Precision Nutrition for Targeting Lipid Metabolism in Colorectal Cancer


Abstract
1. Introduction
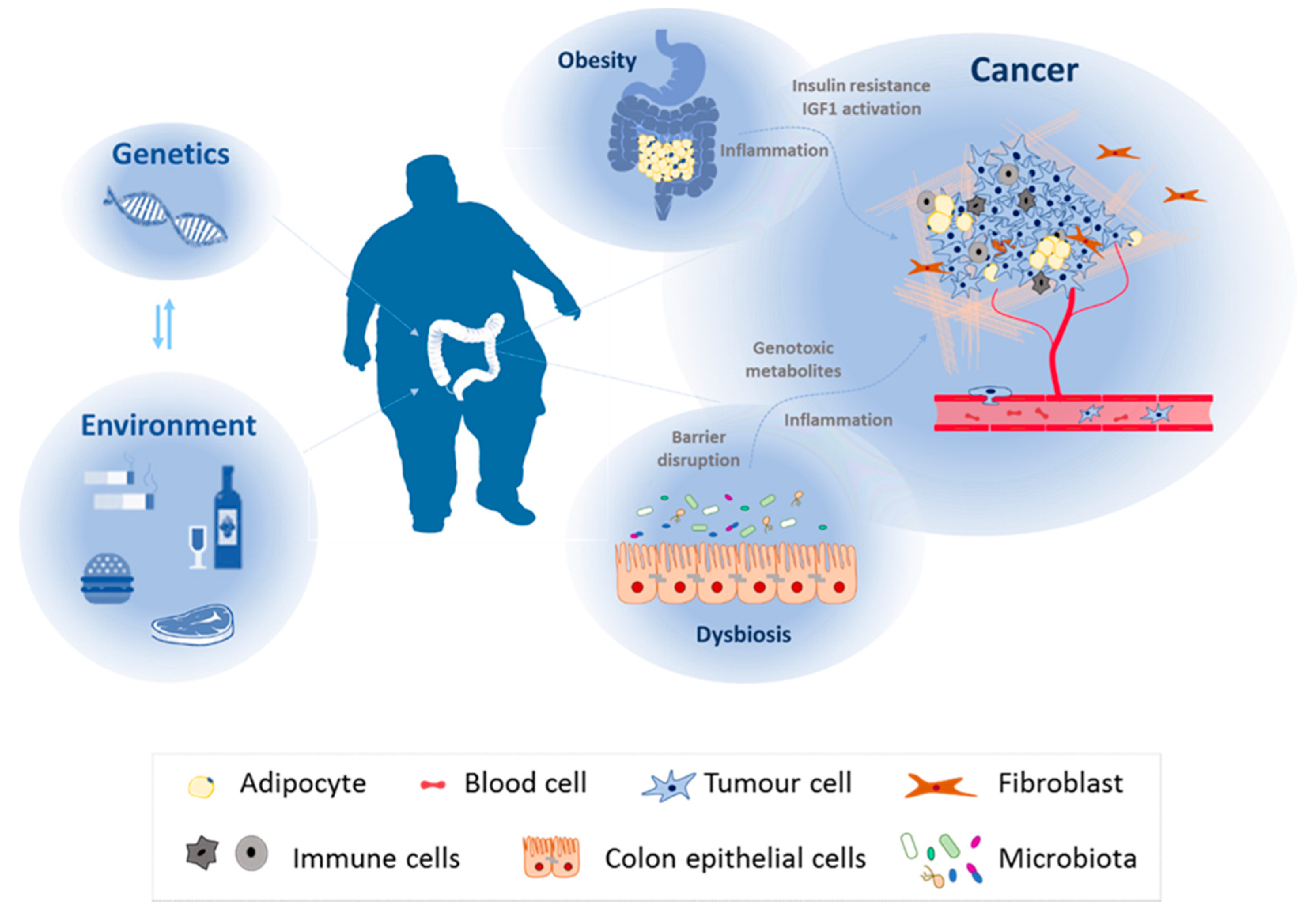
2. Lipid Metabolism, Diet and Colorectal Cancer
2.1. Diet, Obesity, and CRC
2.2. Diet, Dysbiosis, and Colorectal Cancer
3. Nutrition and Colorectal Cancer
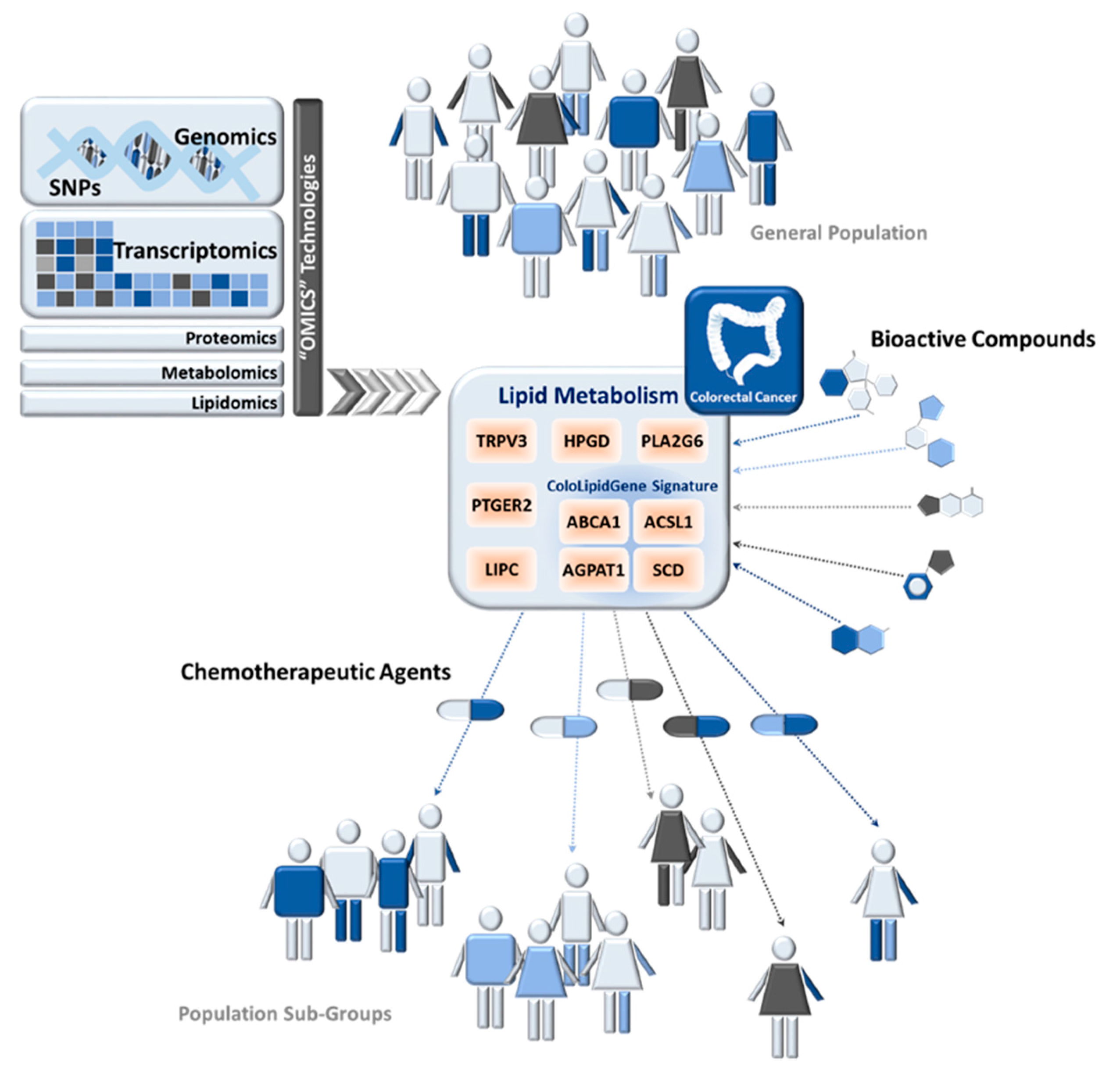
Precision Nutrition in Colorectal Cancer
4. Precision Nutrition and Lipid Metabolism in Colorectal Cancer
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G.; Thompson, C.B. Q&A: Cancer: Clues from cell metabolism. Nature 2010, 465, 562–564. [Google Scholar] [PubMed]
- Jiang, L.; Xiao, L.; Sugiura, H.; Huang, X.; Ali, A.; Kuro-o, M.; Deberardinis, R.J.; Boothman, D.A. Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene 2015, 34, 3908–3916. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.E.; Pop, A.; Koh, W.; Anthis, N.J.; Saunders, W.B.; Davis, G.E. Tumor cell invasion of collagen matrices requires coordinate lipid agonist-induced G-protein and membrane-type matrix metalloproteinase-1-dependent signaling. Mol. Cancer 2006. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martínez, R.; Cruz-Gil, S.; Gómez de Cedrón, M.; Álvarez-Fernández, M.; Vargas, T.; Molina, S.; García, B.; Herranz, J.; Moreno-Rubio, J.; Reglero, G.; et al. A link between lipid metabolism and epithelial-mesenchymal transition provides a target for colon cancer therapy. Oncotarget 2015, 6, 38719–38736. [Google Scholar] [CrossRef] [PubMed]
- English, D.; Brindley, D.N.; Spiegel, S.; Garcia, J.G.N. Lipid mediators of angiogenesis and the signalling pathways they initiate. Biochim. Biophys. Acta 2002, 1582, 228–239. [Google Scholar] [CrossRef]
- Wong, B.W.; Wang, X.; Zecchin, A.; Thienpont, B.; Cornelissen, I.; Kalucka, J.; García-Caballero, M.; Missiaen, R.; Huang, H.; Brüning, U.; et al. The role of fatty acid β-oxidation in lymphangiogenesis. Nature 2017, 542, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017. [Google Scholar] [CrossRef] [PubMed]
- Vargas, T.; Moreno-Rubio, J.; Herranz, J.; Cejas, P.; Molina, S.; González-Vallinas, M.; Mendiola, M.; Burgos, E.; Aguayo, C.; Custodio, A.B.; et al. ColoLipidGene: Signature of lipid metabolism-related genes to predict prognosis in stage-II colon cancer patients. Oncotarget 2015, 6, 7348–7363. [Google Scholar] [CrossRef] [PubMed]
- Vargas, T.; Moreno-Rubio, J.; Herranz, J.; Cejas, P.; Molina, S.; Mendiola, M.; Burgos, E.; Custodio, A.B.; De Miguel, M.; Martín-Hernández, R.; et al. 3’UTR Polymorphism in ACSL1 Gene Correlates with Expression Levels and Poor Clinical Outcome in Colon Cancer Patients. PLoS ONE 2016, 11, e0168423. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Blatter, S.; Rottenberg, S. Minimal residual disease in cancer therapy—Small things make all the difference. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother. 2015, 21–22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Simonds, N.I.; Ghazarian, A.A.; Pimentel, C.B.; Schully, S.D.; Ellison, G.L.; Gillanders, E.M.; Mechanic, L.E. Review of the Gene-Environment Interaction Literature in Cancer: What Do We Know? Genet. Epidemiol. 2016, 40, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D. Gene—Environment-wide association studies: Emerging approaches. Nat. Rev. Genet. 2010, 11, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Biro, F.M.; Wien, M. Childhood obesity and adult morbidities. Am. J. Clin. Nutr. 2010, 91, 1499S–1505S. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, B.; Doll, R. Environmental factors and cancer incidence and mortality in different countries, with special reference to dietary practices. Int. J. Cancer 1975, 15, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, D.P. Epidemiology of cancer of the colon and rectum. Cancer 1971, 28, 3–13. [Google Scholar] [CrossRef]
- Moghaddam, A.A.; Woodward, M.; Huxley, R. Obesity and Risk of Colorectal Cancer: A Meta-analysis of 31 Studies with 70,000 Events. Cancer Epidemiol. Prev. Biomark. 2007, 16, 2533–2547. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.R.; De Caterina, R.; Görman, U.; Allayee, H.; Kohlmeier, M.; Prasad, C.; Choi, M.S.; Curi, R.; de Luis, D.A.; Gil, Á.; et al. Guide and Position of the International Society of Nutrigenetics/Nutrigenomics on Personalised Nutrition: Part 1—Fields of Precision Nutrition. J. Nutr. Nutr. 2016, 9, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Thorisson, G.A.; Stein, L.D. The SNP Consortium website: Past, present and future. Nucleic Acids Res. 2003, 31, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, J. Personalised nutrition: How far has nutrigenomics progressed? Eur. J. Clin. Nutr. 2013, 67, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Gottlieb, E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim. Biophys. Acta 2009, 1788, 2022–2031. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Prijic, S.; Urban, B.C.; Tisza, M.J.; Zuo, Y.; Li, L.; Tan, Z.; Chen, X.; Mani, S.A.; Chang, J.T. Candidate Antimetastasis Drugs Suppress the Metastatic Capacity of Breast Cancer Cells by Reducing Membrane Fluidity. Cancer Res. 2016, 76, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Carito, V.; Bonuccelli, G.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Caroleo, M.C.; Cione, E.; Howell, A.; Pestell, R.G.; Lisanti, M.P.; Sotgia, F. Metabolic remodeling of the tumor microenvironment: Migration stimulating factor (MSF) reprograms myofibroblasts toward lactate production, fueling anabolic tumor growth. Cell Cycle Georget. Tex 2012, 11, 3403–3414. [Google Scholar] [CrossRef] [PubMed]
- Guaita-Esteruelas, S.; Gumà, J.; Masana, L.; Borràs, J. The peritumoural adipose tissue microenvironment and cancer. The roles of fatty acid binding protein 4 and fatty acid binding protein 5. Mol. Cell. Endocrinol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Roy, A.; Dwarakanath, B.S. Metabolic Cooperation and Competition in the Tumor Microenvironment: Implications for Therapy. Front. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ventura, R.; Mordec, K.; Waszczuk, J.; Wang, Z.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G.; Heuer, T.S. Inhibition of de novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression. EBioMedicine 2015, 2, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Tiper, I.V.; East, J.E.; Subrahmanyam, P.B.; Webb, T.J. Sphingosine 1-phosphate signaling impacts lymphocyte migration, inflammation and infection. Pathog. Dis. 2016, 74. [Google Scholar] [CrossRef] [PubMed]
- Baró, L.; Hermoso, J.C.; Núñez, M.C.; Jiménez-Rios, J.A.; Gil, A. Abnormalities in plasma and red blood cell fatty acid profiles of patients with colorectal cancer. Br. J. Cancer 1998, 77, 1978–1983. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Ma, Z.; Rasenick, M.M.; Yeh, S.; Yu, J. N-3 poly-unsaturated fatty acids shift estrogen signaling to inhibit human breast cancer cell growth. PLoS ONE 2012, 7, e52838. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Han, C.; Dai, Y.; Shen, M.; Wu, T. Omega-3 polyunsaturated fatty acids inhibit hepatocellular carcinoma cell growth through blocking beta-catenin and cyclooxygenase-2. Mol. Cancer Ther. 2009, 8, 3046–3055. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.-J.; Wu, J.-M.; Tsai, H.-L.; Huang, C.-W.; Lu, C.-Y.; Sun, L.-C.; Shih, Y.-L.; Chen, C.-W.; Chuang, J.-F.; Wu, M.-H.; et al. Prospective double-blind randomized study on the efficacy and safety of an n-3 fatty acid enriched intravenous fat emulsion in postsurgical gastric and colorectal cancer patients. Nutr. J. 2015, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Whelan, J.; McEntee, M.F. Dietary (n-6) PUFA and intestinal tumorigenesis. J. Nutr. 2004, 134, 3421S–3426S. [Google Scholar] [PubMed]
- Kim, E.K.; Ha, J.M.; Kim, Y.W.; Jin, S.Y.; Ha, H.K.; Bae, S.S. Inhibitory role of polyunsaturated fatty acids on lysophosphatidic acid-induced cancer cell migration and adhesion. FEBS Lett. 2014, 588, 2971–2977. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Ma, S.; Wang, S.; Sun, G. Meta-Analysis of Saturated Fatty Acid Intake and Breast Cancer Risk. Medicine 2015, 94, e2391. [Google Scholar] [CrossRef] [PubMed]
- Bayerdörffer, E.; Mannes, G.A.; Richter, W.O.; Ochsenkühn, T.; Seeholzer, G.; Köpcke, W.; Wiebecke, B.; Paumgartner, G. Decreased high-density lipoprotein cholesterol and increased low-density cholesterol levels in patients with colorectal adenomas. Ann. Intern. Med. 1993, 118, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Morad, S.A.F.; Madigan, J.P.; Levin, J.C.; Abdelmageed, N.; Karimi, R.; Rosenberg, D.W.; Kester, M.; Shanmugavelandy, S.S.; Cabot, M.C. Tamoxifen magnifies therapeutic impact of ceramide in human colorectal cancer cells independent of p53. Biochem. Pharmacol. 2013, 85, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, T.; Tsukahara, R.; Fujiwara, Y.; Yue, J.; Cheng, Y.; Guo, H.; Bolen, A.; Zhang, C.; Balazs, L.; Re, F.; et al. Phospholipase D2-dependent inhibition of the nuclear hormone receptor PPARgamma by cyclic phosphatidic acid. Mol. Cell 2010, 39, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Kurabe, N.; Hayasaka, T.; Ogawa, M.; Masaki, N.; Ide, Y.; Waki, M.; Nakamura, T.; Kurachi, K.; Kahyo, T.; Shinmura, K.; et al. Accumulated phosphatidylcholine (16:0/16:1) in human colorectal cancer; possible involvement of LPCAT4. Cancer Sci. 2013, 104, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Mjabri, B.; Boucrot, P.; Aubry, J. The 1-O-octadecyl-2-O-methyl-sn-glycero-3-phosphocholine causes a differential incorporation of hexadecanol into neutral ether ester glycerolipids of 2 variant cell lines of rat colon carcinoma. Arch. Int. Physiol. Biochim. Biophys. 1992, 100, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Mayne, S.T.; Playdon, M.C.; Rock, C.L. Diet, nutrition, and cancer: Past, present and future. Nat. Rev. Clin. Oncol. 2016, 13, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Martín-Timón, I.; Sevillano-Collantes, C.; Segura-Galindo, A.; del Cañizo-Gómez, F.J. Type 2 diabetes and cardiovascular disease: Have all risk factors the same strength? World J. Diabetes 2014, 5, 444–470. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, M.; Decorby, K.; Choi, B.C.K. The Association between Obesity and Cancer Risk: A Meta-Analysis of Observational Studies from 1985 to 2011. Int. Sch. Res. Not. 2013, 2013, e680536. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, G.; Sun, B.; Zhao, G.; Liu, D.; Sun, J.; Liu, C.; Guo, H. Impact of obesity upon prostate cancer-associated mortality: A meta-analysis of 17 cohort studies. Oncol. Lett. 2015, 9, 1307–1312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, H.; Yang, S.; Zhang, J.; Qian, L.; Chen, X. Overweight, obesity and endometrial cancer risk: Results from a systematic review and meta-analysis. Int. J. Biol. Markers 2014, 29, e21–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, T.-T.; Zhao, J.-J.; Qi, S.-F.; Du, P.; Liu, D.-W.; Tian, Q.-B. The association between overweight, obesity and ovarian cancer: A meta-analysis. Jpn. J. Clin. Oncol. 2015, 45, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Xu, X.; Wang, X.; Zheng, X.-Y. Obesity and risk of bladder cancer: A meta-analysis of cohort studies. Asian Pac. J. Cancer Prev. 2013, 14, 3117–3121. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X.; Wang, J.; Yan, Z.; Luo, J. Excess body weight and the risk of primary liver cancer: An updated meta-analysis of prospective studies. Eur. J. Cancer 2012, 48, 2137–2145. [Google Scholar] [CrossRef] [PubMed]
- Turati, F.; Tramacere, I.; La Vecchia, C.; Negri, E. A meta-analysis of body mass index and esophageal and gastric cardia adenocarcinoma. Ann. Oncol. 2013, 24, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Wolk, A. Overweight, obesity and risk of liver cancer: A meta-analysis of cohort studies. Br. J. Cancer 2007, 97, 1005–1008. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xu, Y. Body mass index and risk of renal cell cancer: A dose-response meta-analysis of published cohort studies. Int. J. Cancer 2014, 135, 1673–1686. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Huang, M.; Wang, L.; Ye, W.; Tong, Y.; Wang, H. Obesity and Risk of Thyroid Cancer: Evidence from a Meta-Analysis of 21 Observational Studies. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2015, 21, 283–291. [Google Scholar]
- Heianza, Y.; Qi, L. Gene-Diet Interaction and Precision Nutrition in Obesity. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.-H.; Byeon, J.-S.; Myung, S.-J.; Yang, S.-K.; Choi, K.-S.; Chung, J.-W.; Kim, B.; Lee, D.; Byun, J.H.; Jang, S.J.; et al. Visceral obesity as a risk factor for colorectal neoplasm. J. Gastroenterol. Hepatol. 2008, 23, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Clayton, P.E.; Banerjee, I.; Murray, P.G.; Renehan, A.G. Growth hormone, the insulin-like growth factor axis, insulin and cancer risk. Nat. Rev. Endocrinol. 2011, 7, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Khandekar, M.J.; Cohen, P.; Spiegelman, B.M. Molecular mechanisms of cancer development in obesity. Nat. Rev. Cancer 2011, 11, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Teoh, S.L.; Das, S. Tumour biology of obesity-related cancers: Understanding the molecular concept for better diagnosis and treatment. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2016, 37, 14363–14380. [Google Scholar] [CrossRef] [PubMed]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current understanding of dysbiosis in disease in human and animal models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Font-Burgada, J.; Sun, B.; Karin, M. Obesity and Cancer: The Oil that Feeds the Flame. Cell Metab. 2016, 23, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Sheflin, A.M.; Whitney, A.K.; Weir, T.L. Cancer-Promoting Effects of Microbial Dysbiosis. Curr. Oncol. Rep. 2014, 16, 406. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Carbonero, F.; Zoetendal, E.G.; DeLany, J.P.; Wang, M.; Newton, K.; Gaskins, H.R.; O’Keefe, S.J.D. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am. J. Clin. Nutr. 2013, 98, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Ambalam, P.; Kondepudi, K.K.; Pithva, S.; Kothari, C.; Patel, A.T.; Purama, R.K.; Dave, J.M.; Vyas, B.R.M. Potential of probiotics, prebiotics and synbiotics for management of colorectal cancer. Gut Microbes 2013, 4, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, D.V. The human gutome: Nutrigenomics of the host-microbiome interactions. Omics J. Integr. Biol. 2011, 15, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Gagnière, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.-A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Kich, D.M.; Vincenzi, A.; Majolo, F.; Volken de Souza, C.F.; Goettert, M.I. Probiotic: Effectiveness nutrition in cancer treatment and prevention. Nutr. Hosp. 2016, 33, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Schirripa, M.; Antoniotti, C.; Moretto, R.; Salvatore, L.; Masi, G.; Falcone, A.; Loupakis, F. First-line chemotherapy for mCRC—A review and evidence-based algorithm. Nat. Rev. Clin. Oncol. 2015, 12, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Rattray, N.J.W.; Charkoftaki, G.; Rattray, Z.; Hansen, J.E.; Vasiliou, V.; Johnson, C.H. Environmental influences in the etiology of colorectal cancer: The premise of metabolomics. Curr. Pharmacol. Rep. 2017, 3, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.E.; Narang, T.; Schnoll-Sussman, F.H.; Pochapin, M.B.; Christos, P.J.; Sherr, D.L. Increasing incidence of rectal cancer in patients aged younger than 40 years: An analysis of the surveillance, epidemiology, and end results database. Cancer 2010, 116, 4354–4359. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Keum, N.; Giovannucci, E.L. Colorectal Cancer Epidemiology in the Nurses’ Health Study. Am. J. Public Health 2016, 106, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Raay, T.V.; Allen-Vercoe, E. Microbial Interactions and Interventions in Colorectal Cancer. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Domingo, J.L.; Nadal, M. Carcinogenicity of consumption of red meat and processed meat: A review of scientific news since the IARC decision. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2017, 105, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, A.; Dissabandara, L.; Gopalan, V. A critical overview on the biological and molecular features of red and processed meat in colorectal carcinogenesis. J. Gastroenterol. 2017, 52, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Loomis, D.; Guyton, K.Z.; Grosse, Y.; Ghissassi, F.E.; Benbrahim-Tallaa, L.; Guha, N.; Mattock, H.; Straif, K.; International Agency for Research on Cancer Monograph Working Group. Carcinogenicity of consumption of red and processed meat. Lancet Oncol. 2015, 16, 1599–1600. [Google Scholar] [CrossRef]
- Carr, P.R.; Walter, V.; Brenner, H.; Hoffmeister, M. Meat subtypes and their association with colorectal cancer: Systematic review and meta-analysis. Int. J. Cancer 2016, 138, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Carr, P.R.; Jansen, L.; Walter, V.; Kloor, M.; Roth, W.; Bläker, H.; Chang-Claude, J.; Brenner, H.; Hoffmeister, M. Associations of red and processed meat with survival after colorectal cancer and differences according to timing of dietary assessment. Am. J. Clin. Nutr. 2016, 103, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Ward, H.A.; Norat, T.; Overvad, K.; Dahm, C.C.; Bueno-de-Mesquita, H.B.; Jenab, M.; Fedirko, V.; van Duijnhoven, F.J.B.; Skeie, G.; Romaguera-Bosch, D.; et al. Pre-diagnostic meat and fibre intakes in relation to colorectal cancer survival in the European Prospective Investigation into Cancer and Nutrition. Br. J. Nutr. 2016, 116, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Helmus, D.S.; Thompson, C.L.; Zelenskiy, S.; Tucker, T.C.; Li, L. Red meat-derived heterocyclic amines increase risk of colon cancer: A population-based case-control study. Nutr. Cancer 2013, 65, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Hunter, D.J.; Spiegelman, D.; Bergkvist, L.; Berrino, F.; van den Brandt, P.A.; Buring, J.E.; Colditz, G.A.; Freudenheim, J.L.; Fuchs, C.S.; et al. Dietary fiber intake and risk of colorectal cancer: A pooled analysis of prospective cohort studies. JAMA 2005, 294, 2849–2857. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Stampfer, M.J.; Colditz, G.A.; Hunter, D.J.; Fuchs, C.; Rosner, B.A.; Speizer, F.E.; Willett, W.C. Multivitamin use, folate, and colon cancer in women in the Nurses’ Health Study. Ann. Intern. Med. 1998, 129, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.B.; Tang, S.Y. Folate status and colorectal cancer risk: A 2016 update. Mol. Aspects Med. 2017, 53, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Bariol, C.; Suter, C.; Cheong, K.; Ku, S.-L.; Meagher, A.; Hawkins, N.; Ward, R. The Relationship between Hypomethylation and CpG Island Methylation in Colorectal Neoplasia. Am. J. Pathol. 2003, 162, 1361–1371. [Google Scholar] [CrossRef]
- Gibson, T.M.; Weinstein, S.J.; Pfeiffer, R.M.; Hollenbeck, A.R.; Subar, A.F.; Schatzkin, A.; Mayne, S.T.; Stolzenberg-Solomon, R. Pre- and postfortification intake of folate and risk of colorectal cancer in a large prospective cohort study in the United States123. Am. J. Clin. Nutr. 2011, 94, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Smith-Warner, S.A.; Spiegelman, D.; Beeson, W.L.; van den Brandt, P.A.; Colditz, G.A.; Folsom, A.R.; Fraser, G.E.; Freudenheim, J.L.; Giovannucci, E.; et al. Dairy foods, calcium, and colorectal cancer: A pooled analysis of 10 cohort studies. J. Natl. Cancer Inst. 2004, 96, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Huncharek, M.; Muscat, J.; Kupelnick, B. Colorectal Cancer Risk and Dietary Intake of Calcium, Vitamin D, and Dairy Products: A Meta-Analysis of 26,335 Cases from 60 Observational Studies. Nutr. Cancer 2008, 61, 47–69. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Morley, T.S.; Kim, M.; Clegg, D.J.; Scherer, P.E. Obesity and cancer--mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 2014, 10, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Eussen, S.J.P.M.; Vollset, S.E.; Igland, J.; Meyer, K.; Fredriksen, A.; Ueland, P.M.; Jenab, M.; Slimani, N.; Boffetta, P.; Overvad, K.; et al. Plasma folate, related genetic variants, and colorectal cancer risk in EPIC. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2010, 19, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, C.M.; Kampman, E.; Bigler, J.; Schwartz, S.M.; Chen, C.; Bostick, R.; Fosdick, L.; Beresford, S.A.; Yasui, Y.; Potter, J.D. Colorectal adenomas and the C677T MTHFR polymorphism: Evidence for gene-environment interaction? Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 1999, 8, 659–668. [Google Scholar]
- Kennedy, D.A.; Stern, S.J.; Matok, I.; Moretti, M.E.; Sarkar, M.; Adams-Webber, T.; Koren, G. Folate Intake, MTHFR Polymorphisms, and the Risk of Colorectal Cancer: A Systematic Review and Meta-Analysis. J. Cancer Epidemiol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Economopoulos, K.P.; Sergentanis, T.N. GSTM1, GSTT1, GSTP1, GSTA1 and colorectal cancer risk: A comprehensive meta-analysis. Eur. J. Cancer Oxf. Engl. 1990 2010, 46, 1617–1631. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Zhou, Y.; Chen, Y.; Li, N.; Chen, B.; Yang, P.; Wu, X. Glutathione S-transferase T1 gene polymorphism and colorectal cancer risk: An updated analysis. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Riscuta, G.; Dumitrescu, R.G. Nutrigenomics: Implications for breast and colon cancer prevention. Methods Mol. Biol. Clifton NJ 2012, 863, 343–358. [Google Scholar]
- Murtaugh, M.A.; Sweeney, C.; Ma, K.-N.; Potter, J.D.; Caan, B.J.; Wolff, R.K.; Slattery, M.L. Vitamin D receptor gene polymorphisms, dietary promotion of insulin resistance, and colon and rectal cancer. Nutr. Cancer 2006, 55, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.A. Nutritional genomic approaches to cancer prevention research. Exp. Oncol. 2007, 29, 250–256. [Google Scholar] [PubMed]
- Park, Y.; Kim, J. Association of Dietary Vitamin D and Calcium with Genetic Polymorphisms in Colorectal Neoplasia. J. Cancer Prev. 2015, 20, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Sheng, S.; Chen, Y.; Shen, Z. Correlation between polymorphism of vitamin D receptor TaqI and susceptibility to colorectal cancer. Medicine (Baltimore) 2017, 96. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Shrubsole, M.J.; Ness, R.M.; Schlundt, D.; Cai, Q.; Smalley, W.E.; Li, M.; Shyr, Y.; Zheng, W. The relation of magnesium and calcium intakes and a genetic polymorphism in the magnesium transporter to colorectal neoplasia risk. Am. J. Clin. Nutr. 2007, 86, 743–751. [Google Scholar] [PubMed]
- Brevik, A.; Joshi, A.D.; Corral, R.; Onland-Moret, N.C.; Siegmund, K.D.; Le Marchand, L.; Baron, J.A.; Martinez, M.E.; Haile, R.W.; Ahnen, D.J.; et al. Polymorphisms in base excision repair genes as colorectal cancer risk factors and modifiers of the effect of diets high in red meat. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2010, 19, 3167–3173. [Google Scholar] [CrossRef] [PubMed]
- Sharafeldin, N.; Slattery, M.L.; Liu, Q.; Franco-Villalobos, C.; Caan, B.J.; Potter, J.D.; Yasui, Y. A Candidate-Pathway Approach to Identify Gene-Environment Interactions: Analyses of Colon Cancer Risk and Survival. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.F. Nutriomes and nutrient arrays—The key to personalised nutrition for DNA damage prevention and cancer growth control. Genome Integr. 2010, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Ordovas, J.M. Nutrigenetics, plasma lipids, and cardiovascular risk. J. Am. Diet. Assoc. 2006, 106, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.C.; Ong, E.; Fukuda, M. Molecular cloning and expression of a novel beta-1, 6-N-acetylglucosaminyltransferase that forms core 2, core 4, and I branches. J. Biol. Chem. 1999, 274, 3215–3221. [Google Scholar] [CrossRef] [PubMed]
- González-Vallinas, M.; Vargas, T.; Moreno-Rubio, J.; Molina, S.; Herranz, J.; Cejas, P.; Burgos, E.; Aguayo, C.; Custodio, A.; Reglero, G.; et al. Clinical relevance of the differential expression of the glycosyltransferase gene GCNT3 in colon cancer. Eur. J. Cancer Oxf. Engl. 1990 2015, 51, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-C.; Chen, H.-Y.; Huang, H.-C.; Huang, J.; Liang, J.-T.; Shen, T.-L.; Lin, N.-Y.; Ho, C.-C.; Cho, I.-M.; Hsu, S.-M. C2GnT-M is downregulated in colorectal cancer and its re-expression causes growth inhibition of colon cancer cells. Oncogene 2006, 25, 3267–3276. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, S.; Chen, J.; Jiang, K.; Zhang, Q.; Guo, K.; Liu, Y. The transcriptional profiling of glycogenes associated with hepatocellular carcinoma metastasis. PLoS ONE 2014, 9, e107941. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V.; Janakiram, N.B.; Madka, V.; Kumar, G.; Scott, E.J.; Pathuri, G.; Bryant, T.; Kutche, H.; Zhang, Y.; Biddick, L.; et al. Small-Molecule Inhibition of GCNT3 Disrupts Mucin Biosynthesis and Malignant Cellular Behaviors in Pancreatic Cancer. Cancer Res. 2016, 76, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, C.; Abrams, T.R.; Brigham, A.; Ceurvels, J.; Clubb, J.; Curtiss, W.; Kirkwood, C.D.; Giese, N.; Hoehn, K.; Iovin, R.; et al. An evidence-based systematic review of rosemary (Rosmarinus officinalis) by the Natural Standard Research Collaboration. J. Diet. Suppl. 2010, 7, 351–413. [Google Scholar] [CrossRef] [PubMed]
- González-Vallinas, M.; Reglero, G.; Molina, A.R. de Rosemary (Rosmarinus officinalis L.) Extract as a Potential Complementary Agent in Anticancer Therapy. Nutr. Cancer 2015, 67, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- González-Vallinas, M.; Molina, S.; Vicente, G.; Zarza, V.; Martín-Hernández, R.; García-Risco, M.R.; Fornari, T.; Reglero, G.; Ramírez de Molina, A. Expression of microRNA-15b and the glycosyltransferase GCNT3 correlates with antitumor efficacy of Rosemary diterpenes in colon and pancreatic cancer. PLoS ONE 2014, 9, e98556. [Google Scholar] [CrossRef] [PubMed]
- Giráldez, M.D.; Lozano, J.J.; Ramírez, G.; Hijona, E.; Bujanda, L.; Castells, A.; Gironella, M. Circulating microRNAs as biomarkers of colorectal cancer: Results from a genome-wide profiling and validation study. Clin. Gastroenterol. Hepatol. 2013, 11, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Gonsalves, W.I.; Mahoney, M.R.; Sargent, D.J.; Nelson, G.D.; Alberts, S.R.; Sinicrope, F.A.; Goldberg, R.M.; Limburg, P.J.; Thibodeau, S.N.; Grothey, A.; et al. Patient and Tumor Characteristics and BRAF and KRAS Mutations in Colon Cancer, NCCTG/Alliance N0147. JNCI J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Xia, G.; Zhang, C.; Sun, Y. Correlation between smoking history and molecular pathways in sporadic colorectal cancer: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 3241–3257. [Google Scholar] [PubMed]
- Porta, M.; Crous-Bou, M.; Wark, P.A.; Vineis, P.; Real, F.X.; Malats, N.; Kampman, E. Cigarette smoking and K-ras mutations in pancreas, lung and colorectal adenocarcinomas: Etiopathogenic similarities, differences and paradoxes. Mutat. Res. 2009, 682, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Jayasekara, H.; MacInnis, R.J.; Williamson, E.J.; Hodge, A.M.; Clendenning, M.; Rosty, C.; Walters, R.; Room, R.; Southey, M.C.; Jenkins, M.A.; et al. Lifetime alcohol intake is associated with an increased risk of KRAS+ and BRAF-/KRAS- but not BRAF+ colorectal cancer. Int. J. Cancer 2017, 140, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Gilsing, A.M.J.; Fransen, F.; de Kok, T.M.; Goldbohm, A.R.; Schouten, L.J.; de Bruïne, A.P.; van Engeland, M.; van den Brandt, P.A.; de Goeij, A.F.P.M.; Weijenberg, M.P. Dietary heme iron and the risk of colorectal cancer with specific mutations in KRAS and APC. Carcinogenesis 2013, 34, 2757–2766. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Hoeft, B.; Linseisen, J.; Beckmann, L.; Müller-Decker, K.; Canzian, F.; Hüsing, A.; Kaaks, R.; Vogel, U.; Jakobsen, M.U.; Overvad, K.; et al. Polymorphisms in fatty acid metabolism-related genes are associated with colorectal cancer risk. Carcinogenesis 2010, 31, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Crous-Bou, M.; Rennert, G.; Salazar, R.; Rodriguez-Moranta, F.; Rennert, H.S.; Lejbkowicz, F.; Kopelovich, L.; Lipkin, S.M.; Gruber, S.B.; Moreno, V. Genetic polymorphisms in fatty acid metabolism genes and colorectal cancer. Mutagenesis 2012, 27, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Frohlich, J.; Ignaszewski, A.P. The impact of dietary changes and dietary supplements on lipid profile. Can. J. Cardiol. 2011, 27, 488–505. [Google Scholar] [CrossRef] [PubMed]
- Daimiel, L.; Vargas, T.; Ramírez de Molina, A. Nutritional genomics for the characterization of the effect of bioactive molecules in lipid metabolism and related pathways. Electrophoresis 2012, 33, 2266–2289. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Butler, E.; McLoughlin, J. Prostate cancer and diet: Food for thought? BJU Int. 2011, 107, 1348–1359. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-Y.; Cai, Y.-Z.; Zhang, Y. Natural phenolic compounds from medicinal herbs and dietary plants: Potential use for cancer prevention. Nutr. Cancer 2010, 62, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Nishiumi, S.; Miyamoto, S.; Kawabata, K.; Ohnishi, K.; Mukai, R.; Murakami, A.; Ashida, H.; Terao, J. Dietary flavonoids as cancer-preventive and therapeutic biofactors. Front. Biosci. Sch. Ed. 2011, 3, 1332–1362. [Google Scholar] [CrossRef]
- Gillies, P.J.; Bhatia, S.K.; Belcher, L.A.; Hannon, D.B.; Thompson, J.T.; Heuvel, J.P.V. Regulation of inflammatory and lipid metabolism genes by eicosapentaenoic acid-rich oil. J. Lipid Res. 2012, 53, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; McClure, D.; Jimenez, L.A.; Megson, I.L.; Rahman, I. Curcumin induces glutathione biosynthesis and inhibits NF-kappaB activation and interleukin-8 release in alveolar epithelial cells: Mechanism of free radical scavenging activity. Antioxid. Redox Signal. 2005, 7, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Iio, A.; Ohguchi, K.; Iinuma, M.; Nozawa, Y.; Ito, M. Hesperetin upregulates ABCA1 expression and promotes cholesterol efflux from THP-1 macrophages. J. Nat. Prod. 2012, 75, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wesemann, S.; Krenn, L.; Ladurner, A.; Heiss, E.H.; Dirsch, V.M.; Atanasov, A.G. Erythrodiol, an Olive Oil Constituent, Increases the Half-Life of ABCA1 and Enhances Cholesterol Efflux from THP-1-Derived Macrophages. Front. Pharmacol. 2017, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.; Hoang, M.-H.; Yeo, S.-K.; Jia, Y.; Lee, S.-J. Induction of ABCA1 and ABCG1 expression by the liver X receptor modulator cineole in macrophages. Bioorg. Med. Chem. Lett. 2013, 23, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Palme, V.; Rotter, S.; Schilcher, N.; Cukaj, M.; Wang, D.; Ladurner, A.; Heiss, E.H.; Stangl, H.; Dirsch, V.M.; Atanasov, A.G. Piperine inhibits ABCA1 degradation and promotes cholesterol efflux from THP-1-derived macrophages. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Rotter, S.; Ladurner, A.; Heiss, E.H.; Oberlies, N.H.; Dirsch, V.M.; Atanasov, A.G. Silymarin Constituents Enhance ABCA1 Expression in THP-1 Macrophages. Mol. Basel Switz. 2015, 21. [Google Scholar] [CrossRef] [PubMed]
- Madden, A.J.; Krehbiel, M.D.; Clarke, S.L. Garlic-derived Compounds Increase Expression of ABCA1 mRNA in RAW 264.7 Murine Macrophages. FASEB J. 2017, 31, 973.1. [Google Scholar]



{kind=link}
{kind=link}
| Gene Symbol | Gene Name | SNP | CRC Risk | Interactors | Reference | |
|---|---|---|---|---|---|---|
| MTHFR | Methylenetetrahydrofolate reductase enzyme | rs1801133 | Cys677Thr | Reduced | High folate intake | [100,101,102] |
| GSTM1 | Glutathione S -transferase M1 | - | Null Phenotype | Increased | - | [103,104] |
| GSTT1 | Glutathione S -transferase T1 | - | Null Phenotype | Increased | - | [103,104] |
| APEX1 | Ascorbate peroxidase | rs1048945 | Glu51His | Reduced | - | [111] |
| PARP | Poly ADP ribose polymerase | rs1136410 | Val762Ala | Modifier of rs1048945 | High-temperature cooked red meat | [111] |
| FLT1 | Vascular endothelial growth factor receptor 1 | rs678714 | Reduced | Smoking | [112] | |
| rs2387632 | Reduced | Animal protein intake | ||||
| KDR | Vascular endothelial growth factor receptor 2 | rs6838752 | Increased | Alcohol | [112] | |
| BMP4 | Bone morphogenetic protein 4 | rs17563 | Reduced | Smoking | [112] | |
| Gene Symbol | Gene Name | SNP | CRC Cases | Controls | Model | CRC Risk | Measure of Risk | (95% CI) | p-Value | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| HPGD | Hydroxyprostaglandin dehydrogenase 15-(NAD) | rs2612656 | 1225 | 2032 | Dom. | Increased risk of developing CRC | OR: 1.24 | (1.07–1.44) | 0.005 | [130] |
| rs8752 | 1225 | 2032 | Dom. | Increased risk of developing CRC | OR: 1.22 | (1.05–1.43) | 0.009 | [130] | ||
| PLA2G6 | Phospholipase A2 group VI | rs4821737 | 1225 | 2032 | Rec. | Increased risk of developing CRC | OR: 1.26 | (1.06–1.50) | 0.009 | [130] |
| TRPV3 | Transient receptor potential vanilloid 3 | rs11078458 | 1225 | 2032 | Rec. | Increased risk of developing CRC | OR: 1.32 | (1.10–1.59) | 0.003 | [130] |
| PTGER2 | Prostaglandin E receptor 2 | rs17831718 | 1225 | 2032 | Dom. | Reduced risk of developing CRC | OR: 0.73 | (0.58–0.91) | 0.006 | [130] |
| LIPC | Hepatic triglyceride lipase | rs9652472 | 1780 | 1864 | Log-add. | Increased risk of developing CRC | OR: 1.52 | (1.20–1.92) | 0.0005 | [131] |
| ACSL1 | Acyl-CoA synthetase 1 | rs8086 | 284 | - | Rec. | Increased risk of CRC relapse | HR: 3.08 | (1.69–5.63) | 0.046 | [11] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguirre-Portolés, C.; Fernández, L.P.; Ramírez de Molina, A. Precision Nutrition for Targeting Lipid Metabolism in Colorectal Cancer. Nutrients 2017, 9, 1076. https://doi.org/10.3390/nu9101076
Aguirre-Portolés C, Fernández LP, Ramírez de Molina A. Precision Nutrition for Targeting Lipid Metabolism in Colorectal Cancer. Nutrients. 2017; 9(10):1076. https://doi.org/10.3390/nu9101076
Chicago/Turabian StyleAguirre-Portolés, Cristina, Lara P. Fernández, and Ana Ramírez de Molina. 2017. "Precision Nutrition for Targeting Lipid Metabolism in Colorectal Cancer" Nutrients 9, no. 10: 1076. https://doi.org/10.3390/nu9101076
APA StyleAguirre-Portolés, C., Fernández, L. P., & Ramírez de Molina, A. (2017). Precision Nutrition for Targeting Lipid Metabolism in Colorectal Cancer. Nutrients, 9(10), 1076. https://doi.org/10.3390/nu9101076