Nutritional Needs and Support for Children with Chronic Liver Disease

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mechanisms of Malnutrition in Children with Cld
2.1. Decreased Energy Intake
2.2. Increased Energy Needs
2.3. Endocrine Dysfunction
2.4. Malabsorption and Disordered Substrate Metabolism
2.4.1. Carbohydrates
2.4.2. Proteins
2.4.3. Fats
2.5. Fat-Soluble Vitamins
2.5.1. Vitamin A
2.5.2. Vitamin D
2.5.3. Vitamin E
2.5.4. Vitamin K
2.6. Trace Elements and Metals
3. Strategies to Manage Malnutrition in Cld
3.1. Nutritional Assessment
3.2. Supplementation of Specific Macronutrients and Micronutrients
3.2.1. Carbohydrates
3.2.2. Proteins
3.2.3. Fats
3.3. Fat-Soluble Vitamins
3.3.1. Vitamin A
3.3.2. Vitamin D
3.3.3. Vitamin E
3.3.4. Vitamin K
3.4. Water-Soluble Vitamins and Minerals
3.5. Mode of Delivery
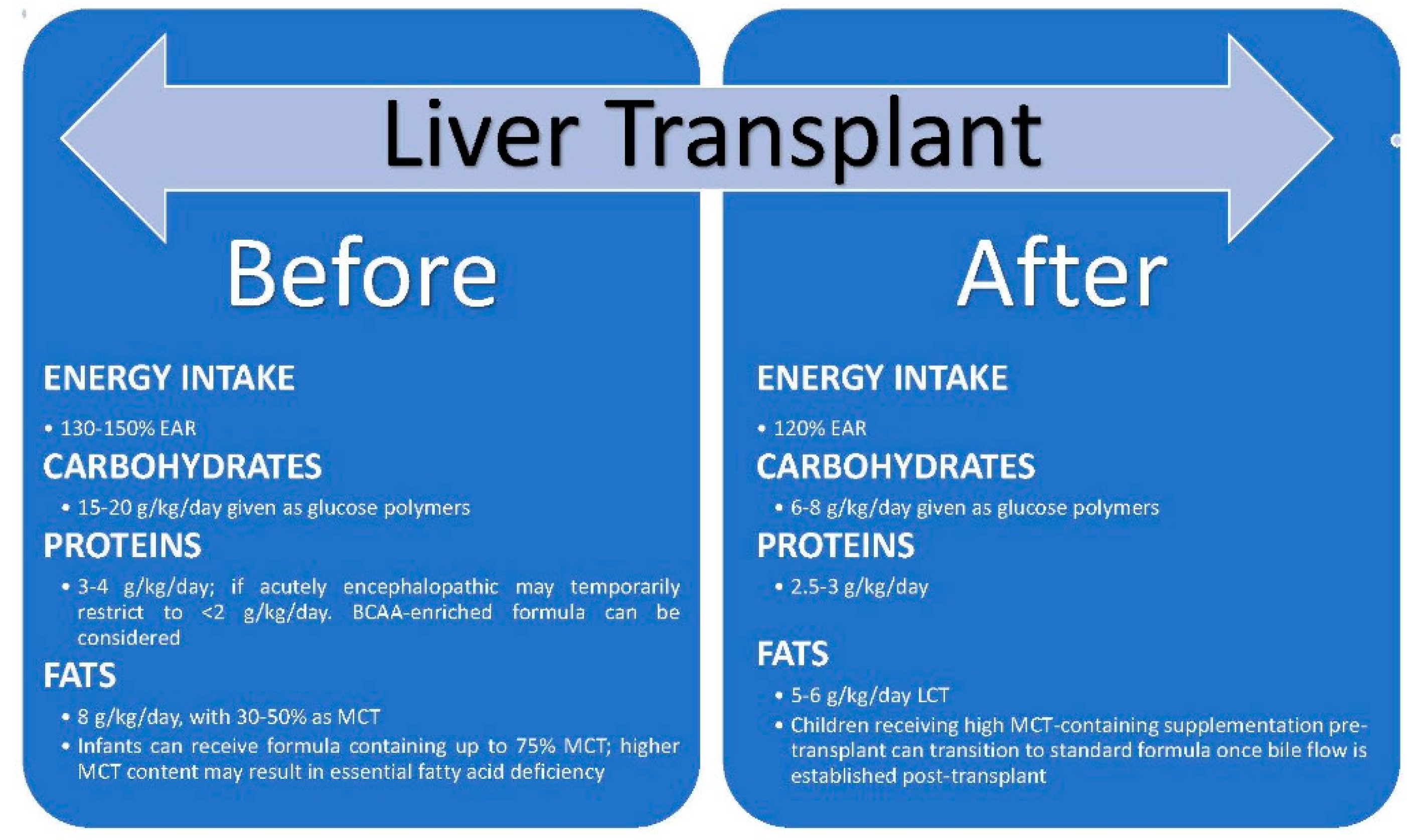
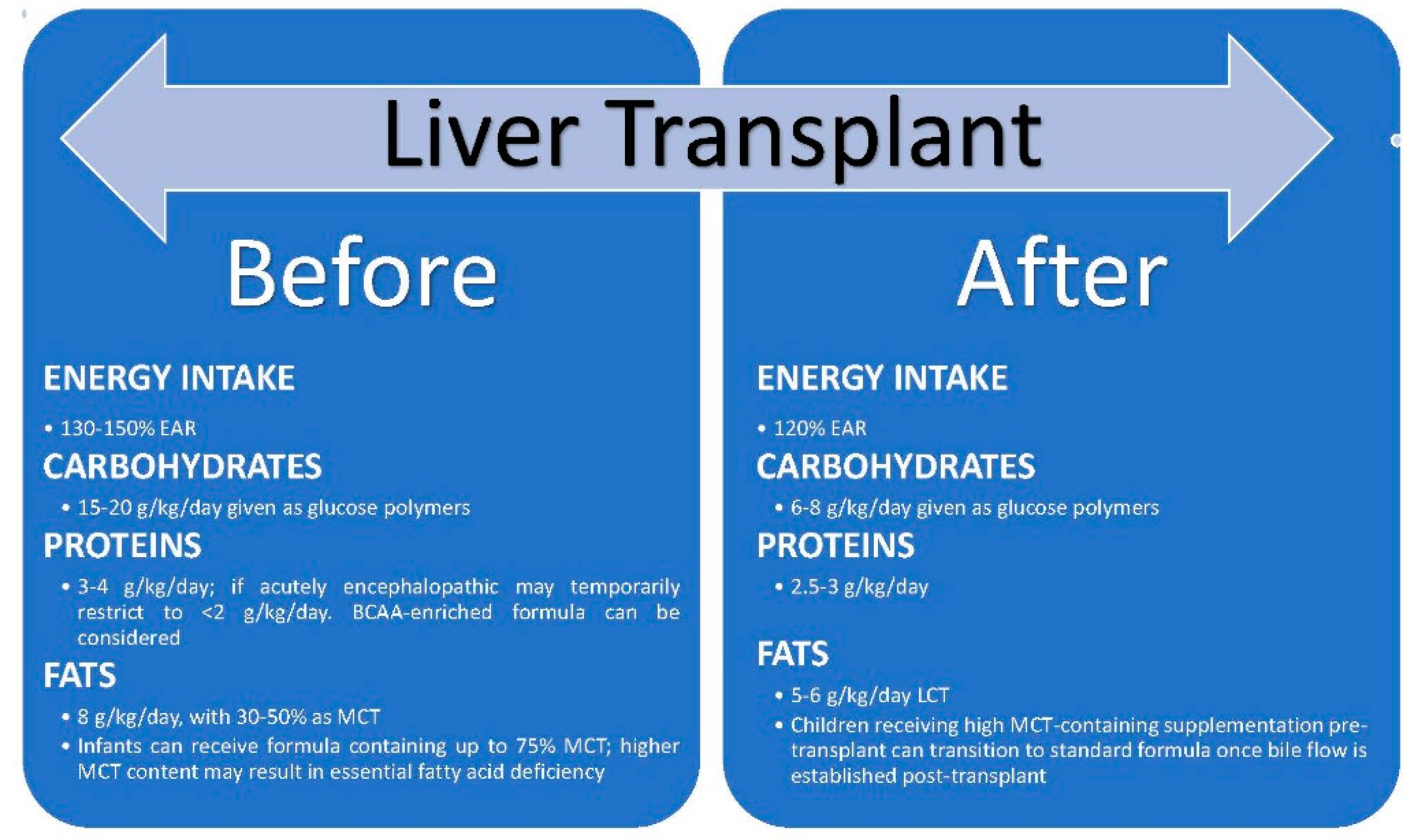
4. Effect of Liver Transplantation on Nutritional Status [Figure 3]
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Pratt, C.A.; Garcia, M.G.; Kerner, J.A., Jr. Nutritional management of neonatal and infant liver disease. NeoReviews 2001, 2, e215–e222. [Google Scholar] [CrossRef]
- Widodo, A.D.; Soelaeman, E.J.; Dwinanda, N.; Narendraswari, P.P.; Purnomo, B. Chronic liver disease is a risk factor for malnutrition and growth retardation in children. Asia Pac. J. Clin. Nutr. 2017, 26 (Suppl. 1), S57–S60. [Google Scholar] [PubMed]
- Young, S.; Kwarta, E.; Azzam, R.; Sentongo, T. Nutrition assessment and support in children with end-stage liver disease. Nutr. Clin. Pract. 2013, 28, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Sokal, E.M.; Goldstein, D.; Ciocca, M.; Lewindon, P.; Ni, Y.H.; Silveira, T.; Sibal, A.; Dhawan, A.; Mack, C.; Bucuvalas, J.; et al. End-stage liver disease and liver transplant: Current situation and key issues. J. Pediatr. Gastroenterol. Nutr. 2008, 47, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Arya, G.; Balistreri, W.F. Pediatric liver disease in the United States: Epidemiology and impact. J. Gastroenterol. Hepatol. 2002, 17, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Baez, N.; Wayman, K.I.; Cox, K.L. Growth and development in chronic liver disease. NeoReviews 2001, 2, e211–e214. [Google Scholar] [CrossRef]
- Danks, D.M.; Campbell, P.E.; Jack, I.; Rogers, J.; Smith, A.L. Studies of the aetiology of neonatal hepatitis and biliary atresia. Arch. Dis. Child. 1977, 52, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Stevenson, R.; Dhawan, A.; Goncalves, I.; Socha, P.; Sokal, E. Guidelines for nutritional care for infants with cholestatic liver disease before liver transplantation. Pediatr. Transplant. 2007, 11, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Smart, K.M.; Alex, G.; Hardikar, W. Feeding the child with liver disease: A review and practical clinical guide. J. Gastroenterol. Hepatol. 2011, 26, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.B.; Schneider, A.C.; da Silveira, T.R. Cirrhosis in children and adolescents: An overview. World J. Hepatol. 2015, 7, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Tahir, A.; Malik, F.R.; Ahmad, I.; Akhtar, P. Aetiological factors of chronic liver disease in children. J. Ayub Med. Coll. Abbottabad 2011, 23, 12–14. [Google Scholar] [PubMed]
- Dhole, S.D.; Kher, A.S.; Ghildiyal, R.G.; Tambse, M.P. Chronic Liver Diseases in Children: Clinical Profile and Histology. J. Clin. Diagn. Res. 2015, 9, SC04–SC07. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, S.; Ng, V.L. Optimizing nutritional management in children with chronic liver disease. Pediatr. Clin. N. Am. 2009, 56, 1161–1183. [Google Scholar] [CrossRef] [PubMed]
- Sokol, R.J.; Stall, C. Anthropometric evaluation of children with chronic liver disease. Am. J. Clin. Nutr. 1990, 52, 203–208. [Google Scholar] [PubMed]
- Chin, S.; Shepherd, R.; Cleghorn, G.; Patrick, M.; Javorsky, G.; Frangoulis, E.; Ong, T.H.; Balderson, G.; Koido, Y.; Matsunami, H.; et al. Survival, growth and quality of life in children after orthotopic liver transplantation: A 5 year experience. J. Paediatr. Child Health 1991, 27, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, R.; Chin, S.; Cleghorn, G.; Patrick, M.; Ong, T.; Lynch, S.; Balderson, G.; Strong, R. Malnutrition in children with chronic liver disease accepted for liver transplanation: Clinical profile and effect on outcome. J. Paediatr. Child Health 1991, 27, 295–299. [Google Scholar] [CrossRef] [PubMed]
- DeRusso, P.; Ye, W.; Shepherd, R.; Haber, B.; Shneider, B.; Whitington, P.; Schwarz, K.B.; Bezerra, J.A.; Rosenthal, P.; Karpen, S.; et al. Growth failure and outcomes in infants with biliary atresia: A report from the Biliary Atresia Research Consortium. Hepatology 2007, 46, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Wayman, K.; Cox, K.; Esquivel, C. Neurodevelopmental outcome of young children with extrahepatic biliary atresia 1 year after liver transplantation. J. Pediatr. 1997, 131, 894–898. [Google Scholar] [CrossRef]
- Stewart, S.; Uauy, R.; Waller, D.; Kennard, B.; Benser, M.; Andrews, W. Mental and motor development, social competence, and growth one year after successful pediatric liver transplantation. J. Pediatr. 1989, 114 (4 Pt 1), 574–581. [Google Scholar] [CrossRef]
- Moukarzel, A.; Najm, I.; Vargas, J.; McDiarmid, S.; Busuttil, R.; Ament, M. Effect of nutritional status on outcome of orthotopic liver transplantation in pediatric patients. Transplant. Proc. 1990, 22, 1560–1563. [Google Scholar] [PubMed]
- Vajro, P.; Maddaluno, S.; Veropalumbo, C. Persistent hypertransaminasemia in asymptomatic children: A stepwise approach. World J. Gastroenterol. 2013, 19, 2740–2751. [Google Scholar] [CrossRef] [PubMed]
- Chin, S.; Shepherd, R.; Thomas, B.; Cleghorn, G.; Patrick, M.; Wilcox, J.; Ong, T.H.; Lynch, S.V.; Strong, R. The nature of malnutrition in children with end-stage liver disease awaiting orthotopic liver transplantation. Am. J. Clin. Nutr. 1992, 56, 164–168. [Google Scholar] [PubMed]
- Laviano, A.; Cangiano, C.; Preziosa, I. Plasma tryptophan levels and anorexia in liver cirrhosis. Int. J. Eat. Disord. 1997, 21, 181–186. [Google Scholar] [CrossRef]
- Aqel, B.; Scolapio, J.; Dickson, R.; Burton, D.; Bouras, E. Contribution of ascites to impaired gastric function and nutritonal intake in patients with cirrhosis and ascites. Clin. Gastroenterol. Hepatol. 2005, 3, 1095–1100. [Google Scholar] [CrossRef]
- Aranda-Michel, J. Nutrition in hepatic failure and liver transplantation. Curr. Gastroenterol. Rep. 2001, 3, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Cortez, A.; de Morais, M.; Speridiao, P.G.; da Motta, M.R.; Calanca, F.; Neto, U. Food intake, growth and body composition of children and adolescents with autoimmune hepatitis. J. Clin. Gastroenterol. 2010, 44, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Pierro, A.K.; Carnielli, V.; Superina, R.; Roberts, E.; Filler, R.; Smith, J.; Heim, T. Resting energy expenditure is increased in infants and children with extrahepatic biliary atresia. J. Pediatr. Surg. 1989, 24, 534–538. [Google Scholar] [CrossRef]
- Protheroe, S.; McKieran, P.; Kelly, D. Can measurement of dietary-induced thermogenesis (DIT) predict response to nutritional intervention in infants with liver disease? Clin. Nutr. 1996, 15, 39. [Google Scholar] [CrossRef]
- Greer, R.; Lehnert, M.; Lewindon, P.; Cleghorn, G.; Shepherd, R. Body composition and components of energy expenditure in children with end-stage liver disease. J. Pediatr. Gastroenterol. Nutr. 2003, 36, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.A.; Proteroe, S.; Clarke, S. Acute and chronic liver disease. In Nutrition in Pediatrics, 5th ed.; Duggan, C., Watkins, J.B., Koletzko, B., Walker, W.A., Eds.; People’s Medical Publishing House-USA: Shelton, CT, USA, 2016; pp. 851–863. [Google Scholar]
- Santetti, D.; Wilasco, M.I.d.A.; Dornelles, C.T.L.; Werlang, I.C.R.; Fontella, F.U.; Kieling, C.O.; Luiz dos Santos, J.; Gonçalves Vieira, S.M.; Sueno Goldani, H.A. Serum proinflammatory cytokines and nutritional status in pediatric chronic liver disease. World J. Gastroenterol. 2015, 21, 8927–8934. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, G.; Dufillot, D.; Antoniazzi, F.; Valentini, R.; Gendrel, D.; Tato, L. Growth hormone-binding proteins and insulin-like grwoth factor-binding proteins in protein-energy malnutrition, before and after nutritonal rehabilitation. Pediatr. Res. 1996, 39, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Romjin, J.; Endert, E.; Suerwein, H. Glucose and fat metablism during short-term starvation in cirrhosis. Gastroenterology 1991, 100, 731–737. [Google Scholar] [CrossRef]
- Hellerstein, M.; Munro, H. Interaction of liver, muscle and adipose tissue in the regulation of metabolism in response to nutritional and other factors. In The Liver: Biology and Pathobiology, 3rd ed.; Arias, I., Boyer, J., Fausto, N., Eds.; Raven Press: New York, NY, USA, 1994; pp. 1169–1191. [Google Scholar]
- Changani, K.; Jalan, R.; Cox, I.; Ala-Korpela, M.; Bhakoo, K.; Taylor-Robinson, S.; Bell, J.D. Evidence for altered hepatic gluconeogenesis in patients with cirrhosis using in vivo 31-phosphorus magnetic resonance spectroscopy. Gut 2001, 49, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Petrides, A.; DeFronzo, R. Glucose and insulin metabolism in cirrhosis. J. Hepatol. 1989, 8, 107–114. [Google Scholar] [CrossRef]
- Nielsen, M.; Caumo, A.; Aagaard, N.; Chandramouli, V.; Schumann, W.; Landau, B.; Schmitz, O.; Vilstrup, H. Contribution of defects in glucose uptake to carbohydrate intolerance in liver cirrhosis: Assessment during physiological glucose and insulin concentrations. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1135–G1143. [Google Scholar] [CrossRef] [PubMed]
- Cleghorn, G. The role of basic nutritional research in pediatric liver disease: A historical perspective. Gastroenterol. Hepatol. 2009, 24, S93–S96. [Google Scholar] [CrossRef] [PubMed]
- Swart, G.R.; van den Berg, J.W.; Wattimena, J.L.; Rietveld, T.; van Vuure, J.K.; Frenkel, M. Elevated protein requirements in cirrhosis of the liver investigated by whole body protein turnover studies. Clin. Sci. 1988, 75, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Protheroe, S.; Jones, R.; Kelly, D. Evaluation of the role of branched chain amino acids in the treatment of protein malnutrition in infants with liver disease. Gut 1995, 37, A350. [Google Scholar]
- Munro, H.N.; Fernstrom, J.D.; Wurtman, R.J. Insulin, plasma amino acid imbalance, and hepatic coma. Lancet 1975, 1, 722–724. [Google Scholar] [CrossRef]
- Charlton, C.P.; Buchanan, E.; Holden, C.E.; Preece, M.A.; Green, A.; Booth, I.W.; Tarlow, M.J. Intensive enteral feeding in advanced cirrhosis: Reversal of malnutrition without precipitation of hepatic encephalopathy. Arch. Dis. Child. 1992, 67, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J. The biology of bile acids. Arch. Intern. Med. 1972, 130, 473–474. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Hamanak, Y.; Suzuki, T. Intestinal microflora and bile acids following biliary tract reconstruction. Nippon Geka Gakkai Zasshi 1991, 92, 1288–1291. [Google Scholar] [PubMed]
- Emerick, K.; Rand, E.; Goldmuntz, E.; Krantz, I.; Spinner, N.; Piccoli, D. Features of Alagille syndrome in 92 patients: Frequency and relation to prognosis. Hepatology 1999, 29, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, N.; Skillman, T. Medium-chain triglycerides. N. Engl. J. Med. 1969, 280, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, J.; Hamilton, J.; Sass-Kortsak, A. Fat absorption in congenital obstructive liver disease. Arch. Dis. Child. 1973, 48, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Phan, C.; Tso, P. Intestinal lipid absorption and transport. Front. Biosci. 2001, 6, D299–D319. [Google Scholar] [CrossRef] [PubMed]
- Sabesin, S.M. Cholestatic lipoproteins—Their pathogenesis and significance. Gastroenterology 1982, 83, 704–709. [Google Scholar] [PubMed]
- Mager, D.R.; McGee, P.L.; Furuya, K.N.; Roberts, E.A. Prevalence of vitamin K deficiency in children with mild to moderate chronic liver disease. J. Pediatr. Gastroenterol. Nutr. 2006, 42, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Shneider, B.L.; Magee, J.C.; Bezerra, J.A.; Haber, B.; Karpen, S.J.; Raghunathan, T.; Rosenthal, P.; Schwarz, K.; Suchy, F.J.; Kerkar, N. Efficacy of fat-soluble vitamin supplementation in infants with biliary atresia. Pediatrics 2012, 130, e607–e614. [Google Scholar] [CrossRef] [PubMed]
- Argao, E.A.; Specker, B.L.; Heubi, J.E. Bone mineral content in infants and children with chronic cholestatic liver disease. Pediatrics 1993, 91, 1151–1154. [Google Scholar] [PubMed]
- Leerbeck, E.; Sondergaard, H. The total content of vitamin D in human milk and cow’s milk. Br. J. Nutr. 1980, 44, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Sokol, R.J.; Heubi, J.E.; Iannaccone, S.; Bove, K.E.; Balistreri, W.F. Mechanism causing vitamin E deficiency during chronic childhood cholestasis. Gastroenterology 1983, 85, 1172–1182. [Google Scholar] [PubMed]
- Usui, Y.; Tanimura, H.; Nishimura, N.; Kobayashi, N.; Okanoue, T.; Ozawa, K. Vitamin K concentrations in the plasma and liver of surgical patients. Am. J. Clin. Nutr. 1990, 51, 846–852. [Google Scholar] [PubMed]
- Grantham-McGregor, S.; Ani, C. A review of studies on the effect of iron deficiency on cognitive development in children. J. Nutr. 2001, 131, 649S–666S. [Google Scholar] [PubMed]
- Stamoulis, I.; Kouraklis, G.; Theocharis, S. Zinc and the liver: An active interaction. Dig. Dis. Sci. 2007, 52, 1595–1612. [Google Scholar] [CrossRef] [PubMed]
- Umusig-Quitain, P.; Gregorio, G.V. High incidence of zinc deficiency among Filipino children with compensated and decompensated liver disease. J. Gastroenterol. Hepatol. 2010, 25, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Goksu, N.; Ozsoylu, S. Hepatic and serum levels of zinc, copper, and magnesium in childhood cirrhosis. J. Pediatr. Gastroenterol. Nutr. 1986, 5, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, E.A.; Hambidge, K.M.; Sokol, R.J.; Stewart, B.; Lilly, J.R. Hepatic concentrations of zinc, copper and manganese in infants with extrahepatic biliary atresia. J. Trace Elem. Med. Biol. 1995, 9, 40–43. [Google Scholar] [CrossRef]
- Fitzgerald, K.; Mikalunas, V.; Rubin, H.; McCarthey, R.; Vanagunas, A.; Craig, R.M. Hypermanganesemia in patients receiving total parenteral nutrition. JPEN J. Parenter. Enter. Nutr. 1999, 23, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Yamaguchi, Y.; Sera, Y.; Ohshiro, H.; Uchino, S.; Yamashita, Y.; Ogawa, M. Manganese deposition in the globus pallidus in patients with biliary atresia. Transplantation 2000, 69, 2339–2343. [Google Scholar] [CrossRef] [PubMed]
- Trocki, O.; Wotton, M.J.; Cleghorn, G.J.; Shepherd, R.W. Value of total body potassium in assessing the nutritional status of children with end-stage liver disease. Ann. N. Y. Acad. Sci. 2000, 904, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Klein, S. The myth of serum albumin as a measure of nutritional status. Gastroenterology 1990, 99, 1845–1846. [Google Scholar] [CrossRef]
- Myron Johnson, A.; Merlini, G.; Sheldon, J.; Ichihara, K. Scientific Division Committee on Plasma Proteins IFoCC, Laboratory M. Clinical indications for plasma protein assays: Transthyretin (prealbumin) in inflammation and malnutrition. Clin. Chem. Lab. Med. 2007, 45, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Bucuvalas, J.C.; Horn, J.A.; Chernausek, S.D. Resistance to growth hormone in children with chronic liver disease. Pediatr. Transplant. 1997, 1, 73–79. [Google Scholar] [PubMed]
- Hogler, W.; Baumann, U.; Kelly, D. Endocrine and bone metabolic complications in chronic liver disease and after liver transplantation in children. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Protheroe, S.M. Feeding the child with chronic liver disease. Nutrition 1998, 14, 796–800. [Google Scholar] [CrossRef]
- Chin, S.E.; Shepherd, R.W.; Thomas, B.J.; Cleghorn, G.J.; Patrick, M.K.; Wilcox, J.A.; Ong, T.H.; Lynch, S.V.; Strong, R. Nutritional support in children with end-stage liver disease: A randomized crossover trial of a branched-chain amino acid supplement. Am. J. Clin. Nutr. 1992, 56, 158–163. [Google Scholar] [PubMed]
- Sokal, E.M.; Baudoux, M.C.; Collette, E.; Hausleithner, V.; Lambotte, L.; Buts, J.P. Branched chain amino acids improve body composition and nitrogen balance in a rat model of extra hepatic biliary atresia. Pediatr. Res. 1996, 40, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Platell, C.; Kong, S.E.; McCauley, R.; Hall, J.C. Branched-chain amino acids. J. Gastroenterol. Hepatol. 2000, 15, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Carey, M.C.; Small, D.M.; Bliss, C.M. Lipid digestion and absorption. Annu. Rev. Physiol. 1983, 45, 651–677. [Google Scholar] [CrossRef] [PubMed]
- Beath, S.V.; Booth, I.W.; Kelly, D.A. Nutritional support in liver disease. Arch. Dis. Child. 1993, 69, 545–547. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, S.S.; Scrivner, D.J.; Murray, N.D.; Vanderhoof, J.A.; Hart, M.H.; Antonson, D.L. Influence of portagen and pregestimil on essential fatty acid status in infantile liver disease. Pediatrics 1992, 89, 151–154. [Google Scholar] [PubMed]
- Koletzko, B.; Agostoni, C.; Carlson, S.E.; Clandinin, T.; Hornstra, G.; Neuringer, M.; Uauy, R.; Yamashiro, Y.; Willatts, P. Long chain polyunsaturated fatty acids (LC-PUFA) and perinatal development. Acta Paediatr. 2001, 90, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Shneider, B.L.; Magee, J.C.; Karpen, S.J.; Rand, E.B.; Narkewicz, M.R.; Bass, L.M.; Schwarz, K.; Whitington, P.F.; Bezerra, J.A.; Kerkar, N.; et al. Total Serum Bilirubin within 3 Months of Hepatoportoenterostomy Predicts Short-Term Outcomes in Biliary Atresia. J. Pediatr. 2016, 170. [Google Scholar] [CrossRef] [PubMed]
- Venkat, V.L.; Shneider, B.L.; Magee, J.C.; Turmelle, Y.; Arnon, R.; Bezerra, J.A.; Hertel, P.M.; Karpen, S.J.; Kerkar, N.; Loomes, K.M. Total serum bilirubin predicts fat-soluble vitamin deficiency better than serum bile acids in infants with biliary atresia. J. Pediatr. Gastroenterol. Nutr. 2014, 59, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Catzola, A.; Vajro, P. Management options for cholestatic liver disease in children. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Guichelaar, M.M.; Kendall, R.; Malinchoc, M.; Hay, J.E. Bone mineral density before and after OLT: Long-term follow-up and predictive factors. Liver Transpl. 2006, 12, 1390–1402. [Google Scholar] [CrossRef] [PubMed]
- Argao, E.A.; Heubi, J.E.; Hollis, B.W.; Tsang, R.C. d-Alpha-tocopheryl polyethylene glycol-1000 succinate enhances the absorption of vitamin D in chronic cholestatic liver disease of infancy and childhood. Pediatr. Res. 1992, 31, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Heubi, J.E.; Hollis, B.W.; Specker, B.; Tsang, R.C. Bone disease in chronic childhood cholestasis. I. Vitamin D absorption and metabolism. Hepatology 1989, 9, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Sokol, R.J.; Heubi, J.E.; Iannaccone, S.T.; Bove, K.E.; Balistreri, W.F. Vitamin E deficiency with normal serum vitamin E concentrations in children with chronic cholestasis. N. Engl. J. Med. 1984, 310, 1209–1212. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, D.H.; Gross, P.; Jones, H.R.; Fulton, A.; Grand, R.J. Intramuscular vitamin E repletion in children with chronic cholestasis. Am. J. Dis. Child. 1987, 141, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Sokol, R.J.; Butler-Simon, N.; Conner, C.; Heubi, J.E.; Sinatra, F.R.; Suchy, F.J.; Heyman, M.B.; Perrault, J.; Rothbaum, R.J.; Levy, J. Multicenter trial of d-alpha-tocopheryl polyethylene glycol 1000 succinate for treatment of vitamin E deficiency in children with chronic cholestasis. Gastroenterology 1993, 104, 1727–1735. [Google Scholar] [CrossRef]
- Ferland, G.; Sadowski, J.A.; O’Brien, M.E. Dietary induced subclinical vitamin K deficiency in normal human subjects. J. Clin. Invest. 1993, 91, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.P.; Shearer, M.J.; Williams, R.; Mieli-Vergani, G. Intestinal absorption of mixed micellar phylloquinone [vitamin K1] is unreliable in infants with conjugated hyperbilirubinaemia: Implications for oral prophylaxis of vitamin K deficiency bleeding. Arch. Dis. Child Fetal Neonatal Ed. 2003, 88, F113–F118. [Google Scholar] [CrossRef] [PubMed]
- Heubi, J.E.; Higgins, J.V.; Argao, E.A.; Sierra, R.I.; Specker, B.L. The role of magnesium in the pathogenesis of bone disease in childhood cholestatic liver disease: A preliminary report. J. Pediatr. Gastroenterol. Nutr. 1997, 25, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Cabre, E.; Gonzalez-Huix, F.; Abad-Lacruz, A.; Esteve, M.; Acero, D.; Fernandez-Banares, F.; Xiol, X.; Gassull, M.A. Effect of total enteral nutrition on the short-term outcome of severely malnourished cirrhotics. A randomized controlled trial. Gastroenterology 1990, 98, 715–720. [Google Scholar] [CrossRef]
- Kearns, P.J.; Young, H.; Garcia, G.; Blaschke, T.; O’Hanlon, G.; Rinki, M.; Sucher, K.; Gregory, P. Accelerated improvement of alcoholic liver disease with enteral nutrition. Gastroenterology 1992, 102, 200–205. [Google Scholar] [CrossRef]
- Cabre, E.; Rodriguez-Iglesias, P.; Caballeria, J.; Quer, J.C.; Sanchez-Lombrana, J.L.; Pares, A.; Papo, M.; Planas, R.; Gassull, M.A. Short- and long-term outcome of severe alcohol-induced hepatitis treated with steroids or enteral nutrition: A multicenter randomized trial. Hepatology 2000, 32, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.A.; Gottrand, F.; Hoden, S.; Turck, D.; Loeuille, G.A.; Farriaux, J.P. Improvement of nutritional status in cholestatic children with supplemental nocturnal enteral nutrition. J. Pediatr. Gastroenterol. Nutr. 1991, 12, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.; Tong, C. Neonatal and paediatric infection. In Viral Hepatitis, 4th ed.; Wiley Blackwell: Hoboken, NJ, USA, 2012. [Google Scholar]
- Fell, J.M.; Reynolds, A.P.; Meadows, N.; Khan, K.; Long, S.G.; Quaghebeur, G.; Taylor, W.J.; Milla, P.J. Manganese toxicity in children receiving long-term parenteral nutrition. Lancet 1996, 347, 1218–1221. [Google Scholar] [CrossRef]
- Huang, C.C.; Chu, N.S.; Lu, C.S.; Wang, J.D.; Tsai, J.L.; Tzeng, J.L.; Wolters, E.C.; Calne, D.B. Chronic manganese intoxication. Arch. Neurol. 1989, 46, 1104–1106. [Google Scholar] [CrossRef] [PubMed]
- Barshes, N.R.; Chang, I.F.; Karpen, S.J.; Carter, B.A.; Goss, J.A. Impact of pretransplant growth retardation in pediatric liver transplantation. J. Pediatr. Gastroenterol. Nutr. 2006, 43, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Rodeck, B.; Melter, M.; Kardorff, R.; Hoyer, P.F.; Ringe, B.; Burdelski, M.; Oldhafer, K.J.; Pichlmayr, R.; Brodehl, J. Liver transplantation in children with chronic end stage liver disease: Factors influencing survival after transplantation. Transplantation 1996, 62, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- SM F, R.J. Feeding patterns in infants post liver transplant. J. Pediatr. Gastroenterol. 2004, 39, S94. [Google Scholar]
- Bartosh, S.M.; Thomas, S.E.; Sutton, M.M.; Brady, L.M.; Whitington, P.F. Linear growth after pediatric liver transplantation. J. Pediatr. 1999, 135, 624–631. [Google Scholar] [CrossRef]
- Cardona, J.; Houssin, D.; Gauthier, F.; Devictor, D.; Losay, J.; Hadchouel, M.; Bernard, O. Liver transplantation in children with Alagille syndrome—A study of twelve cases. Transplantation 1995, 60, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Kamath, B.M.; Yin, W.; Miller, H.; Anand, R.; Rand, E.B.; Alonso, E.; Bucuvalas, J. Outcomes of liver transplantation for patients with Alagille syndrome: The studies of pediatric liver transplantation experience. Liver Transpl. 2012, 18, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.A. Posttransplant growth failure in children. Liver Transpl. Surg. 1997, 3 (5 Suppl. 1), S32–S39. [Google Scholar] [PubMed]
- Van Mourik, I.D.; Beath, S.V.; Brook, G.A.; Cash, A.J.; Mayer, A.D.; Buckels, J.A.; Kelly, D.A. Long-term nutritional and neurodevelopmental outcome of liver transplantation in infants aged less than 12 months. J. Pediatr. Gastroenterol. Nutr. 2000, 30, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Holt, R.I.; Broide, E.; Buchanan, C.R.; Miell, J.P.; Baker, A.J.; Mowat, A.P.; Mieli-Vergani, G. Orthotopic liver transplantation reverses the adverse nutritional changes of end-stage liver disease in children. Am. J. Clin. Nutr. 1997, 65, 534–542. [Google Scholar] [PubMed]
- Amedee-Manesme, O.; Furr, H.C.; Alvarez, F.; Hadchouel, M.; Alagille, D.; Olson, J.A. Biochemical indicators of vitamin A depletion in children with cholestasis. Hepatology 1985, 5, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Lapillonne, A.; Hakme, C.; Mamoux, V.; Chambon, M.; Fournier, V.; Chirouze, V.; Lachaux, A. Effects of liver transplantation on long-chain polyunsaturated fatty acid status in infants with biliary atresia. J. Pediatr. Gastroenterol. Nutr. 2000, 30, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Holt, R.I.; Jones, J.S.; Stone, N.M.; Baker, A.J.; Miell, J.P. Sequential changes in insulin-like growth factor I (IGF-I) and IGF-binding proteins in children with end-stage liver disease before and after successful orthotopic liver transplantation. J. Clin. Endocrinol. Metab. 1996, 81, 160–168. [Google Scholar] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.H.; Perumpail, B.J.; Yoo, E.R.; Ahmed, A.; Kerner Jr., J.A. Nutritional Needs and Support for Children with Chronic Liver Disease. Nutrients 2017, 9, 1127. https://doi.org/10.3390/nu9101127
Yang CH, Perumpail BJ, Yoo ER, Ahmed A, Kerner Jr. JA. Nutritional Needs and Support for Children with Chronic Liver Disease. Nutrients. 2017; 9(10):1127. https://doi.org/10.3390/nu9101127
Chicago/Turabian StyleYang, Christine H., Brandon J. Perumpail, Eric R. Yoo, Aijaz Ahmed, and John A. Kerner Jr. 2017. "Nutritional Needs and Support for Children with Chronic Liver Disease" Nutrients 9, no. 10: 1127. https://doi.org/10.3390/nu9101127
APA StyleYang, C. H., Perumpail, B. J., Yoo, E. R., Ahmed, A., & Kerner Jr., J. A. (2017). Nutritional Needs and Support for Children with Chronic Liver Disease. Nutrients, 9(10), 1127. https://doi.org/10.3390/nu9101127