A New Proposal for the Pathogenic Mechanism of Non-Coeliac/Non-Allergic Gluten/Wheat Sensitivity: Piecing Together the Puzzle of Recent Scientific Evidence

 and
and 
Abstract
1. Introduction
2. Scientific Background
2.1. Diagnostic Difficulties
2.2. ATIs as a Trigger of NCG/WS?
2.3. Microbial Lipopolysaccharide, Intestinal Alkaline Phosphatase and Intestinal Permeability
3. New Hypothesis on the Pathogenic Mechanism of NCG/WS
4. Implications of the New Hypothesis
5. NCG/WS Is a Cancer Risk Factor?
6. Starting Points for Future Research
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ellis, A.; Linaker, B.D. Non-coeliac gluten sensitivity? Lancet 1978, 1, 1358–1359. [Google Scholar] [CrossRef]
- Cooper, B.T.; Holmes, G.K.; Ferguson, R.; Thompson, R.A.; Allan, R.N.; Cooke, W.T. Gluten-sensitive diarrhea without evidence of celiac disease. Gastroenterology 1980, 79, 801–806. [Google Scholar] [PubMed]
- Sapone, A.; Bai, J.C.; Ciacci, C.; Dolinsek, J.; Green, P.H.; Hadjivassiliou, M.; Kaukinen, K.; Rostami, K.; Sanders, D.S.; Schumann, M.; et al. Spectrum of gluten-related disorders: Consensus on new nomenclature and classification. BMC Med. 2012, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Catassi, C.; Bai, J.C.; Bonaz, B.; Bouma, G.; Calabrò, A.; Carroccio, A.; Castillejo, G.; Ciacci, C.; Cristofori, F.; Dolinsek, J.; et al. Non-celiac Gluten Sensitivity: The New Frontier of Gluten Related Disorders. Nutrients 2013, 5, 3839–3853. [Google Scholar] [CrossRef] [PubMed]
- Catassi, C.; Elli, L.; Bonaz, B.; Bouma, G.; Carroccio, A.; Castillejo, G.; Cellier, C.; Cristofori, F.; de Magistris, L.; Dolinsek, J.; et al. Diagnosis of Non-Celiac Gluten Sensitivity (NCGS): The Salerno Experts’ Criteria. Nutrients 2015, 7, 4966–4977. [Google Scholar] [CrossRef] [PubMed]
- Vanga, R.; Leffler, D.A. Gluten sensitivity: Not celiac and not certain. Gastroenterology 2013, 145, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Sapone, A.; Lammers, K.M.; Mazzarella, G.; Mikhailenko, I.; Cartenì, M.; Casolaro, V.; Fasano, A. Differential Mucosal IL-17 Expression in Two Gliadin-Induced Disorders: Gluten Sensitivity and the Autoimmune Enteropathy Celiac Disease. Int. Arch. Allergy Immunol. 2010, 152, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Molina-Infante, J.; Santolaria, S.; Sanders, D.S.; Fernández-Bañares, F. Systematic review: Noncoeliac gluten sensitivity. Aliment. Pharmacol. Ther. 2015, 41, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Volta, U.; Caio, G.; Karunaratne, T.B.; Alaedini, A.; De Giorgio, R. Non-coeliac gluten/wheat sensitivity: Advances in knowledge and relevant questions. Exp. Rev. Gastroenterol. Hepatol. 2017, 11, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Carroccio, A. Searching for the immunological basis of wheat sensitivity. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Hill, I.D.; Fasano, A.; Guandalini, S.; Hoffenberg, E.; Levy, J.; Reilly, N.; Verma, R. NASPGHAN Clinical Report on the Diagnosis and Treatment of Gluten-related Disorders. J. Pediatr. Gastroenterol. Nutr. 2016, 63, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Caio, G.; Riegler, G.; Patturelli, M.; Facchiano, A.; DE Magistris, L.; Sapone, A. Pathophysiology of non-celiac gluten sensitivity: Where are we now? Minerva Gastroenterol. Dietol. 2017, 63, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Molina-Infante, J.; Carroccio, A. Suspected non-celiac gluten sensitivity confirmed in few patients after gluten challenge in double-blind, placebo-controlled trials. Clin. Gastroenterol. Hepatol. 2017, 15, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Bardella, M.T.; Elli, L.; Ferretti, F. Non Celiac Gluten Sensitivity. Curr. Gastroenterol. Rep. 2016, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Elli, L.; Roncoroni, L.; Bardella, M.T. Non-celiac gluten sensitivity: Time for sifting the grain. World J. Gastroenterol. 2015, 21, 8221–8226. [Google Scholar] [CrossRef] [PubMed]
- Di Sabatino, A.; Corazza, G.R. Nonceliac gluten sensitivity: Sense or sensibility? Ann. Intern. Med. 2012, 156, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Elli, L.; Tomba, C.; Branchi, F.; Roncoroni, L.; Lombardo, V.; Bardella, M.T.; Ferretti, F.; Conte, D.; Valiante, F.; Fini, L.; et al. Evidence for the Presence of Non-Celiac Gluten Sensitivity in Patiens with Functional Gastrointestinal Symptoms: Results from a Multicenter Randomized Double-Blind Placebo-Controlled Gluten Challange. Nutrients 2016, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Biesiekierski, J.R.; Peters, S.L.; Newnham, E.D.; Rosella, O.; Muir, J.G.; Gibson, P.R. No Effects of Gluten in Patients With Self-Reported Non-Celiac Gluten Sensitivity After Dietary Reduction of Fermentable, Poorly Absorbed, Short-Chain Carbohydrates. Gastroenterology 2013, 145, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Zevallos, V.F.; Raker, V.; Tenzer, S.; Jimenez-Calvente, C.; Ashfaq-Khan, M.; Rüssel, N.; Pickert, G.; Schild, H.; Steinbrink, K.; Schuppan, D. Nutritional Wheat Amylase-Trypsin Inhibitors Promote Intestinal Inflammation via Activation of Myeloid Cells. Gastroenterology 2017, 152, 1100–1113. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Pickert, G.; Ashfaq-Khan, M.; Zevallos, V. Non-celiac wheat sensitivity: Differential diagnosis, triggers and implications. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Junker, Y.; Zeissig, S.; Kim, S.J.; Barisani, D.; Wieser, H.; Leffler, D.A.; Zevallos, V.; Libermann, T.A.; Dillon, S.; Freitag, T.L.; et al. Wheat amylase trypsin inhibitors drive intestinal inflammation via activation of toll-like receptor 4. J. Exp. Med. 2012, 209, 2395–2408. [Google Scholar] [CrossRef] [PubMed]
- Zanini, B.; Baschè, R.; Ferraresi, A.; Ricci, C.; Lanzarotto, F.; Marullo, M.; Villanacci, V.; Hidalgo, A.; Lanzini, A. Randomised clinical study: Gluten challenge induces symptom recurrence in only a minority of patients who meet clinical criteria for non-coeliac gluten sensitivity. Aliment. Pharmacol. Ther. 2015, 42, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Zingone, F.; Russo, I.; Morra, I.; Tortora, R.; Pogna, N.; Scalia, G.; Iovino, P.; Ciacci, C. Gliadin Does Not Induce Mucosal Inflammation or Basophil Activation in Patiens With Nonceliac Gluten Sensitivity. Clin. Gastroenterol. Hepatol. 2013, 11, 1294–1299. [Google Scholar] [CrossRef] [PubMed]
- Rosinach, M.; Fernández-Bañares, F.; Carrasco, A.; Ibarra, M.; Temiño, R.; Salas, A.; Esteve, M. Double-Blind Randomized Clinical Trial: Gluten versus Placebo Rechallange in Patiens with Lymphocytic Enteritis and Suspected Celiac Disease. PLoS ONE 2016, 11, e0157879. [Google Scholar] [CrossRef] [PubMed]
- Gibson, P.R.; Muir, J.G.; Newnham, E.D. Other Dietary Confounders: FODMAPs et al. Dig. Dis. 2015, 33, 269–276. [Google Scholar] [CrossRef] [PubMed]
- De Punder, K.; Pruimboom, L. The dietary intake of wheat and other cereal grains and their role in inflammation. Nutrients 2013, 5, 771–787. [Google Scholar] [CrossRef] [PubMed]
- Dalla Pellegrina, C.; Perbellini, O.; Scupoli, M.T.; Tomelleri, C.; Zanetti, C.; Zoccatelli, G.; Fusi, M.; Peruffo, A.; Rizzi, C.; Chignola, R. Effects of wheat germ agglutinin on human gastrointestinal epithelium: Insights from an experimental model of immune/epithelial cell interaction. Toxicol. Appl. Pharmacol. 2009, 237, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Pruimboom, L.; de Punder, K. The opioid effects of gluten exorphins: Asymptomatic celiac disease. J. Health Popul. Nutr. 2015, 33, 24. [Google Scholar] [CrossRef] [PubMed]
- Uhde, M.; Ajamian, M.; Caio, G.; De Giorgio, R.; Indart, A.; Green, P.H.; Verna, E.C.; Volta, U.; Alaedini, A. Intestinal cell damage and systemic immune activation in individuals reporting sensitivity to wheat in the absence of coelic disease. Gut 2016, 65, 1930–1937. [Google Scholar] [CrossRef] [PubMed]
- Hollon, J.; Puppa, E.L.; Greenwald, B.; Goldberg, E.; Guerrerio, A.; Fasano, A. Effect of gliadin on permeability of intestinal biopsy explants from celiac disease patients and patients with non-celiac gluten sensitivity. Nutrients 2015, 7, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, M.R.; Cremon, C.; Caio, G.; Bellacosa, L.; De Giorgio, R.; Volta, U.; Stanghellini, V.; Barbara, G. Increased zonulin serum levels and correlation with symptoms in non-celiac gluten sensitivity and irritable bowel syndrome with diarrhea. In Proceedings of the UEG Week 2014, Vienna, Austria, 18–22 October 2014. [Google Scholar]
- Elli, L.; Villalta, D.; Roncoroni, L.; Barisani, D.; Ferrero, S.; Pellegrini, N.; Bardella, M.T.; Valiante, F.; Tomba, C.; Carroccio, A.; et al. Nomenclature and diagnosis of gluten-related disorders: A position statement by the Italian Association of Hospital Gastroenterologists and Endoscopists (AIGO). Dig. Liver Dis. 2017, 49, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Carroccio, A.; Mansueto, P.; D’Alcamo, A.; Iacono, G. Non-celiac wheat sensitivity as an allergic condition: Personal experience and narrative review. Am. J. Gastroenterol. 2013, 108, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Cianferoni, A. Wheat allergy: Diagnosis and management. J. Asthma Allergy 2016, 9, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Katz, K.D.; Rashtak, S.; Lahr, B.D.; Melton, L.J.; Krause, P.K.; Maggi, K.; Talley, N.J.; Murray, J.A. Screening for celiac disease in a North American population: Sequential serology and gastrointestinal symptoms. Am. J. Gastroenterol. 2011, 106, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Leffler, D.A.; Dennis, M.; Hyett, B.; Kelly, E.; Schuppan, D.; Kelly, C.P. Etiologies and predictors of diagnosis in nonresponsive celiac disease. Clin. Gastroenterol. Hepatol. 2007, 5, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Drossman, D.A. Functional gastrointestinal disorders: History, pathophysiology, clinical features, and Rome IV. Gastroenterology 2016, 150, 1262–1279. [Google Scholar] [CrossRef] [PubMed]
- Palsson, O.S.; Whitehead, W.E.; van Tilburg, M.A.; Chang, L.; Chey, W.; Crowell, M.D.; Keefer, L.; Lembo, A.J.; Parkman, H.P.; Rao, S.S.; et al. Development and Validation of the Rome IV Diagnostic Questionnaire for Adults. Gastroenterology 2016, 150, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Makharia, A.; Catassi, C.; Makharia, G.K. The overlap between irritable bowel syndrome and non-celiac gluten sensitivity: A clinical dilemma. Nutrients 2015, 7, 10417–10426. [Google Scholar] [CrossRef] [PubMed]
- De Giorgio, R.; Volta, U.; Gibson, P.R. Sensitivity to wheat, gluten and FODMAPs in IBS: Facts or fiction? Gut 2016, 65, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Biesiekierski, J.R.; Newnham, E.D.; Shepherd, S.J.; Muir, J.G.; Gibson, P.R. Characterization of adults with a self-diagnosis of nonceliac gluten sensitivity. Nutr. Clin. Pract. 2014, 29, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A.; Sapone, A.; Zevallos, V.; Schuppan, D. Nonceliac Gluten Sensitivity. Gastroenterology 2015, 148, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Volta, U.; Pinto-Sanchez, M.I.; Boschetti, E.; Caio, G.; De Giorgio, R.; Verdu, E.F. Dietary Triggers in Irritable Bowel Syndrome: Is There a Role for Gluten? J. Neurogastroenterol. Motil. 2016, 22, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Biesiekierski, J.R.; Rosella, O.; Rose, R.; Liels, K.; Barrett, J.S.; Shepherd, S.J.; Gibson, P.R.; Muir, J.G. Quantification of fructans, galacto-oligosaccharides and other short-chain carbohydrates in processed grains and cereals. J. Hum. Nutr. Diet. 2011, 24, 154–176. [Google Scholar] [CrossRef] [PubMed]
- Varney, J.; Barrett, J.; Scarlata, K.; Catsos, P.; Gibson, P.R.; Muir, J.G. FODMAPs: Food composition, defining cutoff values and international application. J. Gastroenterol. Hepatol. 2017, 32, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Tuck, C.J.; Muir, J.G.; Barrett, J.S.; Gibson, P.R. Fermentable oligo-, di-, monosaccharide and polyols: Role in irritable bowel syndrome. Expert Rev. Gastroenterol. Hepatol. 2014, 8, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Halmos, E.P.; Christophersen, C.T.; Bird, A.R.; Shepherd, S.J.; Gibson, P.R.; Muir, J.G. Diets that differ in their FODMAP content alter the colonic luminal microenvironment. Gut 2015, 64, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Puertollano, E.; Kolida, S.; Yaqoob, P. Biological significance of short-chain fatty acid metabolism by the intestinal microbiome. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Catassi, C. Gluten Sensitivity. Ann. Nutr. Metab. 2015, 67, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Balakireva, A.V.; Zamyatnin, A.A. Properties of Gluten Intolerance: Gluten Structure, Evolution, Pathogenicity and Detoxification Capabilities. Nutrients 2016, 8, 644. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Koch, R.; Moschen, A.R. Proinflammatory Wheat Attacks on the Intestine: Alpha-Amylase Trypsin Inhibitors as New Players. Gastroenterology 2013, 144, 1561–1563. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Gisbert-Schuppan, K. Reply. Gastroenterology 2013, 144, 1563–1564. [Google Scholar] [CrossRef]
- Cuccioloni, M.; Mozzicafreddo, M.; Ali, I.; Bonfili, L.; Cecarini, V.; Eleuteri, A.M.; Angeletti, M. Interaction between wheat alpha-amylase/trypsin bi-functional inhibitor and mammalian digestive enzymes: Kinetic, equilibrium and structural characterization of binding. Food Chem. 2016, 213, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005, 307, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Matysiak-Budnik, T.; Candalh, C.; Dugave, C.; Namane, A.; Cellier, C.; Cerf-Bensussan, N.; Heyman, M. Alterations of the intestinal transport and processing of gliadin peptides in celiac disease. Gastroenterology 2003, 125, 696–707. [Google Scholar] [CrossRef]
- Gribar, S.C.; Richardson, W.M.; Sodhi, C.P.; Hackam, D.J. No longer an innocent bystander: Epithelial toll-like receptor signaling in the development of mucosal inflammation. Mol. Med. 2008, 14, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Al-Sadi, R.; Said, H.M.; Ma, T.Y. Lipopolysaccharide Causes an Increase in Intestinal Tight Junction Permeability in Vitro and in Vivo by Inducing Enterocyte Membrane Expression and Localization of TLR-4 and CD14. Am. J. Pathol. 2013, 182, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Hu, D.; Huo, H.; Zhang, W.; Adiliaghdam, F.; Morrison, S.; Ramirez, J.M.; Gul, S.S.; Hamarneh, S.R.; Hodin, R.A. Intestinal Alkaline Phosphatase Regulates Tight Junction Protein Levels. J. Am. Coll. Surg. 2016, 222, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.T.; Malo, M.S.; Moss, A.K.; Zeller, S.; Johnson, P.; Ebrahimi, F.; Mostafa, G.; Alam, S.N.; Ramasamy, S.; Warren, H.S.; et al. Identification of specific targets for the gut mucosal defense factor intestinal alkaline phosphatase. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G467–G475. [Google Scholar] [CrossRef] [PubMed]
- Lallès, J.P. Intestinal alkaline phosphatase: Novel functions and protective effects. Nutr. Rev. 2014, 72, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Bilski, J.; Mazur-Bialy, A.; Wojcik, D.; Zahradnik-Bilska, J.; Brzozowski, B.; Magierowski, M.; Mach, T.; Magierowska, K.; Brzozowski, T. The Role of Intestinal Alkaline Phosphatase in Inflammatory Disorders of Gastrointestinal Tract. Mediat. Inflamm. 2017, 2017, 9074601. [Google Scholar] [CrossRef] [PubMed]
- Molnár, K.; Vannay, A.; Sziksz, E.; Bánki, N.F.; Győrffy, H.; Arató, A.; Dezsőfi, A.; Veres, G. Decreased mucosal expression of intestinal alkaline phosphatase in children with coeliac disease. Virchows Arch. 2012, 460, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Tuin, A.; Poelstra, K.; de Jager-Krikken, A.; Bok, L.; Raaben, W.; Velders, M.P.; Dijkstra, G. Role of alkaline phosphatase in colitis in man and rats. Gut 2009, 58, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Molnár, K.; Vannay, A.; Szebeni, B.; Bánki, N.F.; Sziksz, E.; Cseh, A.; Győrffy, H.; Lakatos, P.L.; Papp, M.; Arató, A.; et al. Intestinal alkaline phosphatase in the colonic mucosa of children with inflammatory bowel disease. World J. Gastroenterol. 2012, 18, 3254–3259. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.K.; Thapa, B.R.; Nain, C.K.; Sharma, A.K.; Singh, K. Brush border enzyme activities in relation to histological lesion in pediatric celiac disease. J. Gastroenterol. Hepatol. 2008, 23, e348–e352. [Google Scholar] [CrossRef] [PubMed]
- Kaliannan, K.; Hamarneh, S.R.; Economopoulos, K.P.; Nasrin Alam, S.; Moaven, O.; Patel, P.; Malo, N.S.; Ray, M.; Abtahi, S.M.; Muhammad, N.; et al. Intestinal alkaline phosphatase prevents metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 7003–7008. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, S.; Nguyen, D.D.; Eston, M.A.; Alam, S.N.; Moss, A.K.; Ebrahimi, F.; Biswas, B.; Mostafa, G.; Chen, K.T.; Kaliannan, K.; et al. Intestinal alkaline phosphatase has beneficial effects in mouse models of chronic colitis. Inflamm. Bowel Dis. 2011, 17, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Lukas, M.; Drastich, P.; Konecny, M.; Gionchetti, P.; Urban, O.; Cantoni, F.; Bortlik, M.; Duricova, D.; Bulitta, M. Exogenous alkaline phosphatase for the treatment of patients with moderate to severe ulcerative colitis. Inflamm. Bowel Dis. 2010, 16, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.C. Endotoxemia: Methods of detection and clinical correlates. Clin. Microbiol. Rev. 1995, 8, 268–292. [Google Scholar] [PubMed]
- Andreasen, A.S.; Krabbe, K.S.; Krogh-Madsen, R.; Taudorf, S.; Pedersen, B.K.; Møller, K. Human endotoxemia as a model of systemic inflammation. Curr. Med. Chem. 2008, 15, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.C.; Walker, P.M.; Foster, D.M.; Harris, D.; Ribeiro, M.; Paice, J.; Romaschin, A.D.; Derzko, A.N. Measurement of endotoxin activity in critically ill patients using whole blood neutrophil dependent chemiluminescence. Crit. Care 2002, 6, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Wellmann, W.; Fink, P.C.; Benner, F.; Schmidt, F.W. Endotoxaemia in active Crohn’s disease. Treatment with whole gut irrigation and 5-aminosalicylic acid. Gut 1986, 27, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Tepas, J.J., 3rd; Hudak, M.L.; Mollitt, D.L.; Wludyka, P.S.; Teng, R.J.; Premachandra, B.R. Neonatal gut barrier and multiple organ failure: Role of endotoxin and proinflammatory cytokines in sepsis and necrotizing enterocolitis. J. Pediatr. Surg. 2007, 42, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Brock-Utne, J.G.; Gaffin, S.L.; Wells, M.T.; Gathiram, P.; Sohar, E.; James, M.F.; Morrell, D.F.; Norman, R.J. Endotoxaemia in exhausted runners after a long-distance race. S. Afr. Med. J. 1988, 73, 533–536. [Google Scholar] [PubMed]
- Amar, J.; Burcelin, R.; Ruidavets, J.B.; Cani, P.D.; Fauvel, J.; Alessi, M.C.; Chamontin, B.; Ferriéres, J. Energy intake is associated with endotoxemia in apparently healthy men. Am. J. Clin. Nutr. 2008, 87, 1219–1223. [Google Scholar] [PubMed]
- Lira, F.S.; Rosa, J.C.; Pimentel, G.D.; Souza, H.A.; Caperuto, E.C.; Carnevali, L.C., Jr.; Seelaender, M.; Damaso, A.R.; Oyama, L.M.; de Mello, M.T.; et al. Endotoxin levels correlate positively with a sedentary lifestyle and negatively with highly trained subjects. Lipids Health Dis. 2010, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Bosenberg, A.T.; Brock-Utne, J.G.; Gaffin, S.L.; Wells, M.T.; Blake, G.T. Strenuous exercise causes systemic endotoxemia. J. Appl. Physiol. 1988, 65, 106–108. [Google Scholar] [PubMed]
- Liu, H.; Li, W.; Wang, X.; Li, J.; Yu, W. Early gut mucosal dysfunction in patients with acute pancreatitis. Pancreas 2008, 36, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.Y.; Iwamoto, G.K.; Hoa, N.T.; Akotia, V.; Pedram, A.; Boivin, M.A.; Said, H.M. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, R.; Ye, D.; Dokladny, K.; Ma, T.Y. Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction permeability. J. Immunol. 2008, 180, 5653–5661. [Google Scholar] [CrossRef] [PubMed]
- Sapone, A.; Lammers, K.M.; Casolaro, V.; Cammarota, M.; Giuliano, M.T.; De Rosa, M.; Stefanile, R.; Mazzarella, G.; Tolone, C.; Russo, M.I.; et al. Divergence of gut permeability and mucosal immune gene expression in two gluten-associated conditions: Celiac disease and gluten sensitivity. BMC Med. 2011, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Roque, M.I.; Camilleri, M.; Smyrk, T.; Murray, J.A.; Marietta, E.; O’Neill, J.; Carlson, P.; Lamsam, J.; Janzow, D.; Eckert, D.; et al. A controlled trial of gluten-free diet in patients with irritable bowel syndrome-diarrhea: Effects on bowel frequency and intestinal function. Gastroenterology 2013, 144, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Drago, S.; El Asmar, R.; Di Pierro, M.; Grazia Clemente, M.; Tripathi, A.; Sapone, A.; Thakar, M.; Iacono, G.; Carroccio, A.; D’Agate, C.; et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand. J. Gastroenterol. 2006, 41, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Pelsers, M.M.; Hermens, W.T.; Glatz, J.F. Fatty acid-binding proteins as plasma markers of tissue injury. Clin. Chim. Acta 2005, 352, 15–35. [Google Scholar] [CrossRef] [PubMed]
- Pelsers, M.M.; Namiot, Z.; Kisielewski, W.; Namiot, A.; Januszkiewicz, M.; Hermens, W.T.; Glatz, J.F. Intestinal-type and liver-type fatty acid-binding protein in the intestine. Tissue distribution and clinical utility. Clin. Biochem. 2003, 36, 529–535. [Google Scholar] [CrossRef]
- Sandler, N.G.; Koh, C.; Roque, A.; Eccleston, J.L.; Siegel, R.B.; Demino, M.; Kleiner, D.E.; Deeks, S.G.; Liang, T.J.; Heller, T.; et al. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterology 2011, 141, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Ménard, D.; Delvin, E.; Montoudis, A.; Beaulieu, J.F.; Mailhot, G.; Dubé, N.; Sinnett, D.; Seidman, E.; Bendayan, M. Localization, function and regulation of the two intestinal fatty acid-binding protein types. Histochem. Cell Biol. 2009, 132, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Douek, D.C. Microbial translocation across the GI tract. Annu. Rev. Immunol. 2012, 30, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Kitchens, R.L.; Thompson, P.A. Modulatory effects of sCD14 and LBP on LPS-host cell interactions. J. Endotoxin Res. 2005, 11, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Sharifov, O.F.; Xu, X.; Gaggar, A.; Grizzle, W.E.; Mishra, V.K.; Honavar, J.; Litovsky, S.H.; Palgunachari, M.N.; White, C.R.; Anantharamaiah, G.M.; et al. Anti-inflammatory mechanisms of apolipoprotein A-I mimetic peptide in acute respiratory distress syndrome secondary to sepsis. PLoS ONE 2013, 8, e64486. [Google Scholar] [CrossRef] [PubMed]
- Estes, J.D.; Harris, L.D.; Klatt, N.R.; Tabb, B.; Pittaluga, S.; Paiardini, M.; Barclay, G.R.; Smedley, J.; Pung, R.; Oliveira, K.M.; et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 2010, 6, e1001052. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Ajuwon, K.M. Butyrate modifies intestinal barrier function in IPEC-J2 cells through a selective upregulation of tight junction proteins and activation of the Akt signaling pathway. PLoS ONE 2017, 12, e0179586. [Google Scholar] [CrossRef] [PubMed]
- Orchel, A.; Dzierzewicz, Z.; Parfiniewicz, B.; Weglarz, L.; Wilczok, T. Butyrate-Induced Differentiation of Colon Cancer Cells Is PKC and JNK Dependent. Dig. Dis. Sci. 2005, 50, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, K.V.; Sherwin, E.; Schellekens, H.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Feeding the microbiota-gut-brain axis: Diet, microbiome, and neuropsychiatry. Transl. Res. 2016, 179, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.H.; Park, J.H.; Jeon, W.M.; Han, K.S. Butyrate modulates bacterial adherence on LS174T human colorectal cells by stimulating mucin secretion and MAPK signaling pathway. Nutr. Res. Pract. 2015, 9, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed]
- Van den Abbeele, P.; Belzer, C.; Goossens, M.; Kleerebezem, M.; De Vos, W.M.; Thas, O.; De Weirdt, R.; Kerckhof, F.M.; Van de Wiele, T. Butyrate-producing Clostridium cluster XIVa species specifically colonize mucins in an in vitro gut model. ISME J. 2013, 7, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Belenguer, A.; Duncan, S.H.; Calder, A.G.; Holtrop, G.; Louis, P.; Lobley, G.E.; Flint, H.J. Two routes of metabolic cross-feeding between Bifidobacterium adolescentis and butyrate-producing anaerobes from the human gut. Appl. Environ. Microbiol. 2006, 72, 3593–3599. [Google Scholar] [CrossRef] [PubMed]
- Hevia, A.; Delgado, S.; Sánchez, B.; Margolles, A. Molecular Players Involved in the Interaction between Beneficial Bacteria and the Immune System. Front. Microbiol. 2015, 6, 1285. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2010, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Rajilić-Stojanović, M.; Biagi, E.; Heilig, H.G.; Kajander, K.; Kekkonen, R.A.; Tims, S.; de Vos, W.M. Global and deep molecular analysis of microbiota signatures in fecal samples from patients with irritable bowel syndrome. Gastroenterology 2011, 141, 1792–1801. [Google Scholar] [CrossRef] [PubMed]
- Chassard, C.; Dapoigny, M.; Scott, K.P.; Crouzet, L.; Del’homme, C.; Marquet, P.; Martin, J.C.; Pickering, G.; Ardid, D.; Eschalier, A.; et al. Functional dysbiosis within the gut microbiota of patients with constipated-irritable bowel syndrome. Aliment. Pharmacol. Ther. 2012, 35, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Kripke, S.A.; Fox, A.D.; Berman, J.M.; Settle, R.G.; Rombeau, J.L. Stimulation of intestinal mucosal growth with intracolonic infusion of short-chain fatty acids. J. Parent. Enter. Nutr. 1989, 13, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; Busche, R.; Luciano, L.; Reale, E.; Engelhardt, W.V. Lack of butyrate is associated with induction of Bax and subsequent apoptosis in the proximal colon of guinea pig. Gastroenterology 1997, 112, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Cornick, S.; Tawiah, A.; Chadee, K. Roles and regulation of the mucus barrier in the gut. Tissue Barriers 2015, 3, e982426. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, R.F.; Austen, W.G.; Zhang, X., Jr.; Munene, G.; Mostafa, G.; Biswas, S.; McCormack, M.; Eberlin, K.R.; Nguyen, J.T.; Tatlidede, H.S.; et al. Intestinal alkaline phosphatase is a gut mucosal defense factor maintained by enteral nutrition. Proc. Natl. Acad. Sci. USA 2008, 105, 3551–3556. [Google Scholar] [CrossRef] [PubMed]
- Lallès, J.P. Microbiota-host interplay at the gut epithelial level, health and nutrition. J. Anim. Sci. Biotechnol. 2016, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Shifrin, D.A., Jr.; McConnell, R.E.; Nambiar, R.; Higginbotham, J.N.; Coffey, R.J.; Tyska, M.J. Enterocyte microvillus-derived vesicles detoxify bacterial products and regulate epithelial-microbial interactions. Curr. Biol. 2012, 22, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, S.W.; Zhu, J.; Zuo, S.; Ma, Y.Y.; Chen, Z.Y.; Zhang, J.L.; Chen, G.W.; Liu, Y.C.; Wang, P.Y. Intestinal alkaline phosphatase inhibits the translocation of bacteria of gut-origin in mice with peritonitis: Mechanism of action. PLoS ONE 2015, 10, e0124835. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Moya, P.; Ortega-González, M.; González, R.; Anzola, A.; Ocón, B.; Hernández-Chirlaque, C.; López-Posadas, R.; Suárez, M.D.; Zarzuelo, A.; Martínez-Augustin, O.; et al. Exogenous alkaline phosphatase treatment complements endogenous enzyme protection in colonic inflammation and reduces bacterial translocation in rats. Pharmacol. Res. 2012, 66, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Bol-Schoenmakers, M.; Fiechter, D.; Raaben, W.; Hassing, I.; Bleumink, R.; Kruijswijk, D.; Maijoor, K.; Tersteeg-Zijderveld, M.; Brands, R.; Pieters, R. Intestinal alkaline phosphatase contributes to the reduction of severe intestinal epithelial damage. Eur. J. Pharmacol. 2010, 633, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Melo, A.D.; Silveira, H.; Bortoluzzi, C.; Lara, L.J.; Garbossa, C.A.; Preis, G.; Costa, L.B.; Rostagno, M.H. Intestinal alkaline phosphatase and sodium butyrate may be beneficial in attenuating LPS-induced intestinal inflammation. Genet. Mol. Res. 2016, 15, 15048875. [Google Scholar] [CrossRef] [PubMed]
- Malo, M.S.; Biswas, S.; Abedrapo, M.A.; Yeh, L.; Chen, A.; Hodin, R.A. The pro-inflammatory cytokines, IL-1beta and TNF-alpha, inhibit intestinal alkaline phosphatase gene expression. DNA Cell Biol. 2006, 25, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Volta, U.; Tovoli, F.; Cicola, R.; Parisi, C.; Fabbri, A.; Piscaglia, M.; Fiorini, E.; Caio, G. Serological tests in gluten sensitivity (nonceliac gluten intolerance). J. Clin. Gastroenterol. 2012, 46, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Infantino, M.; Manfredi, M.; Meacci, F.; Grossi, V.; Severino, M.; Benucci, M.; Bellio, E.; Bellio, V.; Nucci, A.; Zolfanelli, F.; et al. Diagnostic accuracy of anti-gliadin antibodies in Non Celiac Gluten Sensitivity (NCGS) patients: A dual statistical approach. Clin. Chim. Acta 2015, 451, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Infantino, M.; Meacci, F.; Grossi, V.; Macchia, D.; Manfredi, M. Anti-gliadin antibodies in non-celiac gluten sensitivity. Minerva Gastroenterol. Dietol. 2017, 63, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Caio, G.; Volta, U.; Tovoli, F.; De Giorgio, R. Effect of gluten free diet on immune response to gliadin in patients with non-celiac gluten sensitivity. BMC Gastroenterol. 2014, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Di Liberto, D.; Mansueto, P.; D’Alcamo, A.; Lo Pizzo, M.; Lo Presti, E.; Geraci, G.; Fayer, F.; Guggino, G.; Iacono, G.; Dieli, F.; et al. Predominance of type 1 innate lymphoid cells in the rectal mucosa of patients with non-celiac wheat sensitivity: Reversal after a wheat-free diet. Clin. Transl. Gastroenterol. 2016, 7, e178. [Google Scholar] [CrossRef] [PubMed]
- Barbara, G.; Stanghellini, V.; De Giorgio, R.; Cremon, C.; Cottrell, G.S.; Santini, D.; Pasquinelli, G.; Morselli-Labate, A.M.; Grady, E.F.; Bunnett, N.W.; et al. Activated mast cells in proximity to colonic nerves correlate with abdominal pain in irritable bowel syndrome. Gastroenterology 2004, 126, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Barbara, G.; Cremon, C.; Carini, G.; Bellacosa, L.; Zecchi, L.; De Giorgio, R.; Corinaldesi, R.; Stanghellini, V. The immune system in irritable bowel syndrome. J. Neurogastroenterol. Motil. 2011, 17, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Bennet, S.M.; Ohman, L.; Simren, M. Gut microbiota as potential orchestrators of irritable bowel syndrome. Gut Liver 2015, 9, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Stinson, L.F.; Payne, M.S.; Keelan, J.A. Planting the seed: Origins, composition, and postnatal health significance of the fetal gastrointestinal microbiota. Crit. Rev. Microbiol. 2017, 43, 352–369. [Google Scholar] [CrossRef] [PubMed]
- Thum, C.; Cookson, A.L.; Otter, D.E.; McNabb, W.C.; Hodgkinson, A.J.; Dyer, J.; Roy, N.C. Can nutritional modulation of maternal intestinal microbiota influence the development of the infant gastrointestinal tract? J. Nutr. 2012, 142, 1921–1928. [Google Scholar] [CrossRef] [PubMed]
- Montemurno, E.; Cosola, C.; Dalfino, G.; Daidone, G.; De Angelis, M.; Gobbetti, M.; Gesualdo, L. What would you like to eat, Mr CKD Microbiota? A Mediterranean Diet, please! Kidney Blood Press. Res. 2014, 39, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Alou, M.T.; Lagier, J.C.; Raoult, D. Diet influence on the gut microbiota and dysbiosis related to nutritional disorders. Hum. Microbiome J. 2016, 1, 3–11. [Google Scholar] [CrossRef]
- Cani, P.D.; Delzenne, N.M. The role of the gut microbiota in energy metabolism and metabolic disease. Curr. Pharm. Des. 2009, 15, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Rocha, D.M.; Caldas, A.P.; Oliveira, L.L.; Bressan, J.; Hermsdorff, H.H. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis 2016, 244, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Castaño, G.P.; Caro-Quintero, A.; Reyes, A.; Lizcano, F. Advances in Gut Microbiome Research, Opening New Strategies to Cope with a Western Lifestyle. Front. Genet. 2016, 7, 224. [Google Scholar] [CrossRef] [PubMed]
- Vici, G.; Belli, L.; Biondi, M.; Polzonetti, V. Gluten free diet and nutrient deficiencies: A review. Clin. Nutr. 2016, 35, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Saturni, L.; Ferretti, G.; Bacchetti, T. The gluten-free diet: Safety and nutritional quality. Nutrients 2010, 2, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Robins, G.G.; Burley, V.J.; Howdle, P.D. Evidence of high sugar intake, and low fibre and mineral intake, in the gluten-free diet. Aliment. Pharmacol. Ther. 2010, 32, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, S.J.; Gibson, P.R. Nutritional inadequacies of the gluten-free diet in both recently-diagnosed and long-term patients with coeliac disease. J. Hum. Nutr. Diet. 2013, 26, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Tortora, R.; Capone, P.; De Stefano, G.; Imperatore, N.; Gerbino, N.; Donetto, S.; Monaco, V.; Caporaso, N.; Rispo, A. Metabolic syndrome in patients with coeliac disease on a gluten-free diet. Aliment. Pharmacol. Ther. 2015, 41, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, B.; Cao, Y.; Zong, G.; Hu, F.B.; Green, P.H.R.; Neugut, A.I.; Rimm, E.B.; Sampson, L.; Dougherty, L.W.; Giovannucci, E.; et al. Long term gluten consumption in adults without celiac disease and risk of coronary heart disease: Prospective cohort study. BMJ 2017, 357, 1892. [Google Scholar] [CrossRef] [PubMed]
- De Palma, G.; Nadal, I.; Collado, M.C.; Sanz, Y. Effects of a gluten-free diet on gut microbiota and immune function in healthy adult human subjects. Br. J. Nutr. 2009, 102, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, F.; van Sinderen, D.; O’Toole, P.W.; Stanton, C. Feeding the microbiota: Transducer of nutrient signals for the host. Gut 2017, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Donovan, S.M. Introduction to the special focus issue on the impact of diet on gut microbiota composition and function and future opportunities for nutritional modulation of the gut microbiome to improve human health. Gut Microbes 2017, 8, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Macfarlane, S. Bacteria, colonic fermentation, and gastrointestinal health. J. AOAC Int. 2012, 95, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Quévrain, E.; Maubert, M.A.; Michon, C.; Chain, F.; Marquant, R.; Tailhades, J.; Miquel, S.; Carlier, L.; Bermúdez-Humarán, L.G.; Pigneur, B.; et al. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s diseas. Gut 2016, 65, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Riedel, C.U.; Foata, F.; Philippe, D.; Adolfsson, O.; Eikmanns, B.J.; Blum, S. Anti-inflammatory effects of bifidobacteria by inhibition of LPS-induced NF-kappaB activation. World J. Gastroenterol. 2006, 12, 3729–3735. [Google Scholar] [CrossRef] [PubMed]
- Furrie, E.; Macfarlane, S.; Kennedy, A.; Cummings, J.H.; Walsh, S.V.; O’Neil, D.A.; Macfarlane, G.T. Synbiotic therapy (Bifidobacterium longum/Synergy 1) initiates resolution of inflammation in patients with active ulcerative colitis: A randomised controlled pilot trial. Gut 2005, 54, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Srutkova, D.; Schwarzer, M.; Hudcovic, T.; Zakostelska, Z.; Drab, V.; Spanova, A.; Rittich, B.; Kozakova, H.; Schabussova, I. Bifidobacterium longum CCM 7952 Promotes Epithelial Barrier Function and Prevents Acute DSS-Induced Colitis in Strictly Strain-Specific Manner. PLoS ONE 2015, 10, e0134050. [Google Scholar] [CrossRef] [PubMed]
- Khokhlova, E.V.; Smeianov, V.V.; Efimov, B.A.; Kafarskaia, L.I.; Pavlova, S.I.; Shkoporov, A.N. Anti-inflammatory properties of intestinal Bifidobacterium strains isolated from healthy infants. Microbiol. Immunol. 2012, 56, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Kaneko, T.; Iwana, H.; Taketomo, N.; Tsunoo, H.; Kanno, J.; Ohkusa, T.; Okayasu, I. Inhibitory effects of Bifidobacterium longum on experimental ulcerative colitis induced in mice by synthetic dextran sulfate sodium. Digestion 2003, 67, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Díaz, J.; Ruiz-Ojeda, F.J.; Vilchez-Padial, L.M.; Gil, A. Evidence of the Anti-Inflammatory Effects of Probiotics and Synbiotics in Intestinal Chronic Diseases. Nutrients 2017, 9, 555. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- Madsen, K.L.; Malfair, D.; Gray, D.; Doyle, J.S.; Jewell, L.D.; Fedorak, R.N. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm. Bowel Dis. 1999, 5, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.S.; Ahn, Y.T.; Park, J.C.; Lee, J.H.; Lee, H.; Huh, C.S.; Kim, D.H.; Ryu, D.H.; Hwang, G.S. 1H NMR-based metabonomic assessment of probiotic effects in a colitis mouse model. Arch. Pharm Res. 2010, 33, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Ki Cha, B.; Mun Jung, S.; Hwan Choi, C.; Song, I.D.; Woong Lee, H.; Joon Kim, H.; Hyuk, J.; Kyung Chang, S.; Kim, K.; Chung, W.S.; et al. The effect of a multispecies probiotic mixture on the symptoms and fecal microbiota in diarrhea-dominant irritable bowel syndrome: A randomized, double-blind, placebo-controlled trial. J. Clin. Gastroenterol. 2012, 46, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Bibiloni, R.; Fedorak, R.N.; Tannock, G.W.; Madsen, K.L.; Gionchetti, P.; Campieri, M.; De Simone, C.; Sartor, R.B. VSL#3 probiotic-mixture induces remission in patients with active ulcerative colitis. Am. J. Gastroenterol. 2005, 100, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Miele, E.; Pascarella, F.; Giannetti, E.; Quaglietta, L.; Baldassano, R.N.; Staiano, A. Effect of a probiotic preparation (VSL#3) on induction and maintenance of remission in children with ulcerative colitis. Am. J. Gastroenterol. 2009, 104, 437–443. [Google Scholar] [CrossRef] [PubMed]
- You, H.J.; Oh, D.K.; Ji, G.E. Anticancerogenic effect of a novel chiroinositol-containing polysaccharide from Bifidobacterium bifidum BGN4. FEMS Microbiol. Lett. 2004, 240, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, V.D.; Romeo, M.; Marino Gammazza, A.; Carini, F.; Damiani, P.; Damiano, G.; Buscemi, S.; Lo Monte, A.I.; Gerges-Geagea, A.; Jurjus, A.; et al. The long-term effects of probiotics in the therapy of ulcerative colitis: A clinical study. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czechoslov. 2016, 160, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Donato, K.A.; Gareau, M.G.; Wang, Y.J.; Sherman, P.M. Lactobacillus rhamnosus GG attenuates interferon-{gamma} and tumour necrosis factor-alpha-induced barrier dysfunction and pro-inflammatory signalling. Microbiology 2010, 156, 3288–3297. [Google Scholar] [CrossRef] [PubMed]
- Orlando, A.; Linsalata, M.; Notarnicola, M.; Tutino, V.; Russo, F. Lactobacillus GG restoration of the gliadin induced epithelial barrier disruption: The role of cellular polyamines. BMC Microbiol. 2014, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Mearin, M.L.; Catassi, C.; Brousse, N.; Brand, R.; Collin, P.; Fabiani, E.; Schweizer, J.J.; Abuzakouk, M.; Szajewska, H.; Hallert, C. European multi-centre study on coeliac disease and non-Hodgkin lymphoma. Eur. J. Gastroenterol. Hepatol. 2006, 18, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Corrao, G.; Corazza, G.R.; Bagnardi, V.; Brusco, G.; Ciacci, C.; Cottone, M.; Sategna Guidetti, C.; Usai, P.; Cesari, P.; Pelli, M.A. Mortality in patients with coeliac disease and their relatives: A cohort study. Lancet 2001, 358, 356–361. [Google Scholar] [CrossRef]
- Green, P.H.; Fleischauer, A.T.; Bhagat, G.; Goyal, R.; Jabri, B.; Neugut, A.I. Risk of malignancy in patients with celiac disease. Am. J. Med. 2003, 115, 191–195. [Google Scholar] [CrossRef]
- Askling, J.; Linet, M.; Gridley, G.; Halstensen, T.S.; Ekström, K.; Ekbom, A. Cancer incidence in a population-based cohort of individuals hospitalized with celiac disease or dermatitis herpetiformis. Gastroenterology 2002, 123, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Logan, R.F.; Smith, C.J.; Hubbard, R.B.; Card, T.R. Malignancy and mortality in people with coeliac disease: Population based cohort study. BMJ 2004, 329, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Elfström, P.; Granath, F.; Ye, W.; Ludvigsson, J.F. Low risk of gastrointestinal cancer among patients with celiac disease, inflammation, or latent celiac disease. Clin. Gastroenterol. Hepatol. 2012, 10, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.A.; McMillan, S.A.; Watson, R.G.; Monaghan, P.; Gavin, A.T.; Fox, C.; Murray, L.J. Malignancy and mortality in a population-based cohort of patients with celiac disease or “gluten sensitivity”. World J. Gastroenterol. 2007, 13, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Sheth, P.; Delos Santos, N.; Seth, A.; LaRusso, N.F.; Rao, R.K. Lipopolysaccharide disrupts tight junctions in cholangiocyte monolayers by a c-Src-, TLR4-, and LBP-dependent mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, 308–318. [Google Scholar] [CrossRef] [PubMed]
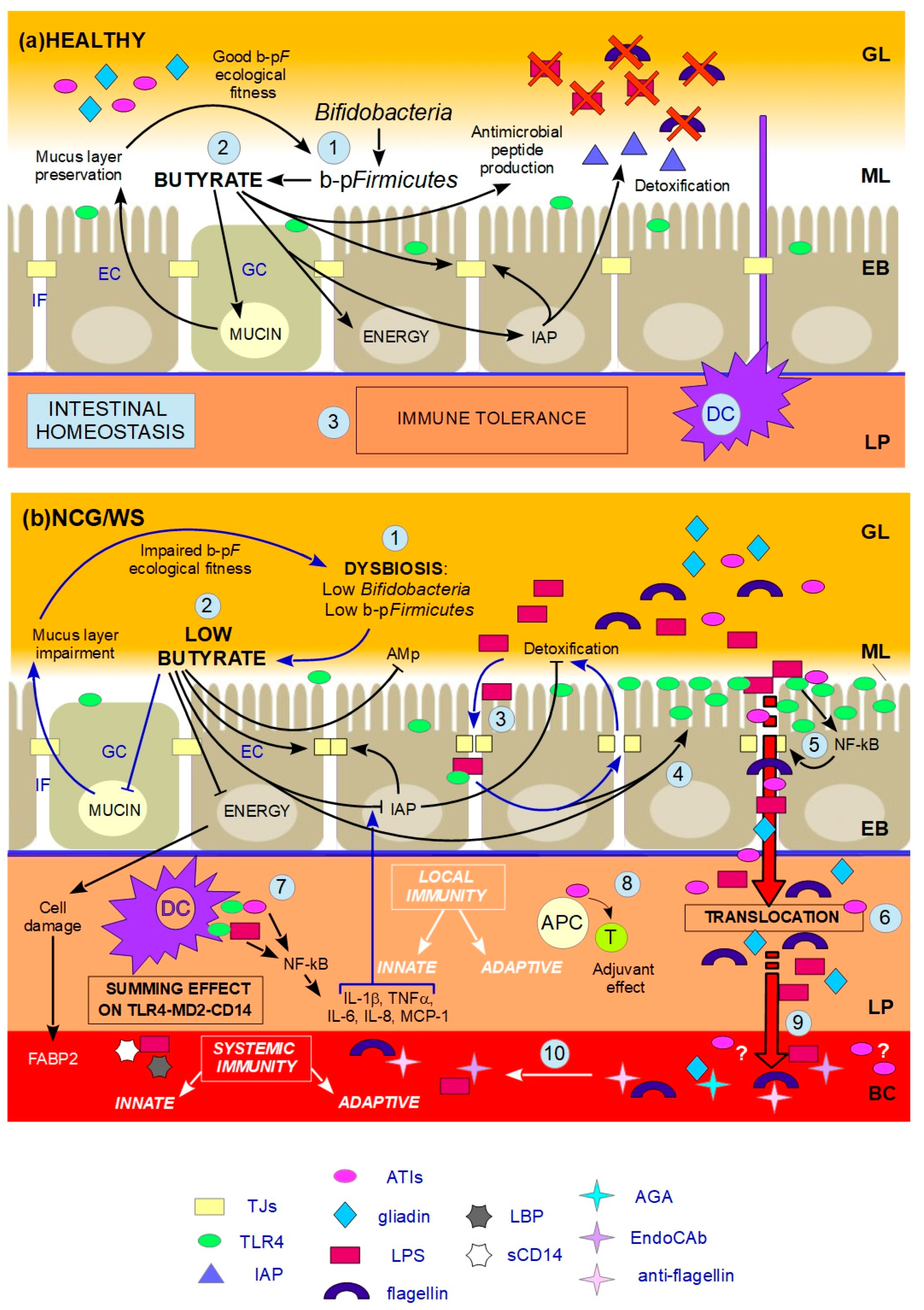
 indicates inhibition. (a) HEALTHY: 1. Butyrate-producing Firmicutes (b-pF) provide adequate levels of butyrate in the ML and Bifidobacteria support the production of butyrate thanks to cross-feeding interactions with b-pF; 2. Butyrate in the ML, close to ECs, plays different trophic and protective functions: it stimulates GCs in the production of mucins, resulting in the preservation of the ML, and thus in a good b-pF ecological fitness. Butyrate constitutes the major energy supply for ECs; it favours the preservation of tight junctions (TJs) integrity by stimulating the expression and membrane co-localization of tight junction proteins (TJPs). Butyrate stimulates the production of antimicrobial peptides (AMp), and the expression and activity of intestinal alkaline phosphatase (IAP), thereby favouring the detoxification of microbial components; 3. All these functions together prevent that the content of the GL directly contacts and/or translocates across the EB, and, together with dendritic cells (DCs) which probe the GL for the presence of antigens, allow gut homeostasis and immune tolerance. (b) NCG/WS: (1) A dysbiosis characterized by low levels of b-pF and/or Bifidobacteria results in not sufficient levels of butyrate in the ML; (2) As a consequence, a chain reaction of events and vicious circles occur: the production of mucins is no longer stimulated, resulting in impairment of the ML. The consequent lowering of b-pF ecological fitness further promotes low levels of butyrate. ECs, without adequate energy source, run into inefficiency and cell damage, resulting in high serum levels of fatty acids binding protein 2 (FABP2). Moreover, TJs integrity is compromised, and the production of AMp is decreased. Low levels of butyrate also cause a decrease in the expression levels and activity of IAP; as a consequence, TJs integrity is further impaired, and the detoxification of microbial components is not sufficient; (3) The failed detoxification enables microbial lipopolysaccharide (LPS) to penetrate in the IF, where it increases paracellular permeability, with a consequent vicious cycle; (4) Furthermore, both LPS in the IF and low levels of butyrate upregulate toll-like receptors 4 (TLR4); (5) Because of the compromised ML, the lumenal content can reach EC surface. LPS and wheat amylase trypsin inhibitors (ATIs) can stimulate overexpressed TLR4, resulting in the production of NF-kB, and then later, inflammatory cytokines, which further damage TJs integrity; (6) Food-borne antigens and microbial components can cross the leaky EB; (7) In the LP, both translocated LPS and ATIs stimulate, at the same time, the TLR4-MD2-CD14 complex on myeloid cells, such as DCs, resulting in a local innate immune response with the production of inflammatory cyokines and chemokines. Among the latter, IL-1β and TNFα further inhibit the activity of IAP, thus maintaining this condition; (8) Moreover, ATIs have an adjuvant effect on possible pre-existing antigenic exposition of antigen-presenting cells (APC) to T-cells (T), triggering an adaptive immune response; (9) Microbial and food-borne antigens translocate in the BC (10), and trigger a systemic innate and adaptive immune response, respectively resulting in high serum levels of lipopolysaccharide-binding protein (LBP) and soluble CD14 (sCD14), and EndoCAb, anti-flagellin and anti-gliadin (AGA) antibodies.
indicates inhibition. (a) HEALTHY: 1. Butyrate-producing Firmicutes (b-pF) provide adequate levels of butyrate in the ML and Bifidobacteria support the production of butyrate thanks to cross-feeding interactions with b-pF; 2. Butyrate in the ML, close to ECs, plays different trophic and protective functions: it stimulates GCs in the production of mucins, resulting in the preservation of the ML, and thus in a good b-pF ecological fitness. Butyrate constitutes the major energy supply for ECs; it favours the preservation of tight junctions (TJs) integrity by stimulating the expression and membrane co-localization of tight junction proteins (TJPs). Butyrate stimulates the production of antimicrobial peptides (AMp), and the expression and activity of intestinal alkaline phosphatase (IAP), thereby favouring the detoxification of microbial components; 3. All these functions together prevent that the content of the GL directly contacts and/or translocates across the EB, and, together with dendritic cells (DCs) which probe the GL for the presence of antigens, allow gut homeostasis and immune tolerance. (b) NCG/WS: (1) A dysbiosis characterized by low levels of b-pF and/or Bifidobacteria results in not sufficient levels of butyrate in the ML; (2) As a consequence, a chain reaction of events and vicious circles occur: the production of mucins is no longer stimulated, resulting in impairment of the ML. The consequent lowering of b-pF ecological fitness further promotes low levels of butyrate. ECs, without adequate energy source, run into inefficiency and cell damage, resulting in high serum levels of fatty acids binding protein 2 (FABP2). Moreover, TJs integrity is compromised, and the production of AMp is decreased. Low levels of butyrate also cause a decrease in the expression levels and activity of IAP; as a consequence, TJs integrity is further impaired, and the detoxification of microbial components is not sufficient; (3) The failed detoxification enables microbial lipopolysaccharide (LPS) to penetrate in the IF, where it increases paracellular permeability, with a consequent vicious cycle; (4) Furthermore, both LPS in the IF and low levels of butyrate upregulate toll-like receptors 4 (TLR4); (5) Because of the compromised ML, the lumenal content can reach EC surface. LPS and wheat amylase trypsin inhibitors (ATIs) can stimulate overexpressed TLR4, resulting in the production of NF-kB, and then later, inflammatory cytokines, which further damage TJs integrity; (6) Food-borne antigens and microbial components can cross the leaky EB; (7) In the LP, both translocated LPS and ATIs stimulate, at the same time, the TLR4-MD2-CD14 complex on myeloid cells, such as DCs, resulting in a local innate immune response with the production of inflammatory cyokines and chemokines. Among the latter, IL-1β and TNFα further inhibit the activity of IAP, thus maintaining this condition; (8) Moreover, ATIs have an adjuvant effect on possible pre-existing antigenic exposition of antigen-presenting cells (APC) to T-cells (T), triggering an adaptive immune response; (9) Microbial and food-borne antigens translocate in the BC (10), and trigger a systemic innate and adaptive immune response, respectively resulting in high serum levels of lipopolysaccharide-binding protein (LBP) and soluble CD14 (sCD14), and EndoCAb, anti-flagellin and anti-gliadin (AGA) antibodies.
indicates inhibition. (a) HEALTHY: 1. Butyrate-producing Firmicutes (b-pF) provide adequate levels of butyrate in the ML and Bifidobacteria support the production of butyrate thanks to cross-feeding interactions with b-pF; 2. Butyrate in the ML, close to ECs, plays different trophic and protective functions: it stimulates GCs in the production of mucins, resulting in the preservation of the ML, and thus in a good b-pF ecological fitness. Butyrate constitutes the major energy supply for ECs; it favours the preservation of tight junctions (TJs) integrity by stimulating the expression and membrane co-localization of tight junction proteins (TJPs). Butyrate stimulates the production of antimicrobial peptides (AMp), and the expression and activity of intestinal alkaline phosphatase (IAP), thereby favouring the detoxification of microbial components; 3. All these functions together prevent that the content of the GL directly contacts and/or translocates across the EB, and, together with dendritic cells (DCs) which probe the GL for the presence of antigens, allow gut homeostasis and immune tolerance. (b) NCG/WS: (1) A dysbiosis characterized by low levels of b-pF and/or Bifidobacteria results in not sufficient levels of butyrate in the ML; (2) As a consequence, a chain reaction of events and vicious circles occur: the production of mucins is no longer stimulated, resulting in impairment of the ML. The consequent lowering of b-pF ecological fitness further promotes low levels of butyrate. ECs, without adequate energy source, run into inefficiency and cell damage, resulting in high serum levels of fatty acids binding protein 2 (FABP2). Moreover, TJs integrity is compromised, and the production of AMp is decreased. Low levels of butyrate also cause a decrease in the expression levels and activity of IAP; as a consequence, TJs integrity is further impaired, and the detoxification of microbial components is not sufficient; (3) The failed detoxification enables microbial lipopolysaccharide (LPS) to penetrate in the IF, where it increases paracellular permeability, with a consequent vicious cycle; (4) Furthermore, both LPS in the IF and low levels of butyrate upregulate toll-like receptors 4 (TLR4); (5) Because of the compromised ML, the lumenal content can reach EC surface. LPS and wheat amylase trypsin inhibitors (ATIs) can stimulate overexpressed TLR4, resulting in the production of NF-kB, and then later, inflammatory cytokines, which further damage TJs integrity; (6) Food-borne antigens and microbial components can cross the leaky EB; (7) In the LP, both translocated LPS and ATIs stimulate, at the same time, the TLR4-MD2-CD14 complex on myeloid cells, such as DCs, resulting in a local innate immune response with the production of inflammatory cyokines and chemokines. Among the latter, IL-1β and TNFα further inhibit the activity of IAP, thus maintaining this condition; (8) Moreover, ATIs have an adjuvant effect on possible pre-existing antigenic exposition of antigen-presenting cells (APC) to T-cells (T), triggering an adaptive immune response; (9) Microbial and food-borne antigens translocate in the BC (10), and trigger a systemic innate and adaptive immune response, respectively resulting in high serum levels of lipopolysaccharide-binding protein (LBP) and soluble CD14 (sCD14), and EndoCAb, anti-flagellin and anti-gliadin (AGA) antibodies.
indicates inhibition. (a) HEALTHY: 1. Butyrate-producing Firmicutes (b-pF) provide adequate levels of butyrate in the ML and Bifidobacteria support the production of butyrate thanks to cross-feeding interactions with b-pF; 2. Butyrate in the ML, close to ECs, plays different trophic and protective functions: it stimulates GCs in the production of mucins, resulting in the preservation of the ML, and thus in a good b-pF ecological fitness. Butyrate constitutes the major energy supply for ECs; it favours the preservation of tight junctions (TJs) integrity by stimulating the expression and membrane co-localization of tight junction proteins (TJPs). Butyrate stimulates the production of antimicrobial peptides (AMp), and the expression and activity of intestinal alkaline phosphatase (IAP), thereby favouring the detoxification of microbial components; 3. All these functions together prevent that the content of the GL directly contacts and/or translocates across the EB, and, together with dendritic cells (DCs) which probe the GL for the presence of antigens, allow gut homeostasis and immune tolerance. (b) NCG/WS: (1) A dysbiosis characterized by low levels of b-pF and/or Bifidobacteria results in not sufficient levels of butyrate in the ML; (2) As a consequence, a chain reaction of events and vicious circles occur: the production of mucins is no longer stimulated, resulting in impairment of the ML. The consequent lowering of b-pF ecological fitness further promotes low levels of butyrate. ECs, without adequate energy source, run into inefficiency and cell damage, resulting in high serum levels of fatty acids binding protein 2 (FABP2). Moreover, TJs integrity is compromised, and the production of AMp is decreased. Low levels of butyrate also cause a decrease in the expression levels and activity of IAP; as a consequence, TJs integrity is further impaired, and the detoxification of microbial components is not sufficient; (3) The failed detoxification enables microbial lipopolysaccharide (LPS) to penetrate in the IF, where it increases paracellular permeability, with a consequent vicious cycle; (4) Furthermore, both LPS in the IF and low levels of butyrate upregulate toll-like receptors 4 (TLR4); (5) Because of the compromised ML, the lumenal content can reach EC surface. LPS and wheat amylase trypsin inhibitors (ATIs) can stimulate overexpressed TLR4, resulting in the production of NF-kB, and then later, inflammatory cytokines, which further damage TJs integrity; (6) Food-borne antigens and microbial components can cross the leaky EB; (7) In the LP, both translocated LPS and ATIs stimulate, at the same time, the TLR4-MD2-CD14 complex on myeloid cells, such as DCs, resulting in a local innate immune response with the production of inflammatory cyokines and chemokines. Among the latter, IL-1β and TNFα further inhibit the activity of IAP, thus maintaining this condition; (8) Moreover, ATIs have an adjuvant effect on possible pre-existing antigenic exposition of antigen-presenting cells (APC) to T-cells (T), triggering an adaptive immune response; (9) Microbial and food-borne antigens translocate in the BC (10), and trigger a systemic innate and adaptive immune response, respectively resulting in high serum levels of lipopolysaccharide-binding protein (LBP) and soluble CD14 (sCD14), and EndoCAb, anti-flagellin and anti-gliadin (AGA) antibodies.

{kind=link}
| Suggested Starting Points For Testing Our Hypothesis On NCG/WS |
|---|
| 1. Association between NCG/WS and dysbiosis, in particular focusing on b-pF and Bifidobacteria levels |
| 2. Association between NCG/WS and an impaired mucus barrier |
| 3. Roles of butyrate and IAP |
| 4. Presence of TJPs co-localization defects and role in the alteration of gut permeability |
| 5. Expression of TJPs and TLR4 at the colonic level |
| 6. Presence of simultaneous stimulation of the TLR4-MD2-CD14 complex by ATIs and LPS |
| 7. Existence of anti-ATIs antibodies |
| 8. Mutations of TLR4 coding gene and related functional studies |
| 9. Association between NCG/WS and HDL levels |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leccioli, V.; Oliveri, M.; Romeo, M.; Berretta, M.; Rossi, P. A New Proposal for the Pathogenic Mechanism of Non-Coeliac/Non-Allergic Gluten/Wheat Sensitivity: Piecing Together the Puzzle of Recent Scientific Evidence. Nutrients 2017, 9, 1203. https://doi.org/10.3390/nu9111203
Leccioli V, Oliveri M, Romeo M, Berretta M, Rossi P. A New Proposal for the Pathogenic Mechanism of Non-Coeliac/Non-Allergic Gluten/Wheat Sensitivity: Piecing Together the Puzzle of Recent Scientific Evidence. Nutrients. 2017; 9(11):1203. https://doi.org/10.3390/nu9111203
Chicago/Turabian StyleLeccioli, Valentina, Mara Oliveri, Marcello Romeo, Massimiliano Berretta, and Paola Rossi. 2017. "A New Proposal for the Pathogenic Mechanism of Non-Coeliac/Non-Allergic Gluten/Wheat Sensitivity: Piecing Together the Puzzle of Recent Scientific Evidence" Nutrients 9, no. 11: 1203. https://doi.org/10.3390/nu9111203
APA StyleLeccioli, V., Oliveri, M., Romeo, M., Berretta, M., & Rossi, P. (2017). A New Proposal for the Pathogenic Mechanism of Non-Coeliac/Non-Allergic Gluten/Wheat Sensitivity: Piecing Together the Puzzle of Recent Scientific Evidence. Nutrients, 9(11), 1203. https://doi.org/10.3390/nu9111203