The Emerging Role of Branched-Chain Amino Acids in Insulin Resistance and Metabolism

{kind=link}
Abstract
:1. Introduction
2. The Positive Effects of BCAAs on Metabolic Health
3. The Negative Effects of BCAAs on Metabolism
4. Mechanism of IR by mTOR
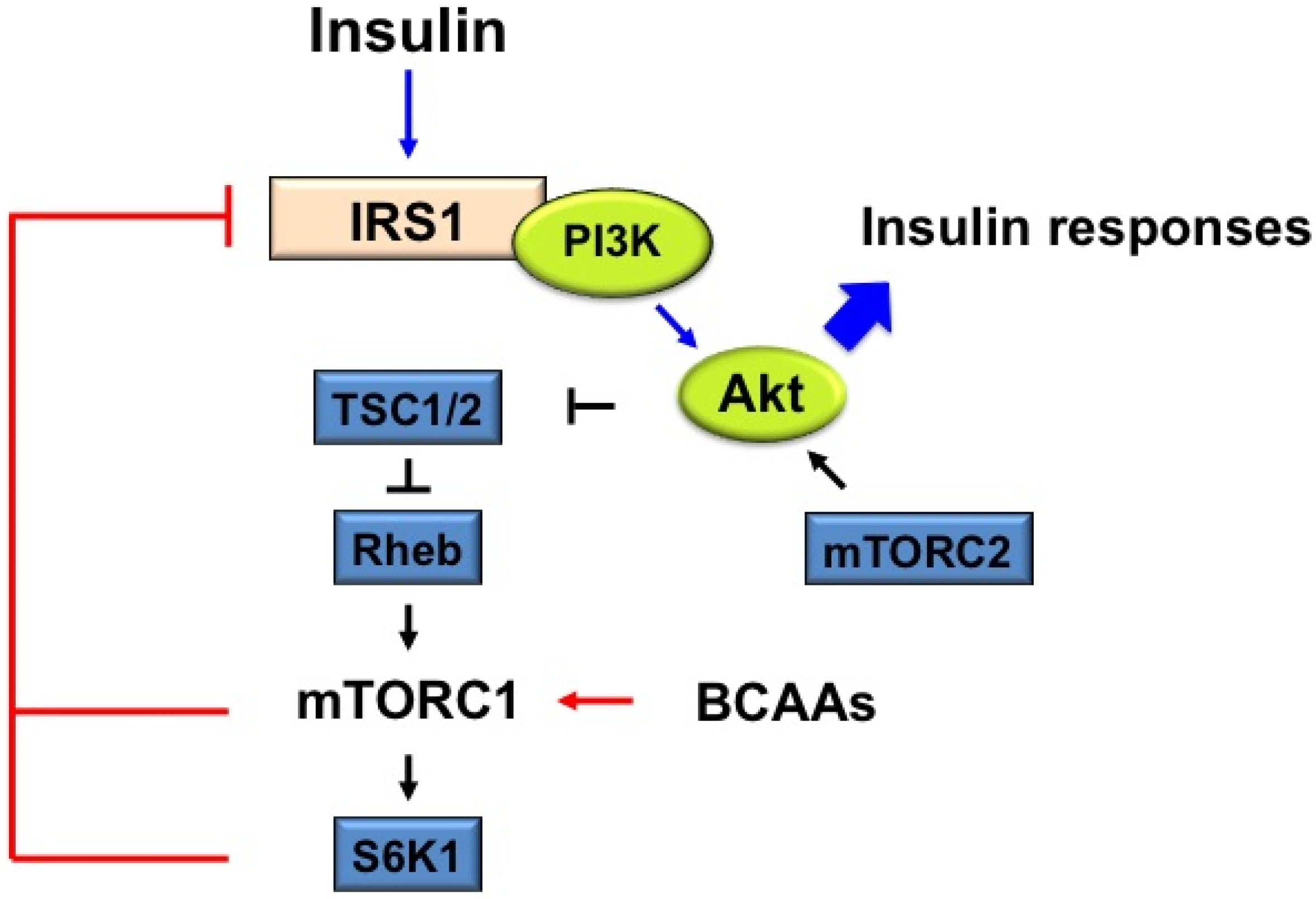
4.1. The Mechanism of Amino Acid-Induced mTORC1 Activation
4.2. The Proposed Mechanism Underlying IR by BCAAs-Induced mTORC1 Activation: Some of the Possible Mechanisms
5. The Controversy over the Role of mTORC1 in IR
6. The Processes That Affect the Level of BCAAs
7. BCAA Dysmetabolism
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Lu, J.; Xie, G.; Jia, W.; Jia, W. Insulin resistance and the metabolism of branched-chain amino acids. Front. Med. 2013, 7, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.; Larson, M.G.; Vasan, R.S.; Cheng, S.; Rhee, E.P.; McCabe, E.; Lewis, G.D.; Fox, C.S.; Jacques, P.F.; Fernandez, C.; et al. Metabolite profiles and the risk of developing diabetes. Nat. Med. 2011, 17, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Giesbertz, P.; Daniel, H. Branched-chain amino acids as biomarkers in diabetes. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Cota, D.; Proulx, K.; Smith, K.A.; Kozma, S.C.; Thomas, G.; Woods, S.C.; Seeley, R.J. Hypothalamic mTOR signaling regulates food intake. Science 2006, 312, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Blouet, C.; Jo, Y.H.; Li, X.; Schwartz, G.J. Mediobasal hypothalamic leucine sensing regulates food intake through activation of a hypothalamus-brainstem circuit. J. Neurosci. 2009, 29, 8302–8311. [Google Scholar] [CrossRef] [PubMed]
- Torres-Leal, F.L.; Fonseca-Alaniz, M.H.; Teodoro, G.F.; de Capitani, M.D.; Vianna, D.; Pantaleão, L.C.; Matos-Neto, E.M.; Rogero, M.M.; Donato, J.; Tirapegui, J. Leucine supplementation improves adiponectin and total cholesterol concentrations despite the lack of changes in adiposity or glucose homeostasis in rats previously exposed to a high-fat diet. Nutr. Metab. 2011, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Reimer, R.A. Dairy protein and leucine alter GLP-1 release and mRNA of genes involved in intestinal lipid metabolism in vitro. Nutrition 2009, 25, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Holst, J.J.; Bjorck, I.M. Metabolic effects of amino acid mixtures and whey protein in healthy subjects: Studies using glucose-equivalent drinks. Am. J. Clin. Nutr. 2007, 85, 996–1004. [Google Scholar] [PubMed]
- Sener, A.; Malaisse, W.J. The stimulus-secretion coupling of amino acid-induced insulin release: Insulinotropic action of branched-chain amino acids at physiological concentrations of glucose and glutamine. Eur. J. Clin. Invest. 1981, 11, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Vary, T.C.; Lynch, C.J. Nutrient signaling components controlling protein synthesis in striated muscle. J. Nutr. 2007, 137, 1835–1843. [Google Scholar] [PubMed]
- Lynch, C.J.; Patson, B.J.; Anthony, J.; Vaval, A.; Jefferson, L.S.; Vary, T.C. Leucine is a direct-acting nutrient signal that regulates protein synthesis in adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E503–E513. [Google Scholar] [CrossRef] [PubMed]
- Dodd, K.M.; Tee, A.R. Leucine and mTORC1: A complex relationship. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1329–E1342. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.; Castellino, P. Correlation between amino acid induced changes in energy expenditure and protein metabolism in humans. Nutrition 1997, 13, 309–312. [Google Scholar] [CrossRef]
- Holecek, M. Three targets of branched-chain amino acid supplementation in the treatment of liver disease. Nutrition 2010, 26, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Macotela, Y.; Emanuelli, B.; Bang, A.M.; Espinoza, D.O.; Boucher, J.; Beebe, K.; Gall, W.; Kahn, C.R. Dietary leucine—An environmental modifier of insulin resistance acting on multiple levels of metabolism. PLoS ONE 2011, 6, e21187. [Google Scholar] [CrossRef] [PubMed]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Newgard, C.B. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012, 15, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Huffman, K.M.; Shah, S.H.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.; Slentz, C.A.; Tanner, C.J.; Kuchibhatla, M.; Houmard, J.A.; Newgard, C.B.; et al. Relationships between circulating metabolic intermediates and insulin action in overweight to obese, inactive men and women. Diabetes Care 2009, 32, 1678–1683. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Ni, Y.; Ma, X.; Bao, Y.; Liu, J.; Huang, F.; Hu, C.; Xie, G.; Zhao, A.; Jia, W.; et al. Branched-chain and aromatic amino acid profiles and diabetes risk in Chinese populations. Sci. Rep. 2016, 6, 20594. [Google Scholar] [CrossRef] [PubMed]
- Knebel, B.; Strassburger, K.; Szendroedi, J.; Kotzka, J.; Scheer, M.; Nowotny, B.; Mussig, K.; Lehr, S.; Pacini, G.; Finner, H.; et al. Specific metabolic profiles and their relationship to insulin resistance in recent-onset type-1 and type-2 diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 2130–2140. [Google Scholar] [CrossRef] [PubMed]
- Shaham, O.; Wei, R.; Wang, T.J.; Ricciardi, C.; Lewis, G.D.; Vasan, R.S.; Carr, S.A.; Thadhani, R.; Gerszten, R.E.; Mootha, V.K. Metabolic profiling of the human response to a glucose challenge reveals distinct axes of insulin sensitivity. Mol. Syst. Biol. 2008, 4, 214. [Google Scholar] [CrossRef] [PubMed]
- Hattersley, J.G.; Pfeiffer, A.F.; Roden, M.; Petzke, K.J.; Hoffmann, D.; Rudovich, N.N.; Randeva, H.S.; Vatish, M.; Osterhoff, M.; Goegebakan, O.; et al. Modulation of amino acid metabolic signatures by supplemented isoenergetic diets differing in protein and cereal fiber content. J. Clin. Endocrinol. Metab. 2014, 99, E2599–E2609. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Garvey, W.T.; Newman, J.W.; Lok, K.H.; Hoppel, C.L.; Adams, S.H. Plasma metabolomic profiles reflective of glucose homeostasis in non-diabetic and type 2 diabetic obese African-American women. PLoS ONE 2010, 5, e15234. [Google Scholar] [CrossRef] [PubMed]
- McCormack, S.E.; Shaham, O.; McCarthy, M.A.; Deik, A.A.; Wang, T.J.; Gerszten, R.E.; Clish, C.B.; Mootha, V.K.; Grinspoon, S.K.; Fleischman, A. Circulating branched-chain amino acid concentrations are associated with obesity and future insulin resistance in children and adolescents. Pediatr. Obes. 2013, 8, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Fernstrom, J.D. Branched-chain amino acids and brain function. J. Nutr. 2005, 135, 1539S–1546S. [Google Scholar] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sabatini, D.M. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005, 17, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Sabatini, D.M. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014, 24, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator is a GEF for the Rag GTPases that signal amino acid levels to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Tsun, Z.Y.; Bar-Peled, L.; Chantranupong, L.; Zoncu, R.; Wang, T.; Kim, C.; Spooner, E.; Sabatini, D.M. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell 2013, 52, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Jeong, S.J.; Park, M.C.; Kim, G.; Kwon, N.H.; Kim, H.K.; Ha, S.H.; Ryu, S.H.; Kim, S. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 2012, 149, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Chantranupong, L.; Wolfson, R.L.; Orozco, J.M.; Saxton, R.A.; Scaria, S.M.; Bar-Peled, L.; Spooner, E.; Isasa, M.; Gygi, S.P.; Sabatini, D.M. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S.; Du, G.; Backer, J.M.; Frohman, M.A.; Chen, J. Class III Pi-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J. Cell Biol. 2011, 195, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001, 294, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S.; Rosenberger, C.L.; Wu, C.; Truong, N.; Sweedler, J.V.; Chen, J. Rapid mitogenic regulation of the mTORC1 inhibitor, deptor, by phosphatidic acid. Mol. Cell 2015, 58, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yoon, S.O.; Poulogiannis, G.; Yang, Q.; Ma, X.M.; Villen, J.; Kubica, N.; Hoffman, G.R.; Cantley, L.C.; Gygi, S.P.; et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011, 332, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D. Balancing Akt with S6K: Implications for both metabolic diseases and tumorigenesis. J. Cell Biol. 2004, 167, 399–403. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. The IRS-signalling system: A network of docking proteins that mediate insulin action. Mol. Cell. Biochem. 1998, 182, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Kozlowski, M.T.; Weng, Q.P.; Morrice, N.; Avruch, J. 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr. Biol. 1998, 8, 69–81. [Google Scholar] [CrossRef]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 2004, 166, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Ozes, O.N.; Akca, H.; Mayo, L.D.; Gustin, J.A.; Maehama, T.; Dixon, J.E.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc. Natl. Acad. Sci. USA 2001, 98, 4640–4645. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, F.; Brule, S.; Hee Um, S.; Li, Y.; Masuda, K.; Roden, M.; Sun, X.J.; Krebs, M.; Polakiewicz, R.D.; Thomas, G.; et al. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 14056–14061. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.J.; White, M.F.; Rondinone, C.M. Mammalian target of rapamycin regulates IRS-1 serine 307 phosphorylation. Biochem. Biophys. Res. Commun. 2004, 316, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, F.; Krebs, M.; Dombrowski, L.; Brehm, A.; Bernroider, E.; Roth, E.; Nowotny, P.; Waldhausl, W.; Marette, A.; Roden, M. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes 2005, 54, 2674–2684. [Google Scholar] [CrossRef] [PubMed]
- She, P.; Reid, T.M.; Bronson, S.K.; Vary, T.C.; Hajnal, A.; Lynch, C.J.; Hutson, S.M. Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell Metab. 2007, 6, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Weickert, M.O.; Roden, M.; Isken, F.; Hoffmann, D.; Nowotny, P.; Osterhoff, M.; Blaut, M.; Alpert, C.; Gogebakan, O.; Bumke-Vogt, C.; et al. Effects of supplemented isoenergetic diets differing in cereal fiber and protein content on insulin sensitivity in overweight humans. Am. J. Clin. Nutr. 2011, 94, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F.; Bradley, D.; Schweitzer, G.G.; Finck, B.N.; Eagon, J.C.; Ilkayeva, O.; Newgard, C.B.; Klein, S. Effect of Roux-en-Y gastric bypass and laparoscopic adjustable gastric banding on branched-chain amino acid metabolism. Diabetes 2013, 62, 2757–2761. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.H. Emerging perspectives on essential amino acid metabolism in obesity and the insulin-resistant state. Adv. Nutr. 2011, 2, 445–456. [Google Scholar] [CrossRef] [PubMed]
- D’Antona, G.; Ragni, M.; Cardile, A.; Tedesco, L.; Dossena, M.; Bruttini, F.; Caliaro, F.; Corsetti, G.; Bottinelli, R.; Carruba, M.O.; et al. Branched-chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle-aged mice. Cell Metab. 2010, 12, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Mordier, S.; Deval, C.; Bechet, D.; Tassa, A.; Ferrara, M. Leucine limitation induces autophagy and activation of lysosome-dependent proteolysis in C2C12 myotubes through a mammalian target of rapamycin-independent signaling pathway. J. Biol. Chem. 2000, 275, 29900–29906. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Ito, Y.; Nishizawa, N.; Nagasawa, T. Regulation of muscle protein degradation, not synthesis, by dietary leucine in rats fed a protein-deficient diet. Amino Acids 2009, 37, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Baptista, I.L.; Leal, M.L.; Artioli, G.G.; Aoki, M.S.; Fiamoncini, J.; Turri, A.O.; Curi, R.; Miyabara, E.H.; Moriscot, A.S. Leucine attenuates skeletal muscle wasting via inhibition of ubiquitin ligases. Muscle Nerve 2010, 41, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Felig, P.; Marliss, E.; Cahill, G.F., Jr. Plasma amino acid levels and insulin secretion in obesity. N. Engl. J. Med. 1969, 281, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Felig, P.; Marliss, E.; Cahill, G.F., Jr. Are plasma amino acid levels elevated in obesity? N. Engl. J. Med. 1970, 282, 166. [Google Scholar] [PubMed]
- Tai, E.S.; Tan, M.L.; Stevens, R.D.; Low, Y.L.; Muehlbauer, M.J.; Goh, D.L.; Ilkayeva, O.R.; Wenner, B.R.; Bain, J.R.; Lee, J.J.; et al. Insulin resistance is associated with a metabolic profile of altered protein metabolism in Chinese and Asian-Indian men. Diabetologia 2010, 53, 757–767. [Google Scholar] [CrossRef] [PubMed]
- O'Connor, P.M.; Bush, J.A.; Suryawan, A.; Nguyen, H.V.; Davis, T.A. Insulin and amino acids independently stimulate skeletal muscle protein synthesis in neonatal pigs. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E110–E119. [Google Scholar] [CrossRef] [PubMed]
- Fryburg, D.A.; Jahn, L.A.; Hill, S.A.; Oliveras, D.M.; Barrett, E.J. Insulin and insulin-like growth factor-i enhance human skeletal muscle protein anabolism during hyperaminoacidemia by different mechanisms. J. Clin. Invest. 1995, 96, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Greenhaff, P.L.; Karagounis, L.G.; Peirce, N.; Simpson, E.J.; Hazell, M.; Layfield, R.; Wackerhage, H.; Smith, K.; Atherton, P.; Selby, A.; et al. Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in human muscle. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E595–E604. [Google Scholar] [CrossRef] [PubMed]
- Estornell, E.; Cabo, J.; Barber, T. Protein synthesis is stimulated in nutritionally obese rats. J. Nutr. 1995, 125, 1309–1315. [Google Scholar] [PubMed]
- Guillet, C.; Masgrau, A.; Boirie, Y. Is protein metabolism changed with obesity? Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Welle, S.; Barnard, R.R.; Statt, M.; Amatruda, J.M. Increased protein turnover in obese women. Metabolism 1992, 41, 1028–1034. [Google Scholar] [CrossRef]
- Neis, E.P.; Dejong, C.H.; Rensen, S.S. The role of microbial amino acid metabolism in host metabolism. Nutrients 2015, 7, 2930–2946. [Google Scholar] [CrossRef] [PubMed]
- Metges, C.C. Contribution of microbial amino acids to amino acid homeostasis of the host. J. Nutr. 2000, 130, 1857S–1864S. [Google Scholar] [PubMed]
- Krajmalnik-Brown, R.; Ilhan, Z.E.; Kang, D.W.; DiBaise, J.K. Effects of gut microbes on nutrient absorption and energy regulation. Nutr. Clin. Pract. 2012, 27, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Sun, H.; She, P.; Youn, J.Y.; Warburton, S.; Ping, P.; Vondriska, T.M.; Cai, H.; Lynch, C.J.; Wang, Y. Protein phosphatase 2 cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J. Clin. Invest. 2009, 119, 1678–1687. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, H.A.; Olson, K.C.; Chen, G.; Lynch, C.J. Adipose transplant for inborn errors of branched chain amino acid metabolism in mice. Mol. Genet. Metab. 2013, 109, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Oyarzabal, A.; Martinez-Pardo, M.; Merinero, B.; Navarrete, R.; Desviat, L.R.; Ugarte, M.; Rodriguez-Pombo, P. A novel regulatory defect in the branched-chain alpha-keto acid dehydrogenase complex due to a mutation in the PPM1K gene causes a mild variant phenotype of maple syrup urine disease. Hum. Mutat. 2013, 34, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.H.; Singer, T.P. Inactivation of the 2-ketoglutarate and pyruvate dehydrogenase complexes of beef heart by branched chain keto acids. J. Biol. Chem. 1983, 258, 1857–1865. [Google Scholar] [PubMed]
- Walajtys-Rode, E.; Williamson, J.R. Effects of branched chain alpha-ketoacids on the metabolism of isolated rat liver cells. III. Interactions with pyruvate dehydrogenase. J. Biol. Chem. 1980, 255, 413–418. [Google Scholar] [PubMed]
- Lackey, D.E.; Lynch, C.J.; Olson, K.C.; Mostaedi, R.; Ali, M.; Smith, W.H.; Karpe, F.; Humphreys, S.; Bedinger, D.H.; Dunn, T.N.; et al. Regulation of adipose branched-chain amino acid catabolism enzyme expression and cross-adipose amino acid flux in human obesity. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1175–E1187. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; She, P.; Peroni, O.D.; Lynch, C.J.; Kahn, B.B. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J. Biol. Chem. 2010, 285, 11348–11356. [Google Scholar] [CrossRef] [PubMed]
- Lefort, N.; Glancy, B.; Bowen, B.; Willis, W.T.; Bailowitz, Z.; De Filippis, E.A.; Brophy, C.; Meyer, C.; Hojlund, K.; Yi, Z.; et al. Increased reactive oxygen species production and lower abundance of complex i subunits and carnitine palmitoyltransferase 1B protein despite normal mitochondrial respiration in insulin-resistant human skeletal muscle. Diabetes 2010, 59, 2444–2452. [Google Scholar] [CrossRef] [PubMed]
- Shin, A.C.; Fasshauer, M.; Filatova, N.; Grundell, L.A.; Zielinski, E.; Zhou, J.Y.; Scherer, T.; Lindtner, C.; White, P.J.; Lapworth, A.L.; et al. Brain insulin lowers circulating BCAA levels by inducing hepatic BCAA catabolism. Cell Metab. 2014, 20, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Mullen, E.; Ohlendieck, K. Proteomic profiling of non-obese type 2 diabetic skeletal muscle. Int. J. Mol. Med. 2010, 25, 445–458. [Google Scholar] [PubMed]
- Kadota, Y.; Toyoda, T.; Kitaura, Y.; Adams, S.H.; Shimomura, Y. Regulation of hepatic branched-chain alpha-ketoacid dehydrogenase complex in rats fed a high-fat diet. Obes. Res. Clin. Pract. 2013, 2013, 7, e439–e444. [Google Scholar] [CrossRef]
- Tiffin, N.; Adie, E.; Turner, F.; Brunner, H.G.; van Driel, M.A.; Oti, M.; Lopez-Bigas, N.; Ouzounis, C.; Perez-Iratxeta, C.; Andrade-Navarro, M.A.; et al. Computational disease gene identification: A concert of methods prioritizes type 2 diabetes and obesity candidate genes. Nucleic Acids Res. 2006, 34, 3067–3081. [Google Scholar] [CrossRef] [PubMed]
- Taneera, J.; Lang, S.; Sharma, A.; Fadista, J.; Zhou, Y.; Ahlqvist, E.; Jonsson, A.; Lyssenko, V.; Vikman, P.; Hansson, O.; et al. A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab. 2012, 16, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Nagasaki, M.; Obayashi, M.; Sato, Y.; Tamura, T.; Shimomura, Y. Mechanism of activation of branched-chain alpha-keto acid dehydrogenase complex by exercise. Biochem. Biophys. Res. Commun. 2011, 287, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Paxton, R.; Harris, R.A. Regulation of branched-chain alpha-ketoacid dehydrogenase kinase. Arch. Biochem. Biophys. 1984, 231, 48–57. [Google Scholar] [CrossRef]
- Corkey, B.E.; Martin-Requero, A.; Walajtys-Rode, E.; Williams, R.J.; Williamson, J.R. Regulation of the branched chain alpha-ketoacid pathway in liver. J. Biol. Chem. 1982, 257, 9668–9676. [Google Scholar] [PubMed]
- Hu, H.; Jaskiewicz, J.A.; Harris, R.A. Ethanol and oleate inhibition of alpha-ketoisovalerate and 3-hydroxyisobutyrate metabolism by isolated hepatocytes. Arch. Biochem. Biophys. 1992, 299, 57–62. [Google Scholar] [CrossRef]
- Frohnert, B.I.; Sinaiko, A.R.; Serrot, F.J.; Foncea, R.E.; Moran, A.; Ikramuddin, S.; Choudry, U.; Bernlohr, D.A. Increased adipose protein carbonylation in human obesity. Obesity 2011, 19, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Ruskovska, T.; Bernlohr, D.A. Oxidative stress and protein carbonylation in adipose tissue—Implications for insulin resistance and diabetes mellitus. J. Proteom. 2013, 92, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, B.D.; Comerford, K.B.; Karakas, S.E.; Knotts, T.A.; Fiehn, O.; Adams, S.H. Whey protein supplementation does not alter plasma branched-chained amino acid profiles but results in unique metabolomics patterns in obese women enrolled in an 8-week weight loss trial. J. Nutr. 2015, 145, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, B.D.; Graham, J.L.; Stanhope, K.L.; Fiehn, O.; Havel, P.J.; Adams, S.H. Plasma amino acid and metabolite signatures tracking diabetes progression in the UCD-T2DM rat model of type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2016. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.H.; Hoppel, C.L.; Lok, K.H.; Zhao, L.; Wong, S.W.; Minkler, P.E.; Hwang, D.H.; Newman, J.W.; Garvey, W.T. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J. Nutr. 2009, 139, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, M.-S. The Emerging Role of Branched-Chain Amino Acids in Insulin Resistance and Metabolism. Nutrients 2016, 8, 405. https://doi.org/10.3390/nu8070405
Yoon M-S. The Emerging Role of Branched-Chain Amino Acids in Insulin Resistance and Metabolism. Nutrients. 2016; 8(7):405. https://doi.org/10.3390/nu8070405
Chicago/Turabian StyleYoon, Mee-Sup. 2016. "The Emerging Role of Branched-Chain Amino Acids in Insulin Resistance and Metabolism" Nutrients 8, no. 7: 405. https://doi.org/10.3390/nu8070405
APA StyleYoon, M.-S. (2016). The Emerging Role of Branched-Chain Amino Acids in Insulin Resistance and Metabolism. Nutrients, 8(7), 405. https://doi.org/10.3390/nu8070405