Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats

 , ,
, , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Treatments
2.2. Plasma Parameters
2.3. Genomic DNA Isolation
2.4. Quantitative Polymerase Chain Reaction (QPCR)
- mtDNA long fragment (13.4 Kbp): 5′-AAAATCCCCGCAAACAATGACCACCC-3′ (sense)/5′-GGCAATTAAGAGTGGGATGGAGCCAA-3′ (anti-sense);
- mtDNA short fragment (235 bp): 5′-CCTCCCATTCATTATCGCCGCCCTGC-3′ (sense)/5′-GTCTGGGTCTCCTAGTAGGTCTGGGAA-3′ (anti-sense).
2.5. mtDNA Copy Number
- COII: 5′-TGAGCCATCCCTTCACTAGG-3′ (sense)/5′-TGAGCCGCAAATTTCAGAG-3′(anti-sense);
- β-actin: 5′-CTGCTCTTTCCCAGATGAGG-3′ (sense)/5′-CCACAGCACTGTAGGGGTTT-3′ (anti-sense).
2.6. mRNA Expression
- β-actin: 5′-CTGCTCTTTCCCAGATGAGG-3′ (sense)/5′-CCACAGCACTGTAGGGGTTT-3′ (anti-sense);
- Polg: 5′-GAAGAGCGTTACTCTTGGACCAG-3′ (sense)/5′-AACATTGTGCCCCACCACTAAC-3′ (anti-sense);
- Pgc1α: 5′-GTCAACAGCAAAAGCCACAA-3′ (sense)/5′-GTGTGAGGAGGGTCATCGTT-3′ (anti-sense);
- Nrf1: 5′-CTGATGGCCATTACATGTGG-3′ (sense)/5′-GTAAAGCCCGGAAGGTTCTT-3′ (anti-sense);
- Tfam: 5′-CAACAGGGAAGAAACGGAAA-3′ (sense)/5′-GTGGCTCTGAGTTTCCGAAG-3′ (anti-sense).
2.7. Preparation of Hepatic Homogenate and Isolated Mitochondria
2.8. Hepatic Lipid Peroxidation
2.9. Hepatic Myeloperoxidase (MPO) Activity
2.10. Western Blotting
2.11. Statistical Analysis
3. Results
3.1. Effect of Fructose-Rich Diet on Hepatic Functionality
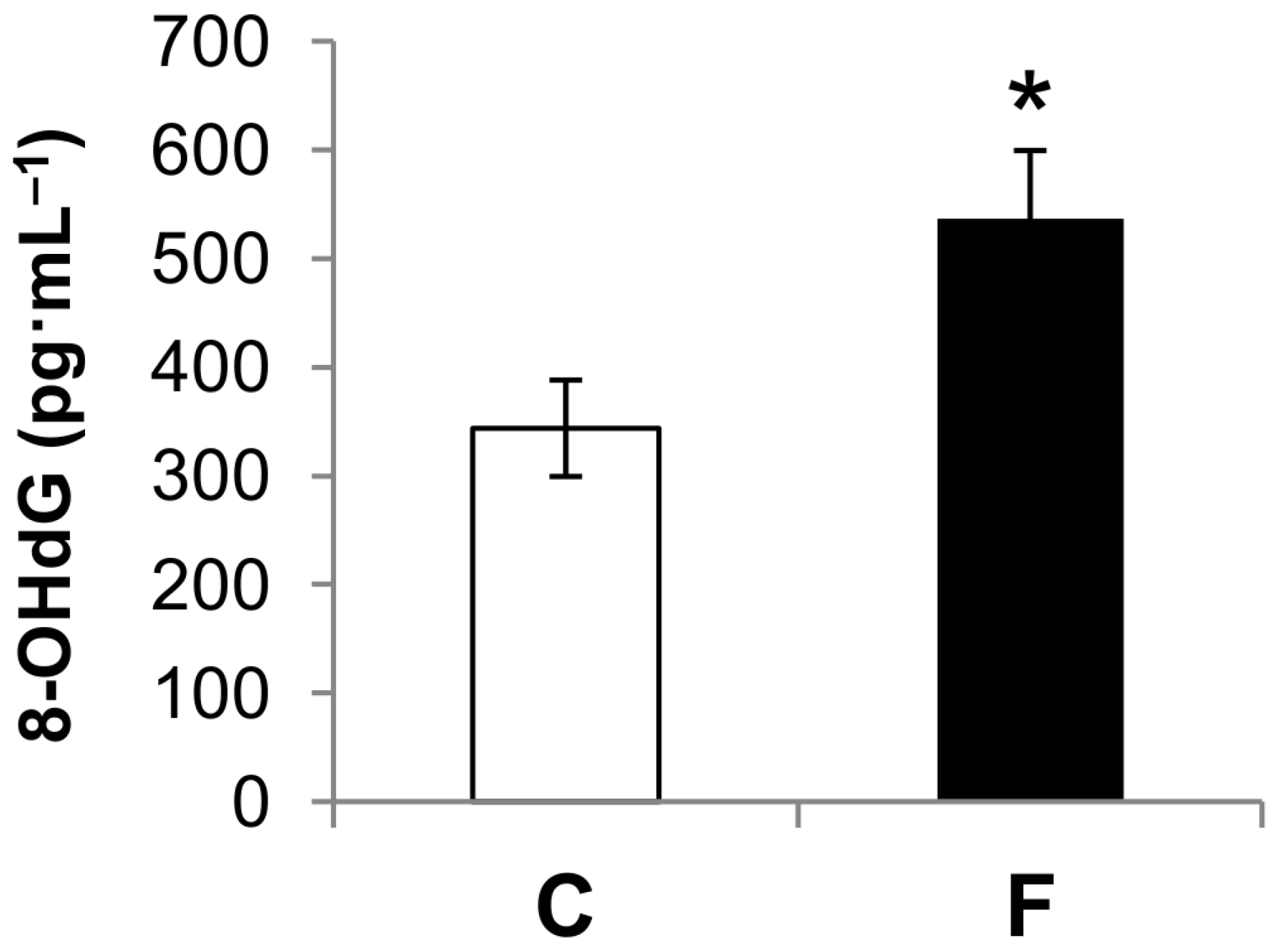
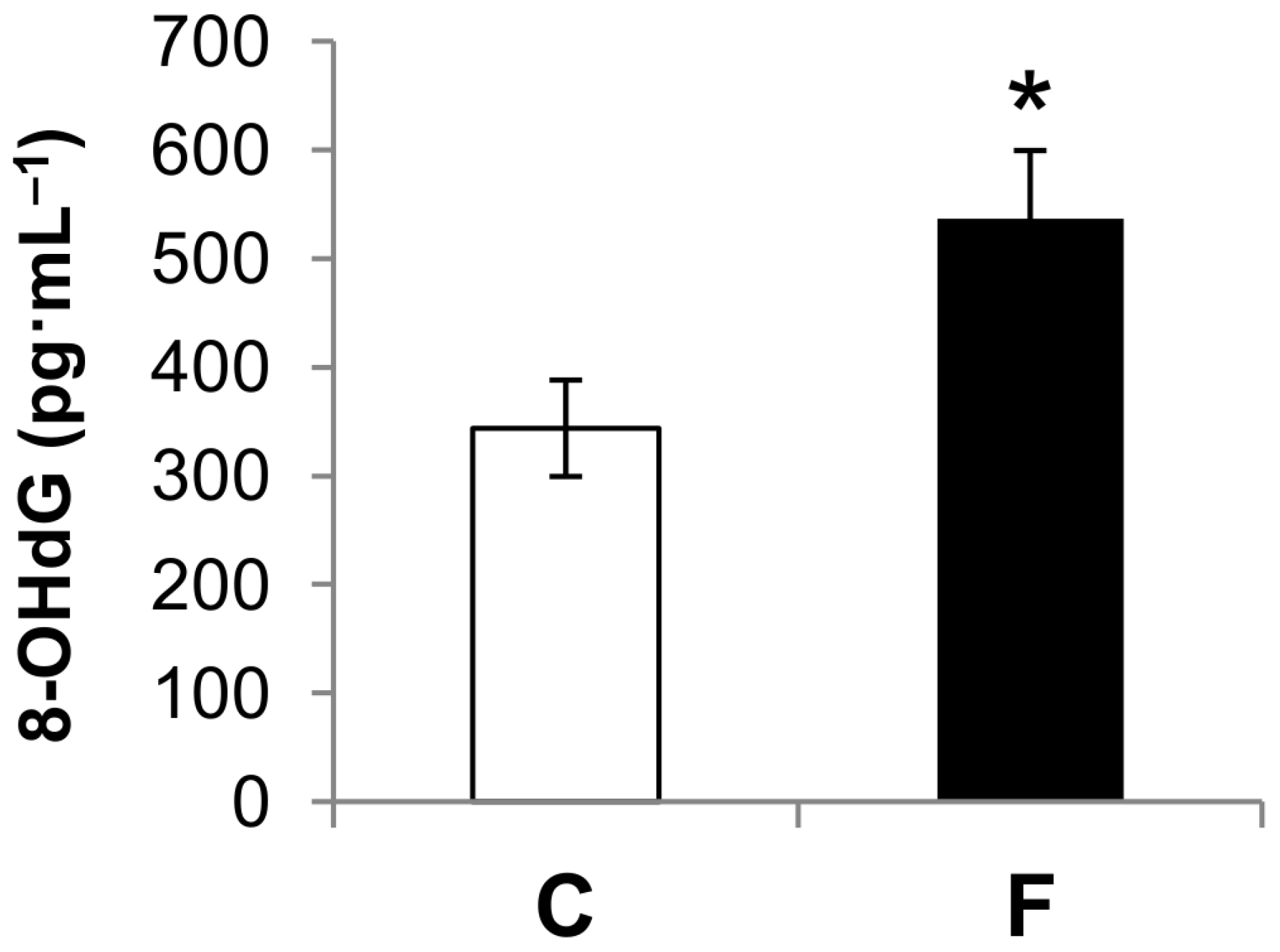
3.2. Effect of Fructose-Rich Diet on Plasma 8-OHdG Concentration
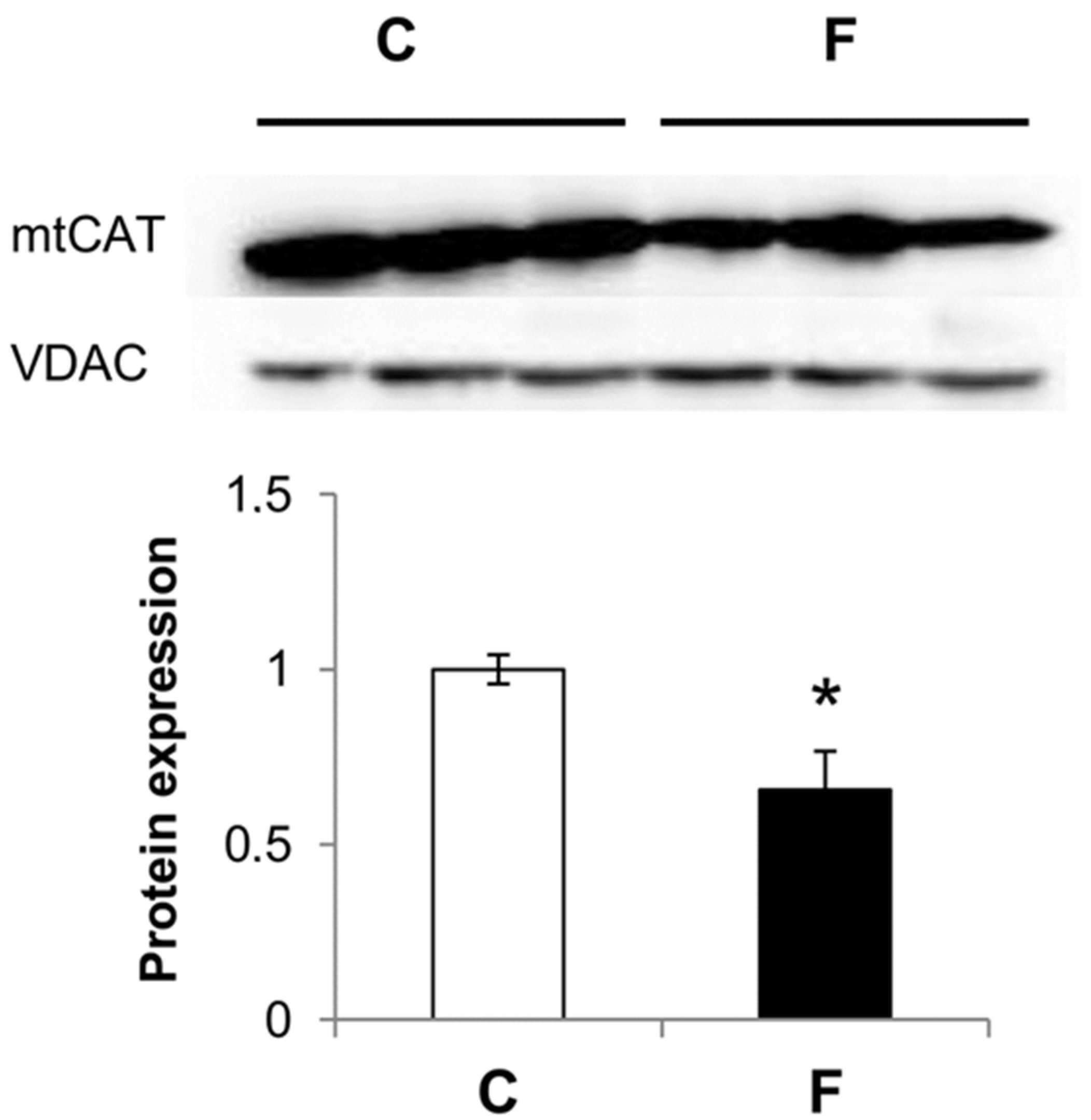
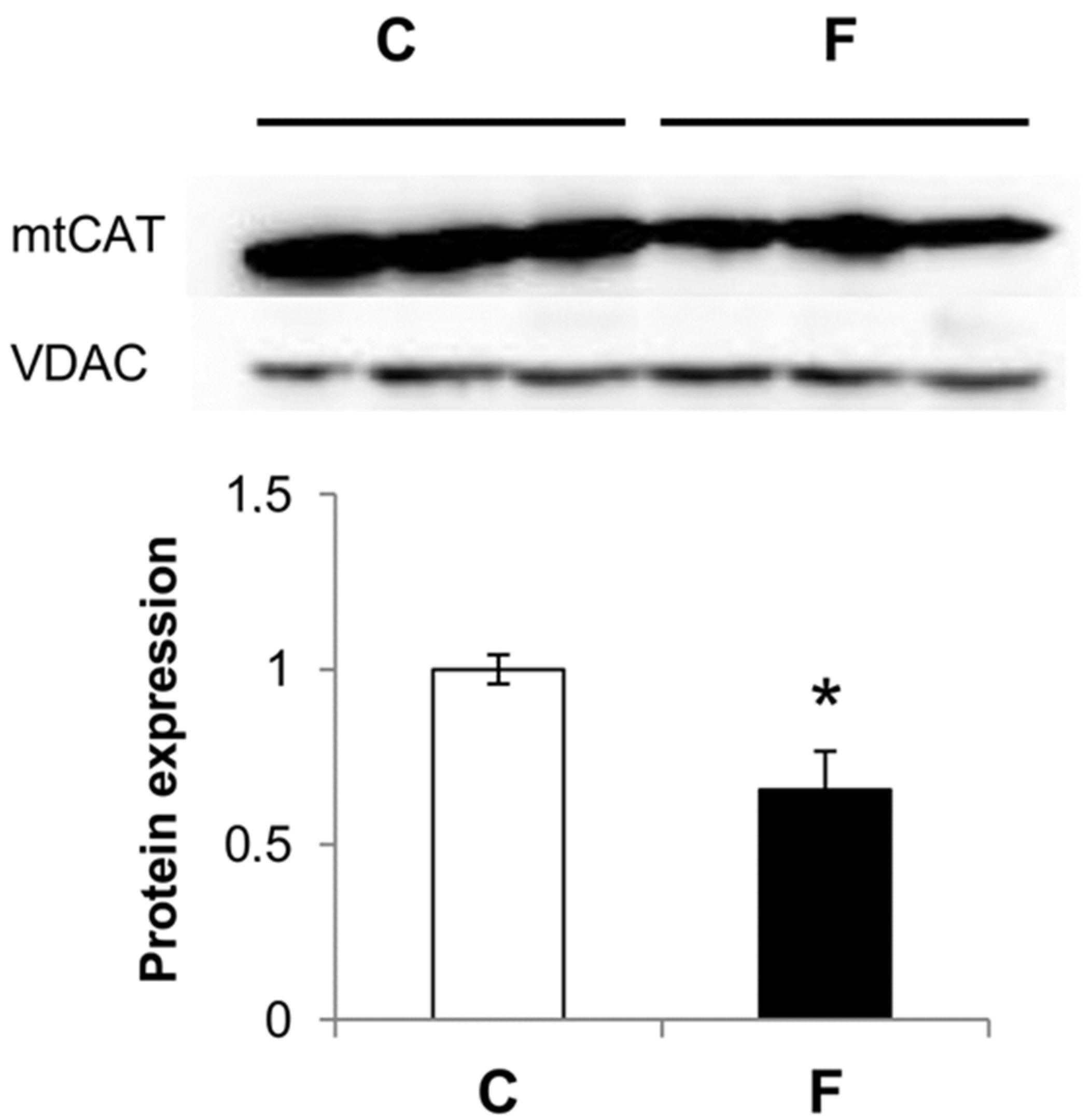
3.3. Effect of Fructose-Rich Diet on Mitochondrial Catalase Expression
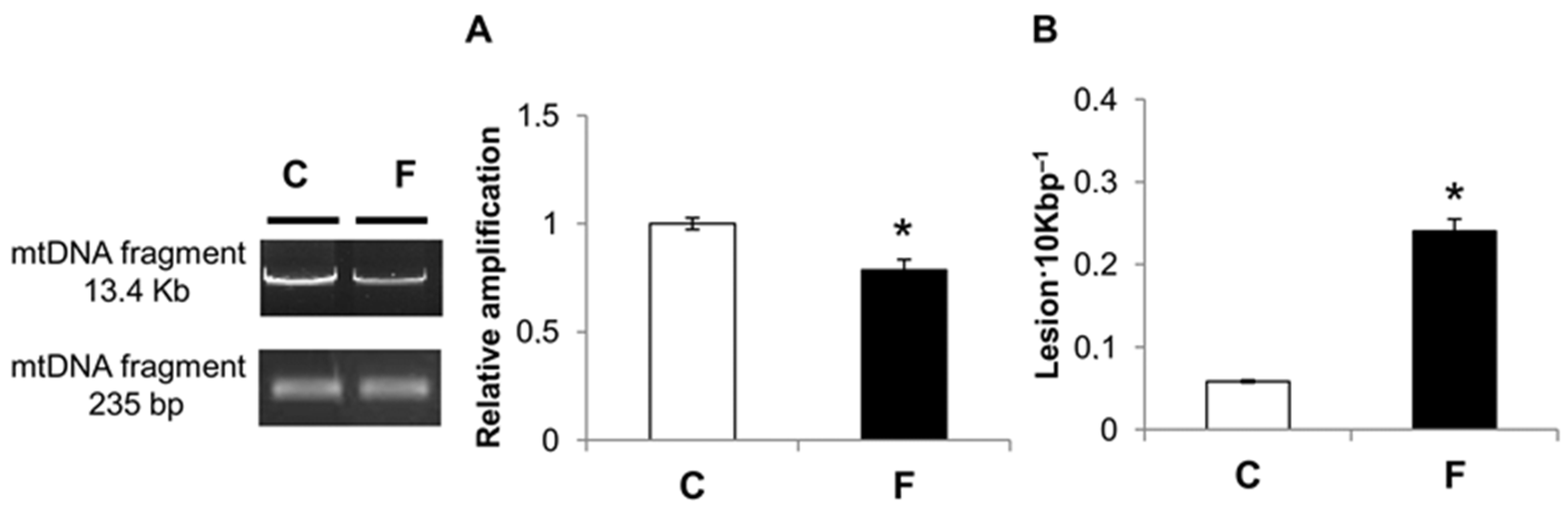
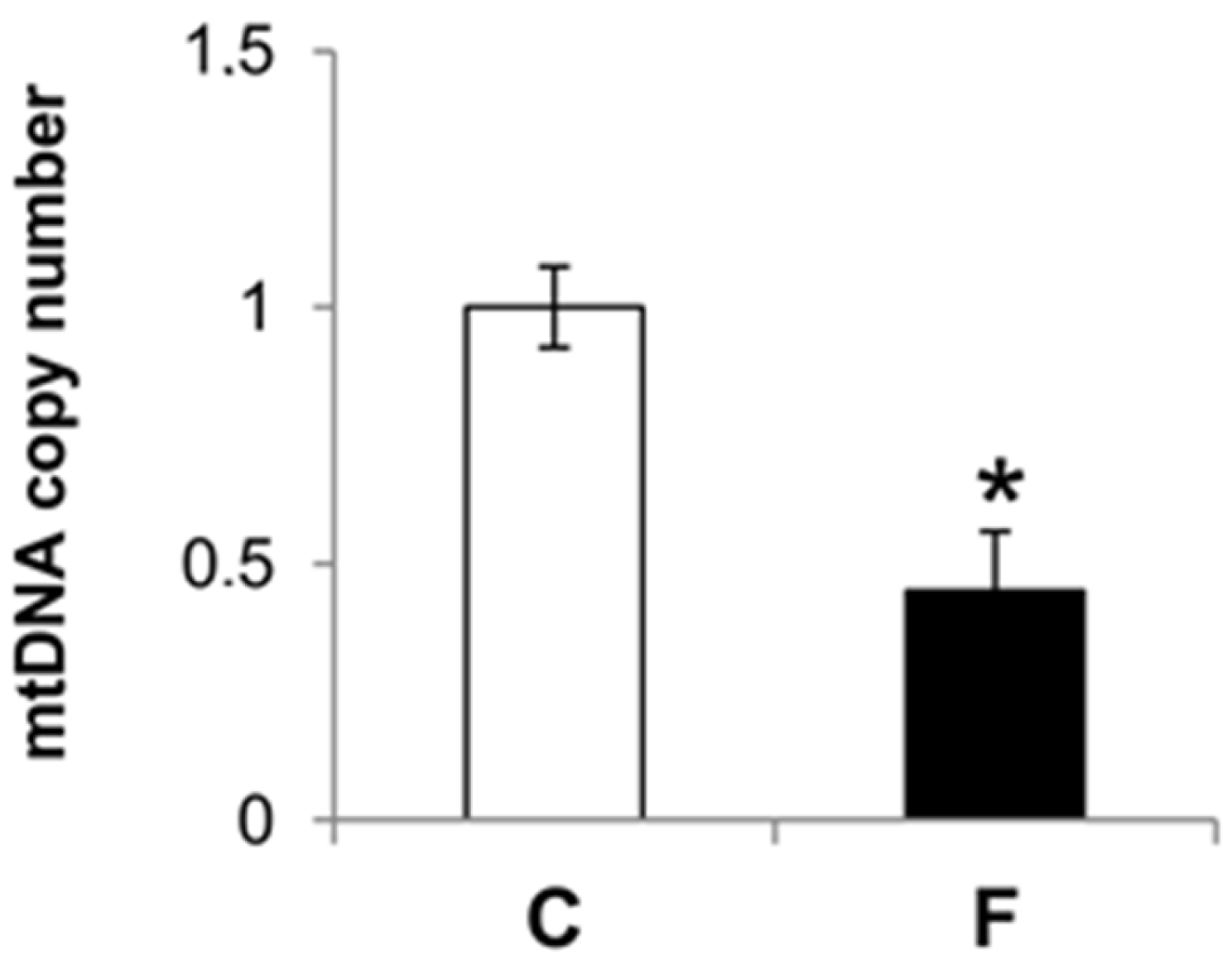
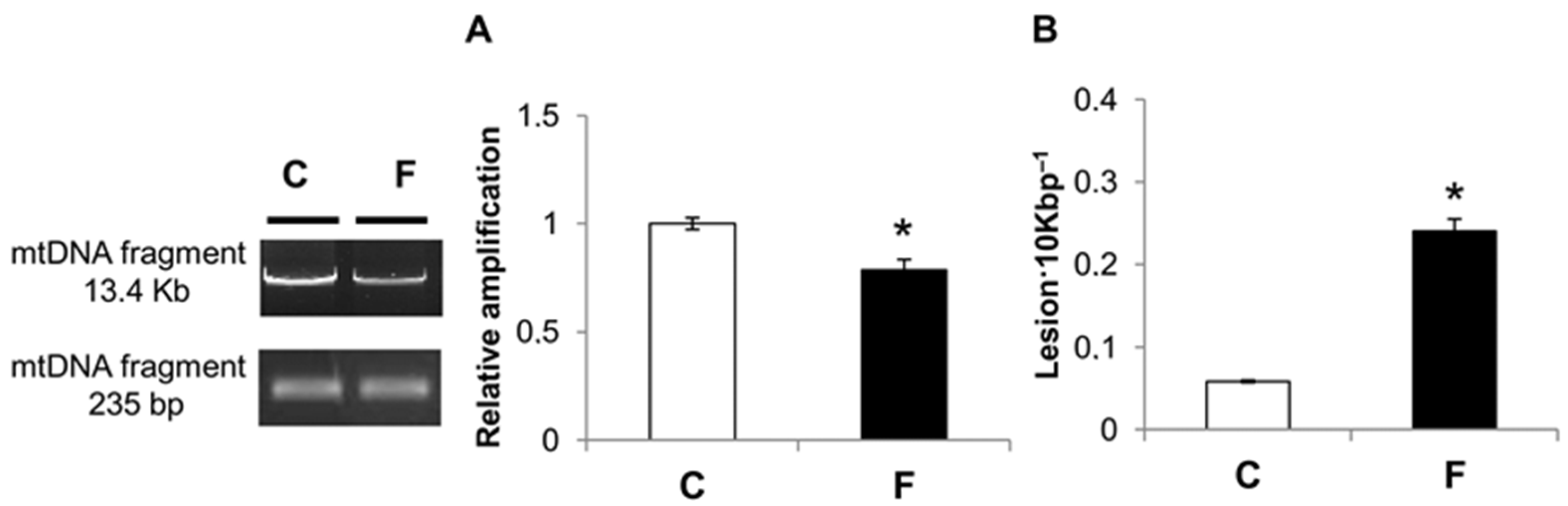
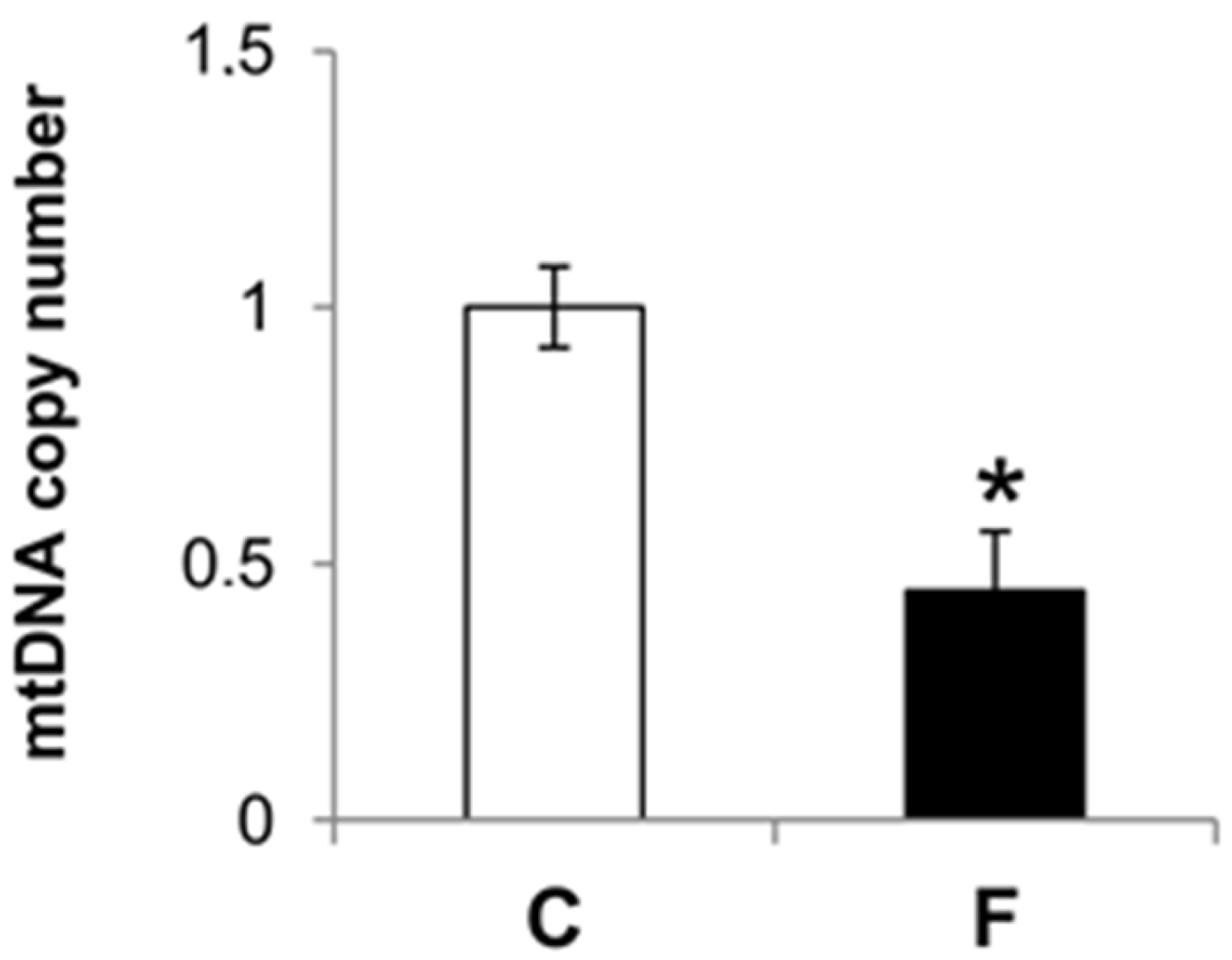
3.4. Effect of Fructose-Rich Diet on mtDNA Damage and Copy Number
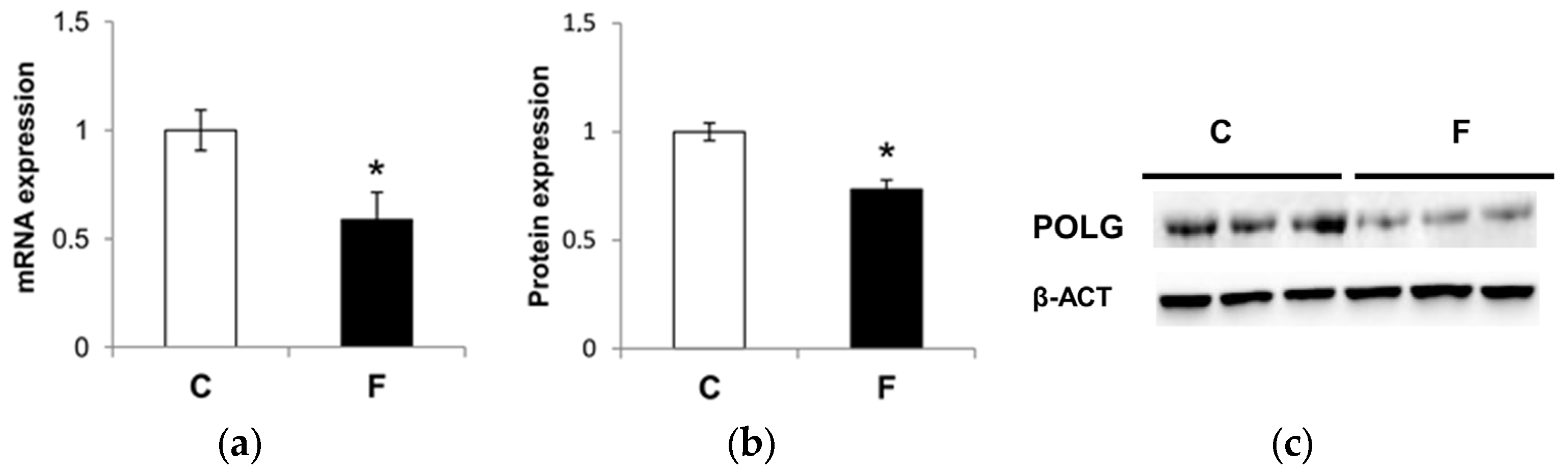
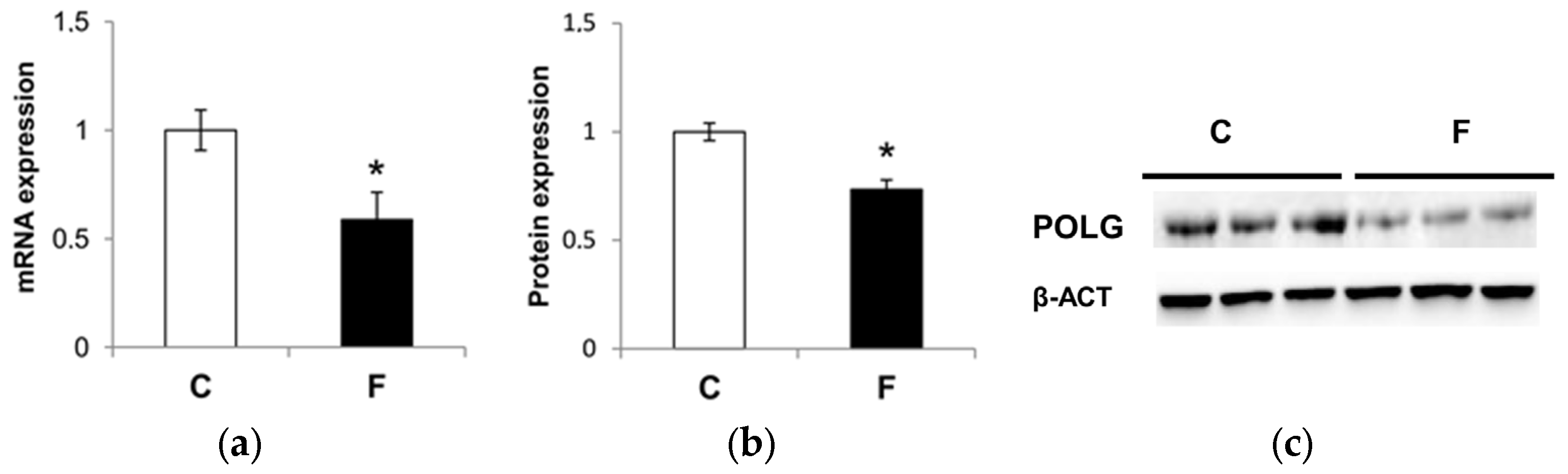
3.5. Effect of Fructose-Rich Diet on Mitochondrial POLG
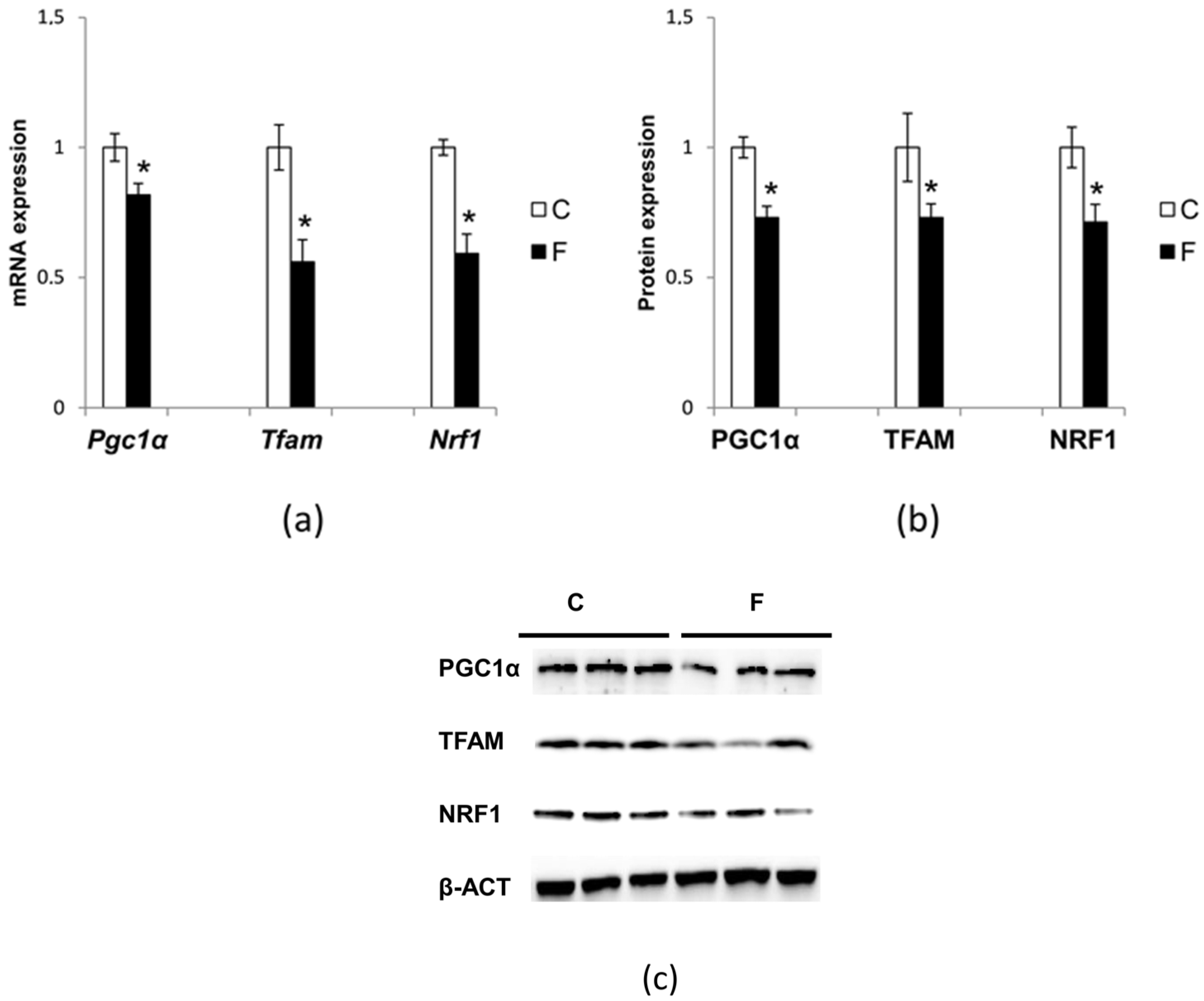
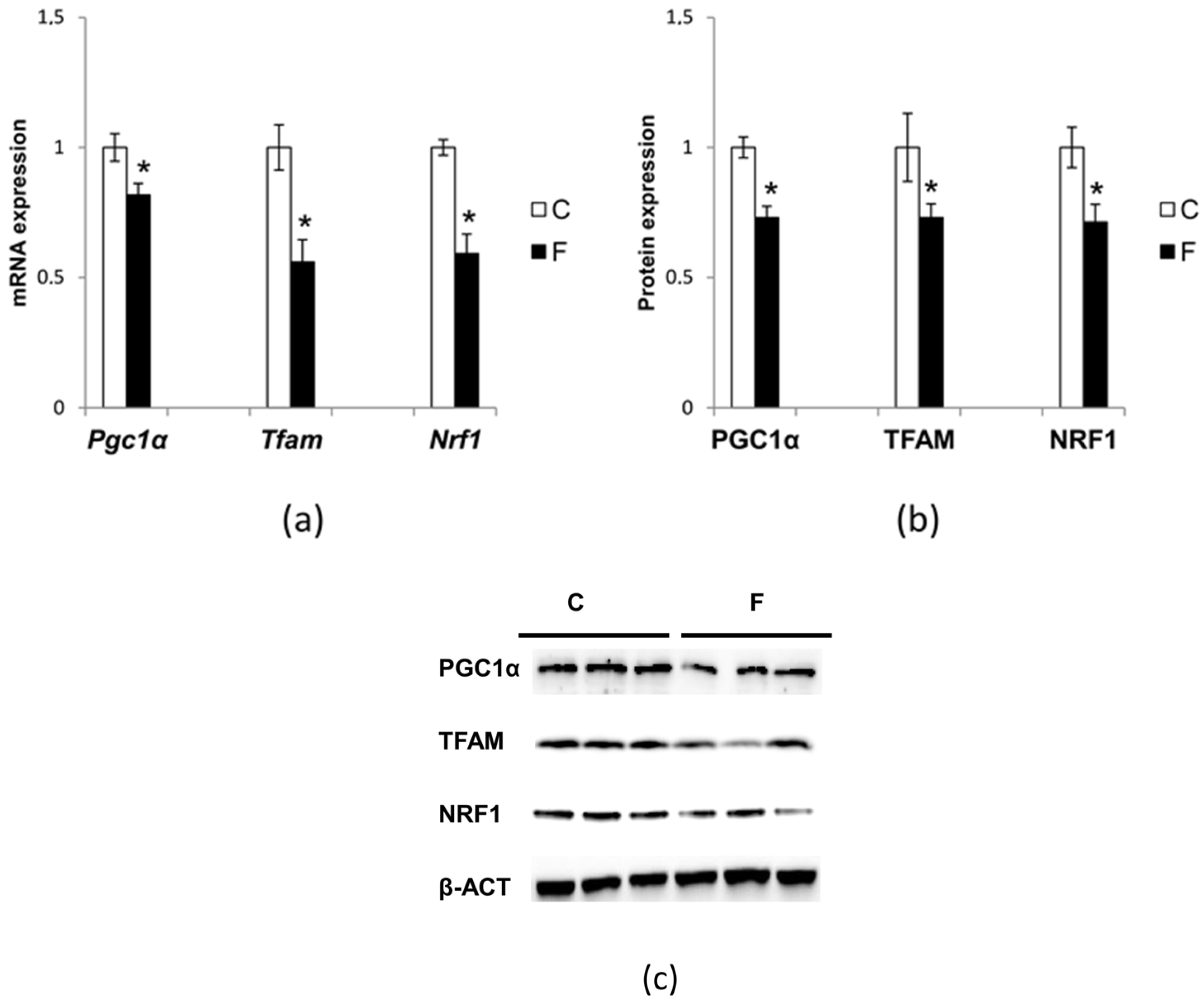
3.6. Effect of Fructose-Rich Diet on Mitochondrial Biogenesis
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marriott, B.P.; Cole, N.; Lee, E. National estimates of dietary fructose intake increased from 1977 to 2004 in the United States. J. Nutr. 2009, 139, 1S–8S. [Google Scholar] [CrossRef] [PubMed]
- Hanover, L.M.; White, J.S. Manufacturing, composition, and applications of fructose. Am. J. Clin. Nutr. 1993, 58, 724S–732S. [Google Scholar] [PubMed]
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [PubMed]
- Hu, F.B.; Malik, V.S. Sugar-sweetened beverages and risk of obesity and type 2 diabetes: Epidemiological evidence. Physiol. Behav. 2010, 100, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Dekker, M.J.; Su, Q.; Baker, C.; Rutledge, A.C.; Adeli, K. Fructose: A highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am. J. Physiol. 2010, 299, E685–E694. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A.; Tran, C.; Paquot, N. Fructose and metabolic diseases: New findings, new questions. Nutrition 2010, 26, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Bianco, F.; Coppola, P.; Mazzoli, A.; Cigliano, L.; Liverini, G.; Iossa, S. Increased skeletal muscle mitochondrial efficiency in rats with fructose-induced alteration in glucose tolerance. Br. J. Nutr. 2013, 110, 1996–2003. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Bianco, F.; Coppola, P.; Mazzoli, A.; Valiante, S.; Liverini, G.; Iossa, S. Adipose tissue remodeling in rats exhibiting fructose-induced obesity. Eur. J. Nutr. 2014, 53, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.H.; Chen, X.Y.; Hu, Q.H.; Wang, M.X.; Jin, R.; Zhang, Q.Y.; Wang, W.; Wang, R.; Kang, L.L.; et al. Reactive oxygen species-induced TXNIP drives fructose-mediated hepatic inflammation and lipid accumulation through NLRP3 inflammasome activation. Antioxid. Redox Signal. 2015, 22, 848–870. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Baker, C.; Christian, P.; Naples, M.; Tong, X.; Zhang, K.; Santha, M.; Adeli, K. Hepatic mitochondrial and ER stress induced by defective PPARα signaling in the pathogenesis of hepatic steatosis. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1264–E1273. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Bianco, F.; Falcone, I.; Coppola, P.; Coppola, P.; Liverini, G.; Iossa, S. Increased hepatic de novo lipogenesis and mitochondrial efficiency in a model of obesity induced by diets rich in fructose. Eur. J. Nutr. 2013, 52, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed]
- Bagul, P.K.; Middela, H.; Matapally, S.; Padiya, R.; Bastia, T.; Madhusudana, K.; Reddy, B.R.; Chakravarty, S.; Banerjee, S.K. Attenuation of insulin resistance, metabolic syndrome and hepatic oxidative stress by resveratrol in fructose-fed rats. Pharmacol. Res. 2012, 66, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.C.; Massa, M.L.; Schinella, G.; Gagliardino, J.J.; Francini, F. Lipoic acid prevents liver metabolic changes induced by administration of a fructose-rich diet. Biochim. Biophys. Acta 2013, 1830, 2226–2232. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.C.; Francini, F.; Gagliardino, J.J.; Massa, M.L. Lipoic acid prevents fructose-induced changes in liver carbohydrate metabolism: Role of oxidative stress. Biochim. Biophys. Acta 2014, 1840, 1145–1151. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.K.; Sindhu, K.K. Oxidative stress and metabolic syndrome. Life Sci. 2009, 84, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Rad Biol Med. 2011, 50, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Hokayem, M.; Blond, E.; Vidal, H.; Lambert, K.; Meugnier, E.; Feillet-Coudray, C.; Coudray, C.; Pesenti, S.; Luyton, C.; Lambert-Porcheron, S.; et al. Grape polyphenols prevent fructose-induced oxidative stress and insulin resistance in first-degree relatives of type 2 diabetic patients. Diabetes Care 2013, 36, 1454–1461. [Google Scholar] [CrossRef] [PubMed]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; Palmieri, V.O.; Portincasa, P.; Moschetta, A.; Palasciano, G. Oxidative stress-induced risk factors associated with the metabolic syndrome: A unifying hypothesis. J. Nutr. Biochem. 2008, 19, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Mamikutty, N.; Thent, Z.C.; Haji Suhaimi, F. Fructose-drinking water induced nonalcoholic fatty liver disease and ultrastructural alteration of hepatocyte mitochondria in male Wistar rat. Biomed. Res. Int. 2015, 2015, 895961–895967. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, A.; Turnu, L.; Porro, B.; Squellerio, I.; Cavalca, V.; Tremoli, E.; Di Minno, M.N. Hydroxy-2-deoxyguanosine levels and cardiovascular disease: A systematic review and meta-analysis of the literature. Antioxid. Redox Signal. 2016, 24, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.H.; Meyer, J.N.; Mandavilli, B.S.; Van Houten, B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 2006, 314, 183–199. [Google Scholar] [PubMed]
- Fernandes, M.A.; Custódio, J.B.; Santos, M.S.; Moreno, A.J.; Vicente, J.A. Tetrandrine concentrations not affecting oxidative phosphorylation protect rat liver mitochondria from oxidative stress. Mitochondrion 2006, 6, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Shajib, M.S.; Manocha, M.M.; Khan, W.I. Investigating intestinal inflammation in DSS-induced model of IBD. J. Vis. Exp. 2012, 60, 3678. [Google Scholar] [CrossRef] [PubMed]
- Krawisz, J.E.; Sharon, P.; Stenson, W.F. Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology 1984, 87, 1344–1350. [Google Scholar] [PubMed]
- Cheng, K.C.; Cahill, D.S.; Kasai, H.; Nishimura, S.; Loeb, L.A. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G–T and A–C substitutions. J. Biol. Chem. 1992, 267, 166–172. [Google Scholar] [PubMed]
- Scarpulla, R.C. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim. Biophys. Acta 2002, 1576, 1–14. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 7, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 6, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Salvi, M.; Battaglia, V.; Brunati, A.M.; La Rocca, N.; Tibaldi, E.; Pietrangeli, P.; Marcocci, L.; Mondovì, B.; Rossi, C.A.; Toninello, A. Catalase takes part in rat liver mitochondria oxidative stress defense. J. Biol. Chem. 2007, 282, 24407–24415. [Google Scholar] [CrossRef] [PubMed]
- Arita, Y.; Harkness, S.H.; Kazzaz, J.A.; Koo, H.C.; Joseph, A.; Melendez, J.A.; Davis, J.M.; Chander, A.; Li, Y. Mitochondrial localization of catalase provides optimal protection from H2O2-induced cell death in lung epithelial cells. Am. J. Physiol. 2005, 290, L978–L986. [Google Scholar] [CrossRef] [PubMed]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Rovenko, B.M.; Kubrak, O.I.; Gospodaryov, D.V.; Yurkevych, I.S.; Sanz, A.; Lushchak, O.V.; Lushchak, V.I. Restriction of glucose and fructose causes mild oxidative stress independently of mitochondrial activity and reactive oxygen species in Drosophila melanogaster. Comp. Biochem. Physiol. 2015, 187, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Kim, S.H.; Hamasaki, N. Mitochondrial transcription factor A (TFAM): Roles in maintenance of mtDNA and cellular functions. Mitochondrion 2007, 7, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, N.; Maurya, C.K.; Arha, D.; Avisetti, D.R.; Prathapan, A.; Raj, P.S.; Raghu, K.G.; Kalivendi, S.V.; Tamrakar, A.K. Fructose induces mitochondrial dysfunction and triggers apoptosis in skeletal muscle cells by provoking oxidative stress. Apoptosis 2015, 20, 930–947. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Munetsuna, E.; Yamada, H.; Ando, Y.; Mizuno, G.; Murase, Y.; Kondo, K.; Ishikawa, H.; Teradaira, R.; Suzuki, K.; et al. Fructose consumption induces hypomethylation of hepatic mitochondrial DNA in rats. Life Sci. 2016, 149, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Tetri, L.H.; Basaranoglu, M.; Brunt, E.M.; Yerian, L.M.; Neuschwander-Tetri, B.A. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G987–G995. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Ford, D.A.; Acharya, S.; Gilkey, G.; Basaranoglu, M.; Tetri, L.H.; Brunt, E.M. Dietary trans-fatty acid induced NASH is normalized following loss of trans-fatty acids from hepatic lipid pools. Lipids 2012, 47, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Bugianesi, E. Carbohydrate intake and nonalcoholic fatty liver disease: Fructose as a weapon of mass destruction. Hepatobiliary Surg. Nutr. 2015, 4, 109–116. [Google Scholar] [PubMed]
- Ackerman, Z.; Oron-Herman, M.; Grozovski, M.; Rosenthal, T.; Pappo, O.; Link, G.; Sela, B.A. Fructose-induced fatty liver disease: Hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension 2005, 45, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Nunes, P.M.; Wright, A.J.; Veltien, A.; van Asten, J.J.; Tack, C.J.; Jones, J.G.; Heerschap, A. Dietary lipids do not contribute to the higher hepatic triglyceride levels of fructose-compared to glucose-fed mice. FASEB J. 2014, 28, 1988–1997. [Google Scholar] [CrossRef] [PubMed]
- Ferder, L.; Ferder, M.D.; Inserra, F. The role of high-fructose corn syrup in metabolic syndrome and hypertension. Curr. Hypertens. Rep. 2010, 12, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.; Ma, J.; Patel, K.; Berger, S.; Lau, J.; Lichtenstein, A.H. Fructose, high-fructose corn syrup, sucrose, and nonalcoholic fatty liver disease or indexes of liver health: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2014, 100, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Mosca, A.; Nobili, V.; De Vito, R.; Crudele, A.; Scorletti, E.; Villani, A.; Alisi, A.; Byrne, C.D. Serum uric acid concentrations and fructose consumption are independently associated with NASH in children and adolescents. J. Hepatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Ichijo, H. Mitochondrial proteolysis: Its emerging roles in stress responses. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Yonemitsu, S.; Erion, D.M.; Iwasaki, T.; Stark, R.; Weismann, D.; Dong, J.; Zhang, D.; Jurczak, M.J.; Löffler, M.G.; et al. The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab. 2009, 9, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Maehara, N.; Kai, T.; Arai, S.; Miyazaki, T. Dietary fructose-induced hepatocellular carcinoma development manifested in mice lacking apoptosis inhibitor of macrophage (AIM). Genes Cells 2016, 21, 1320–1332. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component (g 100 g−1) | Control Diet | Fructose Diet |
|---|---|---|
| Standard chow * | 50.5 | 50.5 |
| Sunflower oil | 1.5 | 1.5 |
| Casein | 9.2 | 9.2 |
| Alphacel | 9.8 | 9.8 |
| Starch | 20.4 | - |
| Fructose | - | 20.4 |
| Water | 6.4 | 6.4 |
| AIN-76 mineral mix | 1.6 | 1.6 |
| AIN-76 vitamin mix | 0.4 | 0.4 |
| Choline | 0.1 | 0.1 |
| Methionine | 0.1 | 0.1 |
| Gross energy density, KJ·g−1 | 17.2 | 17.2 |
| Protein, % metabolisable energy | 29.0 | 29.0 |
| Lipids, % metabolisable energy | 10.6 | 10.6 |
| Carbohydrates, % metabolisable energy | 60.4 | 60.4 |
| Of which: Fructose | - | 30.0 |
| Starch | 52.8 | 22.8 |
| Sugars | 7.6 | 7.6 |
| Item | Control | Fructose |
|---|---|---|
| Initial body weight, g | 470 ± 10 | 470 ± 10 |
| Final body weight, g | 540 ± 23 | 545 ± 15 |
| Food intake, g·day−1 | 32 ± 1.0 | 32 ± 1.0 |
| Plasma ALT, U·L−1 | 16.8 ± 1.0 | 27.3 ± 1.0 * |
| Plasma AST, U·L−1 | 43.0 ± 3.1 | 65.2 ± 3.3 * |
| Hepatic lipid peroxidation, nmol TBARS·g−1 liver | 61.5 ± 2.1 | 75.9 ± 2.0 * |
| Hepatic MPO activity, U·mg−1 liver | 0.31 ± 0.01 | 0.62 ± 0.02 * |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cioffi, F.; Senese, R.; Lasala, P.; Ziello, A.; Mazzoli, A.; Crescenzo, R.; Liverini, G.; Lanni, A.; Goglia, F.; Iossa, S. Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats. Nutrients 2017, 9, 323. https://doi.org/10.3390/nu9040323
Cioffi F, Senese R, Lasala P, Ziello A, Mazzoli A, Crescenzo R, Liverini G, Lanni A, Goglia F, Iossa S. Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats. Nutrients. 2017; 9(4):323. https://doi.org/10.3390/nu9040323
Chicago/Turabian StyleCioffi, Federica, Rosalba Senese, Pasquale Lasala, Angela Ziello, Arianna Mazzoli, Raffaella Crescenzo, Giovanna Liverini, Antonia Lanni, Fernando Goglia, and Susanna Iossa. 2017. "Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats" Nutrients 9, no. 4: 323. https://doi.org/10.3390/nu9040323
APA StyleCioffi, F., Senese, R., Lasala, P., Ziello, A., Mazzoli, A., Crescenzo, R., Liverini, G., Lanni, A., Goglia, F., & Iossa, S. (2017). Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats. Nutrients, 9(4), 323. https://doi.org/10.3390/nu9040323