight (g) | |||
| - Initial body weight | 105.86 ± 2.64 | 109.69 ± 2.50 | 110.75 ± 3.36 |
| - Final body weight | 483.84 ± 11.49 a | 532.21 ± 10.36 b | 571.08 ± 7.52 c |
| - Weight change | 377.98 ± 11.63 a | 422.53 ± 10.65 b | 458.03 ± 8.91 c |
| Food intake (g/day) | 20.66 ± 0.37 b | 18.03 ± 0.43 a | 20.03 ± 0.37 b |
| Energy intake (kcal/day) | 79.43 ± 1.42 a | 85.23 ± 2.02 b | 94.68 ± 1.75 c |
| Food efficiency (g gain/g consumed) | 0.22 ± 0.005 a | 0.28 ± 0.004 b | 0.28 ± 0.004 b |
| Energy efficiency (g gain/kcal consumed) | 0.057 ± 0.002 | 0.060 ± 0.000 | 0.059 ± 0.001 |
| Tissue weight (g/100 g body weight) | |||
| - Liver | 2.87 ± 0.06 | 3.05 ± 0.11 | 3.14 ± 0.11 |
| - Skeletal muscle | 0.68 ± 0.03 | 0.61 ± 0.04 | 0.66 ± 0.05 |
| NOR (n = 9) | HF (n = 7) | HF+LVD (n = 7) | |
|---|---|---|---|
| 25(OH)D (nmol/L) | 102.59 ± 6.75 | 103.46 ± 5.76 | 68.56 ± 7.97 # |
| Glucose (mmol/L) | 12.28 ± 0.98 | 14.03 ± 2.40 | 16.74 ± 1.95 * |
| Insulin (μU/mL) | 39.90 ± 4.19 | 58.77± 5.06 ** | 75.56 ± 8.17 # |
| Lipids (mmol/L) | |||
| - Triglyceride | 0.87 ± 0.10 | 1.22 ± 0.08 * | 1.80 ± 0.28 * |
| - Total cholesterol | 1.97 ± 0.13 | 2.63 ± 0.13 ** | 3.56 ± 0.54 # |
| - HDL cholesterol | 2.13 ± 0.10 | 2.35 ± 0.10 | 2.35 ± 0.21 |
| - LDL cholesterol | 0.21 ± 0.03 | 0.29 ± 0.03 ** | 0.39 ± 0.03 ## |
| AST (IU/L) | 107.50 ± 11.82 | 107.00 ± 13.85 | 107.83 ± 12.97 |
| ALT (IU/L) | 33.63 ± 6.45 | 30.60 ± 4.23 | 30.71 ± 2.56 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, E.; Kim, Y. Vitamin D Insufficiency Exacerbates Adipose Tissue Macrophage Infiltration and Decreases AMPK/SIRT1 Activity in Obese Rats. Nutrients 2017, 9, 338. https://doi.org/10.3390/nu9040338
Chang E, Kim Y. Vitamin D Insufficiency Exacerbates Adipose Tissue Macrophage Infiltration and Decreases AMPK/SIRT1 Activity in Obese Rats. Nutrients. 2017; 9(4):338. https://doi.org/10.3390/nu9040338
Chicago/Turabian StyleChang, Eugene, and Yangha Kim. 2017. "Vitamin D Insufficiency Exacerbates Adipose Tissue Macrophage Infiltration and Decreases AMPK/SIRT1 Activity in Obese Rats" Nutrients 9, no. 4: 338. https://doi.org/10.3390/nu9040338
APA StyleChang, E., & Kim, Y. (2017). Vitamin D Insufficiency Exacerbates Adipose Tissue Macrophage Infiltration and Decreases AMPK/SIRT1 Activity in Obese Rats. Nutrients, 9(4), 338. https://doi.org/10.3390/nu9040338