Early-Life Nutritional Programming of Type 2 Diabetes: Experimental and Quasi-Experimental Evidence

Abstract
:1. Introduction
2. Conceptual Framework for Developmental Nutritional Programming of Type 2 Diabetes (T2D)
3. Evidence from Animal Models
4. Quasi-Experimental Design in Studying the Developmental Origin of T2D
4.1. Dutch Famine of 1944–1945
4.2. Famines in 20th-Century Austria
4.3. Ukrainian Famine of 1932–1933
4.4. Leningrad Siege of 1941–1944
4.5. Chinese Famine of 1959–1961
4.6. Nigerian Famine of 1967–1970
4.7. Holocaust (1939−1945)
5. Seasonality of Birth
6. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Wilmot, E.; Idris, I. Early onset type 2 diabetes: Risk factors, clinical impact and management. Ther. Adv. Chronic. Dis. 2014, 5, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Jaacks, L.M.; Siegel, K.R.; Gujral, U.P.; Narayan, K.M. Type 2 diabetes: A 21st century epidemic. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.H.; Haase, T.N.; Jaksch, C.; Nalla, A.; Søstrup, B.; Nalla, A.A.; Larsen, L.; Rasmussen, M.; Dalgaard, L.T.; Gaarn, L.W. Impact of fetal and neonatal environment on beta cell function and development of diabetes. Acta Obstet. Gynecol. Scand. 2014, 93, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Dabelea, D.; Hanson, R.L.; Lindsay, R.S.; Pettitt, D.J.; Imperatore, G.; Gabir, M.M.; Roumain, J.; Bennett, P.H.; Knowler, W.C. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: A study of discordant sibships. Diabetes 2000, 49, 2208–2211. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.G. Developmental Origins of Health and Disease—From a small body size at birth to epigenetics. Ann. Med. 2016, 48, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B. Dynamic cross talk between metabolic organs in obesity and metabolic diseases. Exp. Mol. Med. 2016, 48, e214. [Google Scholar] [CrossRef] [PubMed]
- Nettle, D.; Bateson, M. Adaptive developmental plasticity: What is it, how can we recognize it and when can it evolve? Proc. Biol. Sci. 2015, 282, 20151005. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.N.; Barker, D.J. Type 2 (non-insulin-dependent) diabetes mellitus: The thrifty phenotype hypothesis. 1992. Int. J. Epidemiol. 2013, 42, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Thorn, S.R.; Rozance, P.J.; Brown, L.D.; Hay, W.W., Jr. The intrauterine growth restriction phenotype: Fetal adaptations and potential implications for later life insulin resistance and diabetes. Semin. Reprod. Med. 2011, 29, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Carolan-Olah, M.; Duarte-Gardea, M.; Lechuga, J. A critical review: Early life nutrition and prenatal programming for adult disease. J. Clin. Nurs. 2015, 24, 3716–3729. [Google Scholar] [CrossRef] [PubMed]
- Tarry-Adkins, J.L.; Ozanne, S.E. Nutrition in early life and age-associated diseases. Ageing Res. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Whincup, P.H.; Kaye, S.J.; Owen, C.G.; Huxley, R.; Cook, D.G.; Anazawa, S.; Barrett-Connor, E.; Bhargava, S.K.; Birgisdottir, B.E.; Carlsson, S.; et al. Birth weight and risk of type 2 diabetes: A systematic review. J. Am. Med. Assoc. 2008, 300, 2886–2897. [Google Scholar] [PubMed]
- Kensara, O.A.; Wootton, S.A.; Phillips, D.I.; Patel, M.; Jackson, A.A.; Elia, M.; Hertfordshire Study Group. Fetal programming of body composition: Relation between birth weight and body composition measured with dual-energy X-ray absorptiometry and anthropometric methods in older Englishmen. Am. J. Clin. Nutr. 2005, 82, 980–987. [Google Scholar] [PubMed]
- Morrison, K.M.; Ramsingh, L.; Gunn, E.; Streiner, D.; van Lieshout, R.; Boyle, M.; Gerstein, H.; Schmidt, L.; Saigal, S. Cardiometabolic health in adults born premature with extremely low birth weight. Pediatrics 2016, 138. [Google Scholar] [CrossRef] [PubMed]
- Stirrat, L.I.; Reynolds, R.M. The effect of fetal growth and nutrient stresses on steroid pathways. J. Steroid. Biochem. Mol. Biol. 2016, 160, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Frankel, S.; Elwood, P.; Sweetnam, P.; Yarnell, J.; Smith, G.D. Birthweight, body-mass index in middle age and incident coronary heart disease. Lancet 1996, 348, 1478–1480. [Google Scholar] [CrossRef]
- Harder, T.; Rodekamp, E.; Schellong, K.; Dudenhausen, J.W.; Plagemann, A. Birth weight and subsequent risk of type 2 diabetes: A meta-analysis. Am. J. Epidemiol. 2007, 165, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Dulloo, A.G. Thrifty energy metabolism in catch-up growth trajectories to insulin and leptin resistance. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.K.; Suh, B.K. Catch-up growth and catch-up fat in children born small for gestational age. Korean J. Pediatr. 2016, 59, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ong, T.P.; Ozanne, S.E. Developmental programming of type 2 diabetes: Early nutrition and epigenetic mechanisms. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Paluch, B.E.; Naqash, A.R.; Brumberger, Z.; Nemeth, M.J.; Griffiths, E.A. Epigenetics: A primer for clinicians. Blood Rev. 2016, 30, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, S.J.; Tellam, R.L.; Morrison, J.L.; Muhlhausler, B.S.; Molloy, P.L. Recent developments on the role of epigenetics in obesity and metabolic disease. Clin. Epigenet. 2015, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A. Epidemiologic evidence for association between adverse environmental exposures in early life and epigenetic variation: A potential link to disease susceptibility? Clin. Epigenet. 2015, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, A.A.; Lindsay, K.L.; Alberdi, G.; McAuliffe, F.M.; Gibney, E.R. Nutrition during pregnancy impacts offspring’s epigenetic status—Evidence from human and animal studies. Nutr. Metab. Insights 2016, 8, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Alam, F.; Islam, M.A.; Gan, S.H.; Mohamed, M.; Sasongko, T.H. DNA methylation: An epigenetic insight into Type 2 diabetes mellitus. Curr. Pharm. Des. 2016, 22, 4398–4419. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.H.; Park, K.S. Recent progress in genetic and epigenetic research on type 2 diabetes. Exp. Mol. Med. 2016, 48, e220. [Google Scholar] [CrossRef] [PubMed]
- Green, A.S.; Rozance, P.J.; Limesand, S.W. Consequences of a compromised intrauterine environment on islet function. J. Endocrinol. 2010, 205, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Portha, B.; Chavey, A.; Movassat, J. Early-life origins of type 2 diabetes: Fetal programming of the beta-cell mass. Exp. Diabetes Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Pinney, S.E. Intrauterine growth retardation–A developmental model of type 2 diabetes. Drug Discov Today Dis. Models. 2013, 10, e71–e77. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, O.; Blondeau, B.; Duvillie, B.; Reusens, B.; Breant, B.; Remacle, C. Different mechanisms operating during different critical time-windows reduce rat fetal beta cell mass due to a maternal low-protein or low-energy diet. Diabetologia 2007, 50, 2495–2503. [Google Scholar] [CrossRef] [PubMed]
- Garofano, A.; Czernichow, P.; Breant, B. In utero undernutrition impairs rat beta-cell development. Diabetologia 1997, 40, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Garofano, A.; Czernichow, P.; Breant, B. Beta-cell mass and proliferation following late fetal and early postnatal malnutrition in the rat. Diabetologia 1998, 41, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Petrik, J.; Reusens, B.; Arany, E.; Remacle, C.; Coelho, C.; Hoet, J.J.; Hill, D.J. A low protein diet alters the balance of islet cell replication and apoptosis in the fetal and neonatal rat and is associated with a reduced pancreatic expression of insulin-like growth factor-II. Endocrinology 1999, 140, 4861–4873. [Google Scholar] [CrossRef] [PubMed]
- Garofano, A.; Czernichow, P.; Breant, B. Effect of ageing on beta-cell mass and function in rats malnourished during the perinatal period. Diabetologia 1999, 42, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Blondeau, B.; Garofano, A.; Czernichow, P.; Breant, B. Age-dependent inability of the endocrine pancreas to adapt to pregnancy: A long-term consequence of perinatal malnutrition in the rat. Endocrinology 1999, 140, 4208–4213. [Google Scholar] [CrossRef] [PubMed]
- Thamotharan, M.; Shin, B.C.; Suddirikku, D.T.; Thamotharan, S.; Garg, M.; Devaskar, S.U. GLUT4 expression and subcellular localization in the intrauterine growth-restricted adult rat female offspring. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E935–E947. [Google Scholar] [CrossRef] [PubMed]
- Matveyenko, A.V.; Singh, I.; Shin, B.C.; Georgia, S.; Devaskar, S.U. Differential effects of prenatal and postnatal nutritional environment on ss-cell mass development and turnover in male and female rats. Endocrinology 2010, 151, 5647–5656. [Google Scholar] [CrossRef] [PubMed]
- Ong, T.P.; Ozanne, S.E. Developmental programming of type 2 diabetes: Early nutrition and epigenetic mechanisms. Curr. Opin. Clin. Nutr. Metab Care 2015, 18, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Simmons, R.A. Developmental origins of diabetes: The role of epigenetic mechanisms. Curr Opin. Endocrinol. Diabetes Obes. 2007, 14, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Simmons, R.A. Developmental origins of adult disease. Pediatr. Clin. N. Am. 2009, 56, 449–466. [Google Scholar] [CrossRef] [PubMed]
- Blondeau, B.; Avril, I.; Duchene, B.; Breant, B. Endocrine pancreas development is altered in foetuses from rats previously showing intra-uterine growth retardation in response to malnutrition. Diabetologia 2002, 45, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Stoffers, D.A.; Nicholls, R.D.; Simmons, R.A. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J. Clin. Investig. 2008, 118, 2316–2324. [Google Scholar] [CrossRef] [PubMed]
- Reusens, B.; Theys, N.; Dumortier, O.; Goosse, K.; Remacle, C. Maternal malnutrition programs the endocrine pancreas in progeny. Am. J. Clin. Nutr. 2011, 94, 1824S–1829S. [Google Scholar] [CrossRef] [PubMed]
- Berends, L.M.; Fernandez-Twinn, D.S.; Martin-Gronert, M.S.; Cripps, R.L.; Ozanne, S.E. Catch-up growth following intra-uterine growth-restriction programmes an insulin-resistant phenotype in adipose tissue. Int. J. Obes. (Lond.) 2013, 37, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Bieswal, F.; Ahn, M.T.; Reusens, B.; Holvoet, P.; Raes, M.; Rees, W.D.; Remacle, C. The importance of catch-up growth after early malnutrition for the programming of obesity in male rat. Obesity (Silver Spring) 2006, 14, 1330–1343. [Google Scholar] [CrossRef] [PubMed]
- Bol, V.V.; Delattre, A.I.; Reusens, B.; Raes, M.; Remacle, C. Forced catch-up growth after fetal protein restriction alters the adipose tissue gene expression program leading to obesity in adult mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R291–R299. [Google Scholar] [CrossRef] [PubMed]
- Last, J.M. A Dictionary of Epidemiology, 3rd ed.; Oxford University Press: New York, NY, USA, 1995. [Google Scholar]
- Heijmans, B.T.; Tobi, E.W.; Lumey, L.H.; Slagboom, P.E. The epigenome: Archive of the prenatal environment. Epigenetics 2009, 4, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Lumey, L.H.; Stein, A.D.; Susser, E. Prenatal famine and adult health. Annu. Rev. Public Health 2011, 32, 237–262. [Google Scholar] [CrossRef] [PubMed]
- Roseboom, T.J.; Painter, R.C.; van Abeelen, A.F.; Veenendaal, M.V.; de Rooij, S.R. Hungry in the womb: What are the consequences? Lessons from the Dutch famine. Maturitas 2011, 70, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed]
- Van Abeelen, A.F.; Elias, S.G.; Bossuyt, P.M.; Grobbee, D.E.; van der Schouw, Y.T.; Roseboom, T.J.; Uiterwaal, C.S. Famine exposure in the young and the risk of type 2 diabetes in adulthood. Diabetes 2012, 61, 2255–2260. [Google Scholar] [CrossRef] [PubMed]
- Portrait, F.; Teeuwiszen, E.; Deeg, D. Early life undernutrition and chronic diseases at older ages: The effects of the Dutch famine on cardiovascular diseases and diabetes. Soc. Sci. Med. 2011, 73, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Lumey, L.H.; Terry, M.B.; Delgado-Cruzata, L.; Liao, Y.; Wang, Q.; Susser, E.; McKeague, I.; Santella, R.M. Adult global DNA methylation in relation to pre-natal nutrition. Int. J. Epidemiol. 2012, 41, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef] [PubMed]
- Thurner, S.; Klimek, P.; Szell, M.; Duftschmid, G.; Endel, G.; Kautzky-Willer, A.; Kasper, D.C. Quantification of excess risk for diabetes for those born in times of hunger, in an entire population of a nation, across a century. Proc. Natl. Acad. Sci. USA 2013, 110, 4703–4707. [Google Scholar] [CrossRef] [PubMed]
- Klitz, W.; Niklasson, B. Viral underpinning to the Austrian record of type 2 diabetes? Proc. Natl. Acad. Sci. USA 2013, 110, E2750. [Google Scholar] [CrossRef] [PubMed]
- Thurner, S.; Klimek, P.; Szell, M.; Duftschmid, G.; Endel, G.; Kautzky-Willer, A.; Kasper, D.C. Reply to Klitz and Niklasson: Can viral infections explain the cross-sectional Austrian diabetes data? Proc. Natl. Acad. Sci. USA 2013, 110, E2751. [Google Scholar] [CrossRef]
- Lumey, L.H.; Khalangot, M.D.; Vaiserman, A.M. Association between type 2 diabetes and prenatal exposure to the Ukraine famine of 1932–33: A retrospective cohort study. Lancet Diabetes Endocrinol. 2015, 3, 787–794. [Google Scholar] [CrossRef]
- Sparén, P.; Vågerö, D.; Shestov, D.B.; Plavinskaja, S.; Parfenova, N.; Hoptiar, V.; Paturot, D.; Galanti, M.R. Long term mortality after severe starvation during the siege of Leningrad: Prospective cohort study. Br. Med. J. 2004, 328, 11. [Google Scholar] [CrossRef] [PubMed]
- Stanner, S.A.; Bulmer, K.; Andrès, C.; Lantseva, O.E.; Borodina, V.; Poteen, V.V.; Yudkin, J.S. Does malnutrition in utero determine diabetes and coronary heart disease in adulthood? Results from the Leningrad siege study: A cross sectional study. Br. Med. J. 1997, 315, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Stanner, S.A.; Yudkin, J.S. Fetal programming and the Leningrad Siege study. Twin Res. 2001, 4, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Bateson, P. Fetal experience and good adult design. Int. J. Epidemiol. 2001, 30, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Khoroshinina, L.P.; Zhavoronkova, N.V. Starving in childhood and diabetes mellitus in elderly age. Adv. Gerontol. 2008, 21, 684–687. [Google Scholar]
- Khoroshinina, L.P. Peculiarities of somatic diseases in people of middle and old age survived Leningrad siege at childhood. Adv. Gerontol. 2004, 14, 55–65. (In Russian) [Google Scholar]
- Koupil, I.; Shestov, D.B.; Sparén, P.; Plavinskaja, S.; Parfenova, N.; Vågerö, D. Blood pressure, hypertension and mortality from circulatory disease in men and women who survived the siege of Leningrad. Eur. J. Epidemiol. 2007, 22, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Jowett, A.J. The demographic responses to famine: The case of China 1958-61. GeoJournal 1991, 23, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lumey, L.H. Exposure to the Chinese famine of 1959–61 in early life and current health conditions: A systematic review and meta-analysis. Lancet 2016, 388, S63. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.; Qi, L.; Jaddoe, V.W.; Feskens, E.J.; Yang, X.; Ma, G.; Hu, F.B. Exposure to the Chinese famine in early life and the risk of hyperglycemia and type 2 diabetes in adulthood. Diabetes 2010, 59, 2400–2406. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, X.; Han, B.; Li, Q.; Chen, Y.; Zhu, C.; Chen, Y.; Xia, F.; Cang, Z.; Zhu, C.; et al. Is exposure to famine in childhood and economic development in adulthood associated with diabetes? J. Clin. Endocrinol. Metab. 2015, 100, 4514–4523. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Cheng, J.; Han, B.; Li, Q.; Chen, Y.; Xia, F.; Jiang, B.; Jensen, M.D.; Lu, Y. Exposure to severe famine in the prenatal or postnatal period and the development of diabetes in adulthood: An observational study. Diabetologia 2017, 60, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y.; Han, X.; Liu, B.; Hu, H.; Wang, F.; Li, X.; Yang, K.; Yuan, J.; Yao, P.; et al. Exposure to the Chinese Famine in childhood increases type 2 diabetes risk in adults. J. Nutr. 2016, 146, 2289–2295. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, S.; Li, S.; Feng, R.; Na, L.; Chu, X.; Wu, X.; Niu, Y.; Sun, Z.; Han, T.; et al. Prenatal exposure to famine and the development of hyperglycemia and type 2 diabetes in adulthood across consecutive generations: A population-based cohort study of families in Suihua, China. Am. J. Clin. Nutr. 2016, 105, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.P. Medical relief in the Nigerian civil war. Lancet 1970, 760, 1330–1334. [Google Scholar] [CrossRef]
- Hult, M.; Tornhammar, P.; Ueda, P.; Chima, C.; Bonamy, A.K.; Ozumba, B.; Norman, M. Hypertension, diabetes and overweight: Looming legacies of the Biafran famine. PLoS ONE 2010, 5, e13582. [Google Scholar] [CrossRef] [PubMed]
- Bercovich, E.; Keinan-Boker, L.; Shasha, S.M. Long-term health effects in adults born during the Holocaust. Isr. Med. Assoc. J. 2014, 16, 203–207. [Google Scholar] [PubMed]
- Keinan-Boker, L.; Shasha-Lavsky, H.; Eilat-Zanani, S.; Edri-Shur, A.; Shasha, S.M. Chronic health conditions in Jewish Holocaust survivors born during World War II. Isr. Med. Assoc. J. 2015, 17, 206–212. [Google Scholar] [PubMed]
- Watson, P.E.; McDonald, B.W. Seasonal variation of nutrient intake in pregnancy: Effects on infant measures and possible influence on diseases related to season of birth. Eur. J. Clin. Nutr. 2007, 61, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Flouris, A.D.; Spiropoulos, Y.; Sakellariou, G.J.; Koutedakis, Y. Effect of seasonal programming on fetal development and longevity: Links with environmental temperature. Am. J. Hum. Biol. 2009, 21, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Finch, C.E.; Crimmins, E.M. Inflammatory exposure and historical changes in human life-spans. Science 2004, 305, 1736–1739. [Google Scholar] [CrossRef] [PubMed]
- Lowell, W.E.; Davis, G.E., Jr. The light of life: Evidence that the sun modulates human lifespan. Med. Hypotheses. 2008, 70, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Crippa, A.; Woodcock, J.; Brage, S. Physical activity and incident type 2 diabetes mellitus: A systematic review and dose-response meta-analysis of prospective cohort studies. Diabetologia 2016, 59, 2527–2545. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.M. Early-life exposure to substance abuse and risk of type 2 diabetes in adulthood. Curr. Diabetes Rep. 2015, 15, 48. [Google Scholar] [CrossRef] [PubMed]
- Chodick, G.; Flash, S.; Deoitch, Y.; Shalev, V. Seasonality in birth weight: Review of global patterns and potential causes. Hum. Biol. 2009, 81, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Banegas, J.R.; Rodríguez-Artalejo, F.; de la Cruz, J.J.; Graciani, A.; Villar, F.; del Rey-Calero, J. Adult men born in spring have lower blood pressure. J. Hypertens. 2000, 18, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.I.; Young, J.B. Birth weight: Climate at birth and the risk of obesity in adult life. Int. J. Obes. Relat. Metab. Disord. 2000, 24, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Wattie, N.; Ardern, C.I.; Baker, J. Season of birth and prevalence of overweight and obesity in Canada. Early Hum. Dev. 2008, 84, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.A.; Davey-Smith, G.; Mitchell, R.; Ebrahim, S. Temperature at birth, coronary heart disease, and insulin resistance: Cross sectional analyses of the British women’s heart and health study. Heart 2004, 90, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z.; Lewy, H.; Wilderman, I.; Casu, A.; Willis, J.; Redondo, M.J.; Libman, I.; White, N.; Craig, M. Seasonality of month of birth of children and adolescents with type 1 diabetes mellitus in homogenous and heterogeneous populations. Isr. Med. Assoc. J. 2005, 7, 381–384. [Google Scholar] [PubMed]
- Grover, V.; Lipton, R.B.; Sclove, S.L. Seasonality of month of birth among African American children with diabetes mellitus in the City of Chicago. J. Pediatr. Endocrinol. Metab. 2004, 17, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Jongbloet, P.H.; van Soestbergen, M.; van der Veen, E.A. Month-of-birth distribution of diabetics and ovopathy: A new aetiological view. Diabetes Res. 1988, 9, 51–58. [Google Scholar] [PubMed]
- Vaiserman, A.M.; Khalangot, M.D.; Carstensen, B.; Tronko, M.D.; Kravchenko, V.I.; Voitenko, V.P.; Mechova, L.V.; Koshel, N.M.; Grigoriev, P.E. Seasonality of birth in adult type 2 diabetic patients in three Ukrainian regions. Diabetologia 2009, 52, 2665–2667. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.M.; Khalangot, M.D. Similar seasonality of birth in type 1 and type 2 diabetes patients: A sign for common etiology? Med. Hypotheses 2008, 71, 604–605. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.B.; Zimmermann, E.; Gamborg, M.; Heitmann, B.L.; Baker, J.L.; Vaag, A.; Sørensen, T.I. No evidence of seasonality of birth in adult type 2 diabetes in Denmark. Diabetologia 2015, 58, 2045–2050. [Google Scholar]
- Lockett, G.A.; Soto-Ramírez, N.; Ray, M.A.; Everson, T.M.; Xu, C.J.; Patil, V.K.; Terry, W.; Kaushal, A.; Rezwan, F.I.; Ewart, S.L.; et al. Association of season of birth with DNA methylation and allergic disease. Allergy 2016, 71, 1314–1324. [Google Scholar] [CrossRef] [PubMed]
- Dugué, P.A.; Geurts, Y.M.; Milne, R.L.; Lockett, G.A.; Zhang, H.; Karmaus, W.; Holloway, J.W. Is there an association between season of birth and blood DNA methylation in adulthood? Allergy 2016, 71, 1501–1504. [Google Scholar] [CrossRef] [PubMed]
- Desiderio, A.; Spinelli, R.; Ciccarelli, M.; Nigro, C.; Miele, C.; Beguinot, F.; Raciti, G.A. Epigenetics: Spotlight on type 2 diabetes and obesity. J. Endocrinol. Investig. 2016, 39, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Sterns, J.D.; Smith, C.B.; Steele, J.R.; Stevenson, K.L.; Gallicano, G.I. Epigenetics and type II diabetes mellitus: Underlying mechanisms of prenatal predisposition. Front. Cell Dev. Biol. 2014, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.C.; van der Meulen, J.H.; Michels, R.P.; Osmond, C.; Barker, D.J.; Hales, C.N.; Bleker, O.P. Glucose tolerance in adults after prenatal exposure to famine. Lancet 1998, 351, 173–177. [Google Scholar] [CrossRef]
- Lussana, F.; Painter, R.C.; Ocke, M.C.; Buller, H.R.; Bossuyt, P.M.; Roseboom, T.J. Prenatal exposure to the Dutch famine is associated with a preference for fatty foods and a more atherogenic lipid profile. Am. J. Clin. Nutr. 2008, 88, 1648–1652. [Google Scholar] [CrossRef] [PubMed]
- Lumey, L.H.; Stein, A.D.; Kahn, H.S.; Romijn, J.A. Lipid profiles in middle-aged men and women after famine exposure during gestation: The dutch hunger winter families study. Am. J. Clin. Nutr. 2009, 89, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Roseboom, T.; de Rooij, S.; Painter, R. The Dutch famine and its long-term consequences for adult health. Early Hum. Dev. 2006, 82, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, W.; Sun, J.; Pang, Z. Association of famine exposure during early life with the risk of type 2 diabetes in adulthood: A meta-analysis. Eur. J. Nutr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Gillman, M.W. Prenatal famine and developmental origins of type 2 diabetes. Lancet Diabetes Endocrinol. 2015, 3, 751–752. [Google Scholar] [CrossRef]
- Kaati, G.; Bygren, L.O.; Edvinsson, S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur. J. Hum. Genet. 2002, 10, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Pembrey, M.E. Male-line transgenerational responses in humans. Hum. Fertil. (Camb.) 2010, 13, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.M.; Pasyukova, E.G. Epigenetic drugs: A novel anti-aging strategy? Front. Genet. 2012, 3, 224. [Google Scholar] [CrossRef] [PubMed]
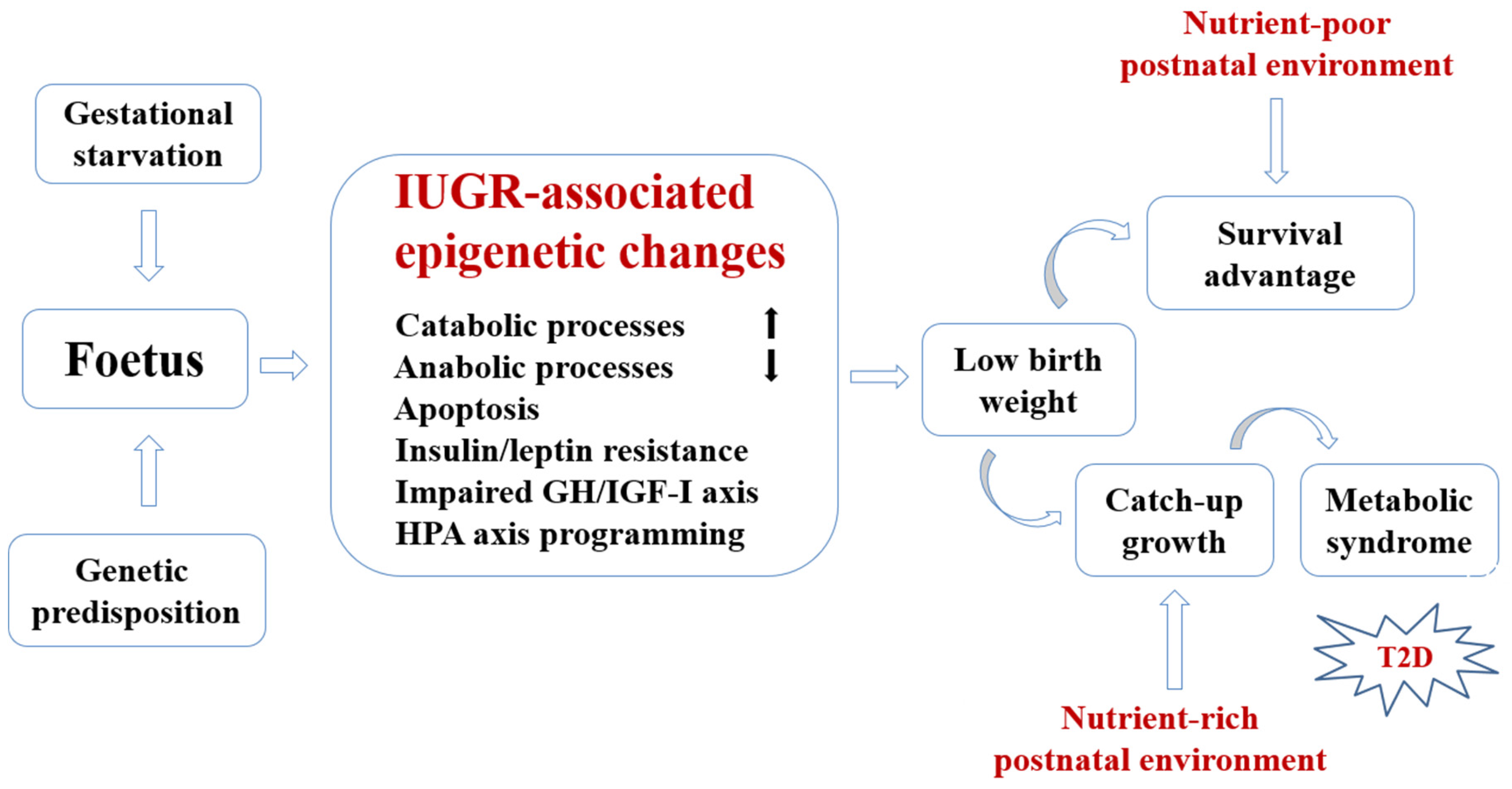

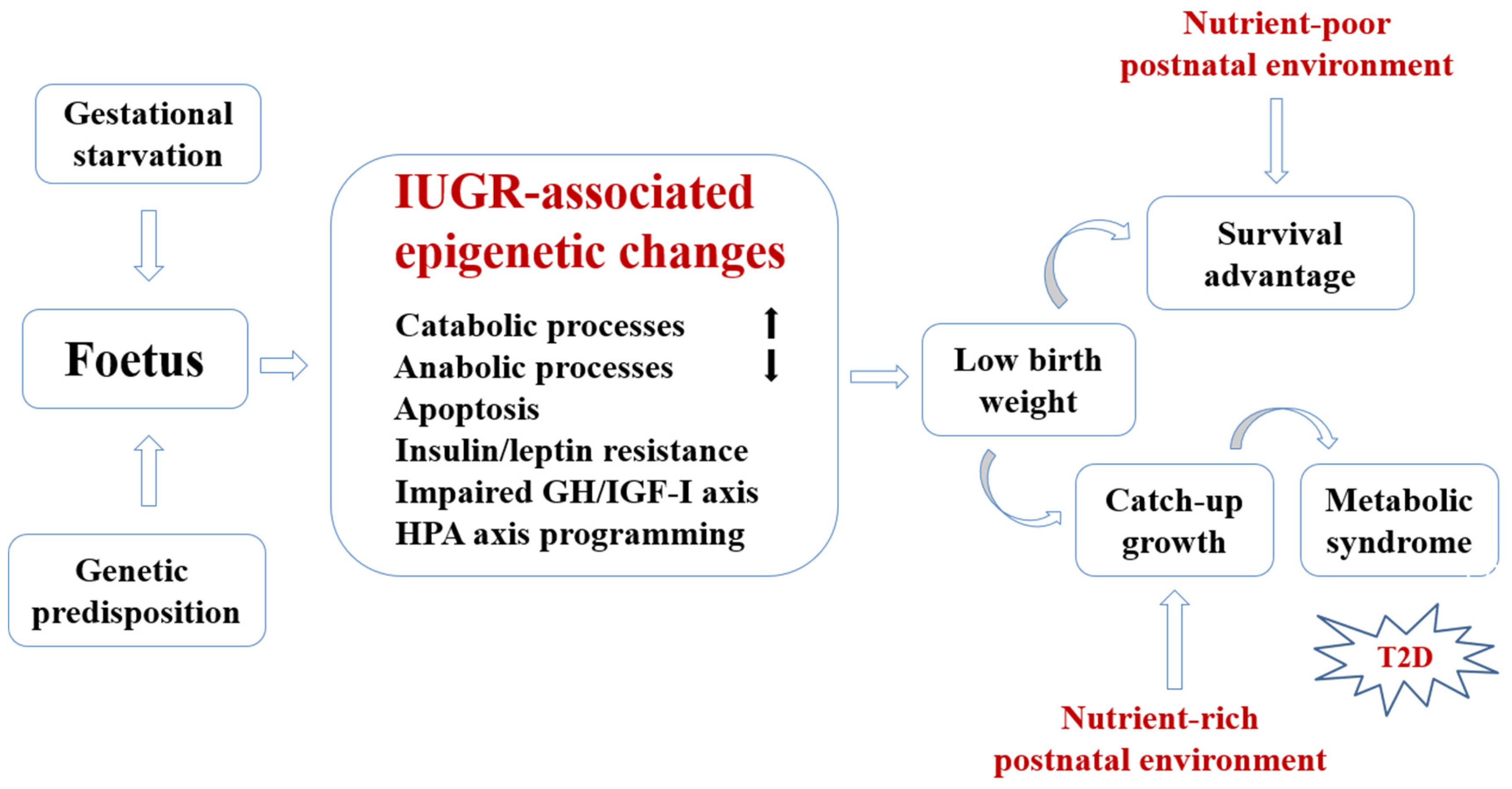{kind=link}
| Country | Cause of Starvation | Period | Adult consequence | Ref. |
|---|---|---|---|---|
| Netherlands (‘Dutch Hunger Winter’) | Nazi food embargo | 1944–1945 | Impaired glucose regulation Atherogenic lipid profiles Obesity, CVD T2D Lower IGF2 methylation Changed methylation of ABCA1, GNASAS, IL10, LEP, INSIGF and MEG3 genes | [78] [79,80,81] [49,81] [49] [51] [55] |
| Austria | Empire’s collapse Nazi food embargo Post-war period | 1918–1919 1938 1946–1947 | High risk of T2D | [56] |
| Ukraine (‘Holodomor’) | Agriculture collectivization | 1932–1933 | High risk of T2D | [59] |
| Russia | Leningrad Siege | 1941–1944 | Endothelial dysfunction, stronger influence of obesity on blood pressure Increasing incidence of T2D Atherosclerosis, arterial hypertension | [61,62] [64] [65,66] |
| China (‘Great Leap Forward Famine’) | Disastrous social agricultural reform | 1959–1961 | Hyperglycemia High risk of T2D | [69,73] [70,71,72,73] |
| Nigeria (‘Biafran Famine’) | Civil war | 1967–1970 | Increased blood pressure, higher levels of p-glucose, increased waist circumference, overweight, high risks of impaired glucose regulation and systolic hypertension | [75] |
| Europe (‘Holocaust’) | Nazi genocide | 1939–1945 | Enhanced BMI, hypertension, dyslipidemia, high risk of T2D and CVD | [76,77] |
| Spain | Seasonal malnutrition | 1935−1954 | High systolic blood pressure | [82] |
| United Kingdom | Seasonal malnutrition | 1920–1930 | Obesity | [83] |
| Canada | Seasonal malnutrition | 1943–1995 | Obesity | [84] |
| United Kingdom | Seasonal malnutrition | 1924–1943 | Dyslipidaemia, insulin resistance and CVD | [85] |
| USA | Seasonal malnutrition | 1968–1995 | High risk of T2D | [86] |
| Netherlands | Seasonal malnutrition | 1920–1948 | High risk of T2D | [87] |
| Ukraine | Seasonal malnutrition | 1930–1938 | High risk of T2D | [88] |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaiserman, A.M. Early-Life Nutritional Programming of Type 2 Diabetes: Experimental and Quasi-Experimental Evidence. Nutrients 2017, 9, 236. https://doi.org/10.3390/nu9030236
Vaiserman AM. Early-Life Nutritional Programming of Type 2 Diabetes: Experimental and Quasi-Experimental Evidence. Nutrients. 2017; 9(3):236. https://doi.org/10.3390/nu9030236
Chicago/Turabian StyleVaiserman, Alexander M. 2017. "Early-Life Nutritional Programming of Type 2 Diabetes: Experimental and Quasi-Experimental Evidence" Nutrients 9, no. 3: 236. https://doi.org/10.3390/nu9030236
APA StyleVaiserman, A. M. (2017). Early-Life Nutritional Programming of Type 2 Diabetes: Experimental and Quasi-Experimental Evidence. Nutrients, 9(3), 236. https://doi.org/10.3390/nu9030236