Retinoic Acid as a Modulator of T Cell Immunity


 , ,
, ,
Abstract
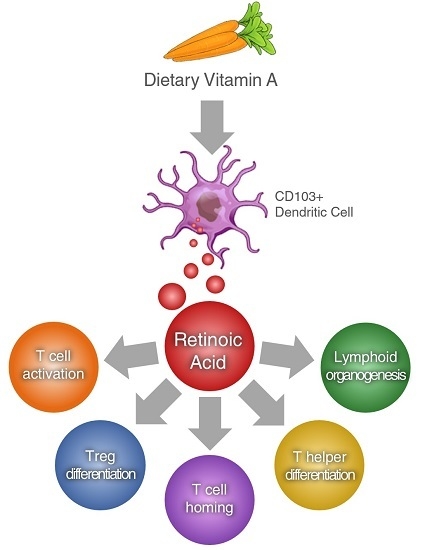
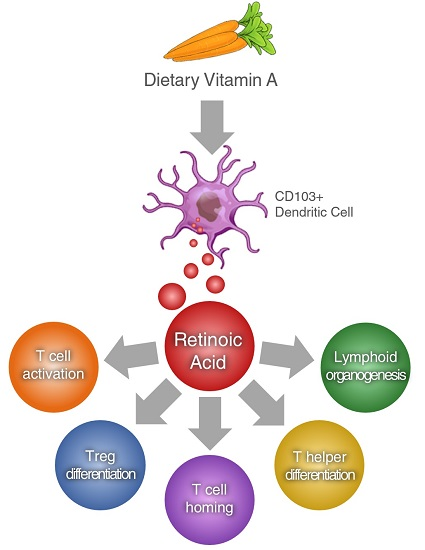
:
1. Introduction
2. Vitamin A Metabolism and Retinoic Acid Signaling
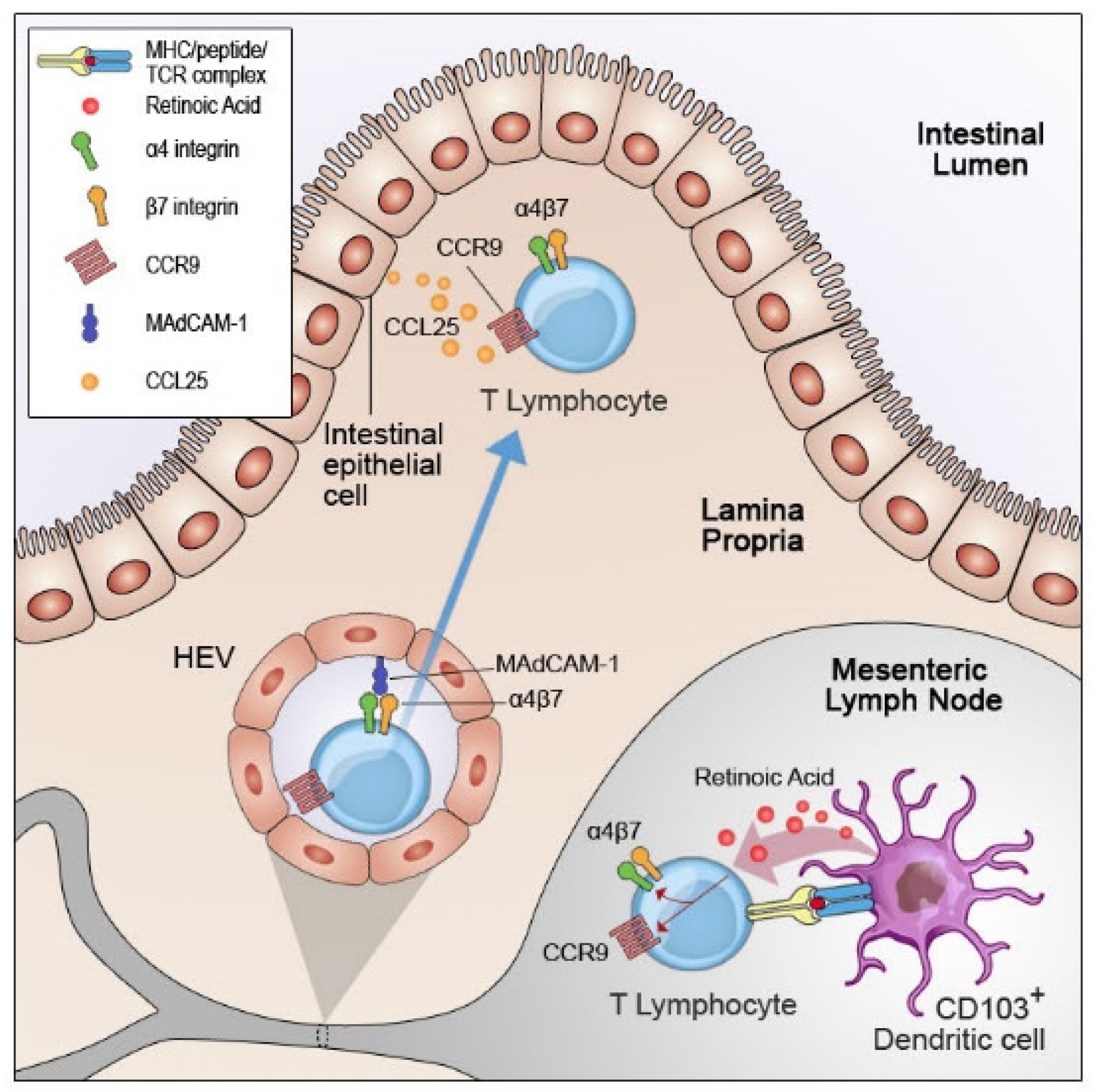
3. Retinoic Acid and T Cell Homing
4. Role of Retinoic Acid in Regulatory T Cell Differentiation
5. Retinoic Acid in T Helper Cell Differentiation and Activation
5.1. Th17/Th1 Cell Differentiation
5.2. Th2 Cell Differentiation
5.3. T Cell Activation
6. Role of Retinoic Acid in Thymus and Lymphoid Organogenesis
7. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix

{kind=link}
{kind=link}
| T Cell Subset | RA Effect | Experimental Setting | Model Organism [Refs] |
|---|---|---|---|
| Treg | Induces Treg differentiation | Cultures in the presence of RA (0.05 nM–10 μM), RAR agonist (AM580) or antagonists (Ro41-5253, LE540, LE135) | Mice [35,45,46,47,48,49,50,58,61,72] Human [47,52,54] |
| Treg | Enhances Treg stability and function | Adoptive transfer of Treg generated in the presence of RA in mice models of inflammation (immunization, transplant, intestinal inflammation, GvHD) | Mice [49,50,58] Human [52,54] |
| Th17 | Induces Th17 differentiation | Cultures in the presence of physiological doses of RA (1 nM) or RAR antagonist (LE540). Cultures of RARα−/− CD4+ T cells | Mice [67,74,76] |
| Th17 | Reduces Th17 differentiation | Cultures in the presence of pharmacological doses of RA (≥10 nM), RAR agonist (TTNBP) or antagonist (LE135) | Mice [46,47,64,61,69,72,74,75,76] Human [73] |
| Th1 | Induces Th1 differentiation | Cultures of RAR deficient CD4+ T cells | Mice [67,70] |
| Th1 | Enhances Th1 stability | Ablation of RA signaling in Th1 committed cells | Mice [70] |
| Th1 | Reduces Th1 differentiation | Cultures in the presence of pharmacological doses of RA (≥10 nM), RXR agonists (HX600, TZ335, PA024), RAR agonists (Am80, Tp80), RXR antagonist (PA452) or RAR antagonists (LE135, LE540) | Mice [72,74,85,87] Human [73] |
| Th2 | Induces Th2 differentiation | Cultures in the presence of 9-cis RA (10 nM–1 μM), RXR agonists (AGN194204, HX600, TZ335, PA024), RAR agonists (Am80, Tp80), RXR antagonist (PA452) or RAR antagonists (LE135, LE540) | Mice [85,87,88] |
| Th2 | Reduces Th2 differentiation | Cultures in the presence of pharmacological doses of RA (≥100 nM) | Mice [72] |
| T Cell Subset | RA Effect | Experimental Setting [Refs] |
|---|---|---|
| Treg | Induces Treg differentiation | In vivo administration RAR antagonist (LE540) in in L. monocytogenes infected mice [64] VAD mice after induction of experimental autoimmune uveitis [65] In vivo administration of RA in mice model of diabetes [66] VAD mice after oral tolerance induction [67] |
| Treg | No effect on frequencies or numbers of endogenous Treg | Frequencies and/or absolute numbers of endogenous Treg in VAD mice [67,69] and RARα−/− mice [67] |
| Th17 | Reduces Th17 differentiation | Frequencies of Th17 cells in small intestine lamina propria after oral administration of RA in L. monocytogenes infected mice [64] Endogenous response to L. monocytogenes in CD4 conditional RARα-deficient mice [70] CD4 conditional RARα-deficient mice in spleen at steady state [70] In vivo differentiation of RARα deficient CD4+ T cells in mice model of intestinal inflammation [70] Frequencies of Th17 cells in spleen after intraperitoneal administration of RA in experimental autoimmune encephalomyelitis mice [61] |
| Th17 | Induces Th17 differentiation | Frequencies of Th17 cells in small intestine [69,75] and Peyer’s patches in VAD mice in steady state [75] Frequencies of Th17 cells in small intestine lamina propria after immunization (OVA + LTR129G) in VAD mice [67] Frequencies of Th17 cells in large intestine lamina propria after infection with C. rodentium in VAD or RAR antagonist BMS493-treated mice [77] Frequencies of Th17 cells in VAD mice after infection with T. gondii [67] In vivo differentiation of RARα deficient CD4+ T cells in mice model of transplantation [37] |
| Th1 | Induces Th1 differentiation | CD4 conditional RARα-deficient mice in spleen at steady state [70] Endogenous response to L. monocytogenes in CD4 conditional RARα-deficient mice [70] In vivo differentiation of RARα deficient CD4+ T cells in mice model of intestinal inflammation [70] In vivo differentiation of RARα deficient CD4+ T cells in mice model of transplantation [37] In vivo differentiation of CD4+ T cells after immunization with cognate antigen in VAD mice [86] |
| Th1 | Impairs Th1 responses | Influenza-specific IgG and IFN-γ production after intranasal inoculation with influenza virus in VAD mice [82] or mice fed with a vitamin A-rich diet [84] Antigen-specific IFN-γ production in mesenteric lymph node and spleen cells after infection with T. spiralis in VAD mice [79,81] |
| Th2 | Induces Th2 differentiation | Influenza-specific IgA and IL-10 production after intranasal inoculation with influenza virus in VAD mice [82] or mice fed with a vitamin A-rich diet [84] Antigen-specific IL-4, IL-5 production in mesenteric lymph node cells after infection with T. spiralis in VAD mice [79,81] In vivo differentiation of CD4+ T cells after immunization with cognate antigen in VAD mice [86] |
References
- Sommer, A. Vitamin A deficiency and clinical disease: An historical overview. J. Nutr. 2008, 138, 1835–1839. [Google Scholar] [PubMed]
- Green, H.N.; Mellanby, E. Vitamin A as an anti-infective agent. Br. Med. J. 1928, 2, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Glasziou, P.P.; Mackerras, D.E. Vitamin A supplementation in infectious diseases: A meta-analysis. BMJ 1993, 306, 366–370. [Google Scholar] [CrossRef] [PubMed]
- WHO IRIS. Available online: http://apps.who.int/iris/handle/10665/61514 (accessed on 2 June 2016).
- WHO. Available online: http://www.who.int/vmnis/database/vitamina/x/en/ (accessed on 2 June 2016).
- Schaible, U.E.; Kaufmann, S.H. Malnutrition and infection: Complex mechanisms and global impacts. PLoS Med. 2007, 4. [Google Scholar] [CrossRef] [PubMed]
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.A.; Grainger, J.R.; Spencer, S.P.; Belkaid, Y. The role of retinoic acid in tolerance and immunity. Immunity 2011, 35, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.E.; Baylor, D.A. Activation, deactivation, and adaptation in vertebrate photoreceptor cells. Annu. Rev. Neurosci. 2001, 24, 779–805. [Google Scholar] [CrossRef] [PubMed]
- Newcomer, M.E.; Ong, D.E. Plasma retinol binding protein: Structure and function of the prototypic lipocalin. Biochim. Biophys. Acta. 2000, 1482, 57–64. [Google Scholar] [CrossRef]
- Napoli, J.L. Retinoic acid: Its biosynthesis and metabolism. Prog. Nucleic Acid Res. Mol. Biol. 1999, 63, 139–188. [Google Scholar] [PubMed]
- Duester, G. Families of retinoid dehydrogenases regulating vitamin A function: Production of visual pigment and retinoic acid. Eur. J. Biochem. 2000, 267, 4315–4324. [Google Scholar] [CrossRef] [PubMed]
- Sandell, L.L.; Sanderson, B.W.; Moiseyev, G.; Johnson, T.; Mushegian, A.; Young, K.; Rey, J.P.; Ma, J.X.; Staehling-Hampton, K.; Trainor, P.A. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 2007, 21, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Brown, C.; Ortiz, C.; Noelle, R.J. Leukocyte homing, fate, and function are controlled by retinoic acid. Physiol. Rev. 2015, 95, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Dolle, P.; Ruberte, E.; Kastner, P.; Petkovich, M.; Stoner, C.M.; Gudas, L.J.; Chambon, P. Differential expression of genes encoding alpha, beta and gamma retinoic acid receptors and crabp in the developing limbs of the mouse. Nature 1989, 342, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Balmer, J.E.; Blomhoff, R. Gene expression regulation by retinoic acid. J. Lipid Res. 2002, 43, 1773–1808. [Google Scholar] [CrossRef] [PubMed]
- Durand, B.; Saunders, M.; Leroy, P.; Leid, M.; Chambon, P. All-trans and 9-cis retinoic acid induction of CRABPII transcription is mediated by RAR-RXR heterodimers bound to DR1 and DR2 repeated motifs. Cell 1992, 71, 73–85. [Google Scholar] [CrossRef]
- Heyman, R.A.; Mangelsdorf, D.J.; Dyck, J.A.; Stein, R.B.; Eichele, G.; Evans, R.M.; Thaller, C. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 1992, 68, 397–406. [Google Scholar] [CrossRef]
- Kurokawa, R.; DiRenzo, J.; Boehm, M.; Sugarman, J.; Gloss, B.; Rosenfeld, M.G.; Heyman, R.A.; Glass, C.K. Regulation of retinoid signalling by receptor polarity and allosteric control of ligand binding. Nature 1994, 371, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, R.; Soderstrom, M.; Horlein, A.; Halachmi, S.; Brown, M.; Rosenfeld, M.G.; Glass, C.K. Polarity-specific activities of retinoic acid receptors determined by a co-repressor. Nature 1995, 377, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Westin, S.; Kurokawa, R.; Nolte, R.T.; Wisely, G.B.; McInerney, E.M.; Rose, D.W.; Milburn, M.V.; Rosenfeld, M.G.; Glass, C.K. Interactions controlling the assembly of nuclear-receptor heterodimers and co-activators. Nature 1998, 395, 199–202. [Google Scholar] [PubMed]
- Raverdeau, M.; Mills, K.H. Modulation of T cell and innate immune responses by retinoic acid. J. Immunol. 2014, 192, 2953–2958. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.R.; von Andrian, U.H. T-cell homing specificity and plasticity: New concepts and future challenges. Trends Immunol. 2006, 27, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Bono, M.R.; Elgueta, R.; Sauma, D.; Pino, K.; Osorio, F.; Michea, P.; Fierro, A.; Rosemblatt, M. The essential role of chemokines in the selective regulation of lymphocyte homing. Cytokine Growth Factor Rev. 2007, 18, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Hosoe, N.; Miura, S.; Watanabe, C.; Tsuzuki, Y.; Hokari, R.; Oyama, T.; Fujiyama, Y.; Nagata, H.; Ishii, H. Demonstration of functional role of TECK/CCL25 in T lymphocyte-endothelium interaction in inflamed and uninflamed intestinal mucosa. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G458–G466. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.R.; Bono, M.R.; Manjunath, N.; Weninger, W.; Cavanagh, L.L.; Rosemblatt, M.; von Andrian, U.H. Selective imprinting of gut-homing T cells by peyer's patch dendritic cells. Nature 2003, 424, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Hirakiyama, A.; Eshima, Y.; Kagechika, H.; Kato, C.; Song, S.Y. Retinoic acid imprints gut-homing specificity on T cells. Immunity 2004, 21, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Svensson, M.; Johansson-Lindbom, B.; Zapata, F.; Jaensson, E.; Austenaa, L.M.; Blomhoff, R.; Agace, W.W. Retinoic acid receptor signaling levels and antigen dose regulate gut homing receptor expression on CD8+ T cells. Mucosal. Immunol. 2008, 1, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.R.; Iwata, M.; Eksteen, B.; Song, S.Y.; Junt, T.; Senman, B.; Otipoby, K.L.; Yokota, A.; Takeuchi, H.; Ricciardi-Castagnoli, P.; et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 2006, 314, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.R.; von Andrian, U.H. Role of retinoic acid in the imprinting of gut-homing IgA-secreting cells. Semin. Immunol. 2009, 21, 28–35. [Google Scholar] [CrossRef] [PubMed]
- DeNucci, C.C.; Pagan, A.J.; Mitchell, J.S.; Shimizu, Y. Control of α4β7 integrin expression and CD4 T cell homing by the β1 integrin subunit. J. Immunol. 2010, 184, 2458–2467. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.G.; Park, J.; Cho, J.Y.; Ulrich, B.; Kim, C.H. Complementary roles of retinoic acid and TGF-β1 in coordinated expression of mucosal integrins by T cells. Mucosal. Immunol. 2011, 4, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, Y.; Yokota, A.; Takeuchi, H.; Maeda, N.; Iwata, M. Retinoic acid-induced CCR9 expression requires transient TCR stimulation and cooperativity between NFATc2 and the retinoic acid receptor/retinoid X receptor complex. J. Immunol. 2011, 186, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Bakdash, G.; Vogelpoel, L.T.; van Capel, T.M.; Kapsenberg, M.L.; de Jong, E.C. Retinoic acid primes human dendritic cells to induce gut-homing, IL-10-producing regulatory T cells. Mucosal. Immunol. 2015, 8, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Coombes, J.L.; Siddiqui, K.R.; Arancibia-Carcamo, C.V.; Hall, J.; Sun, C.M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β-and retinoic acid-dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, S.I.; Ahrendt, M.; Bode, U.; Wahl, B.; Kremmer, E.; Forster, R.; Pabst, O. Stromal mesenteric lymph node cells are essential for the generation of gut-homing T cells in vivo. J. Exp. Med. 2008, 205, 2483–2490. [Google Scholar] [CrossRef] [PubMed]
- Pino-Lagos, K.; Guo, Y.; Brown, C.; Alexander, M.P.; Elgueta, R.; Bennett, K.A.; de Vries, V.; Nowak, E.; Blomhoff, R.; Sockanathan, S.; et al. A retinoic acid-dependent checkpoint in the development of CD4+ T cell-mediated immunity. J. Exp. Med. 2011, 208, 1767–1775. [Google Scholar] [CrossRef] [PubMed]
- Pabst, O.; Mowat, A.M. Oral tolerance to food protein. Mucosal. Immunol. 2012, 5, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Sakaguchi, S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat. Immunol. 2010, 11, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Rudensky, A. Control of regulatory T cell lineage commitment and maintenance. Immunity 2009, 30, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Karim, M.; Kingsley, C.I.; Bushell, A.R.; Sawitzki, B.S.; Wood, K.J. Alloantigen-induced CD25+ CD4+ regulatory T cells can develop in vivo from CD25− CD4+ precursors in a thymus-independent process. J. Immunol. 2004, 172, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Vukmanovic-Stejic, M.; Zhang, Y.; Cook, J.E.; Fletcher, J.M.; McQuaid, A.; Masters, J.E.; Rustin, M.H.; Taams, L.S.; Beverley, P.C.; Macallan, D.C.; et al. Human CD4+ CD25hi Foxp3+ regulatory T cells are derived by rapid turnover of memory populations in vivo. J. Clin. Investig. 2006, 116, 2423–2433. [Google Scholar] [CrossRef] [PubMed]
- Yamagiwa, S.; Gray, J.D.; Hashimoto, S.; Horwitz, D.A. A role for TGF-β in the generation and expansion of CD4+ CD25+ regulatory T cells from human peripheral blood. J. Immunol. 2001, 166, 7282–7289. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.M.; Hall, J.A.; Blank, R.B.; Bouladoux, N.; Oukka, M.; Mora, J.R.; Belkaid, Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 2007, 204, 1775–1785. [Google Scholar] [CrossRef] [PubMed]
- Schambach, F.; Schupp, M.; Lazar, M.A.; Reiner, S.L. Activation of retinoic acid receptor-α favours regulatory T cell induction at the expense of IL-17-secreting T helper cell differentiation. Eur. J. Immunol. 2007, 37, 2396–2399. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.G.; Lim, H.W.; Andrisani, O.M.; Broxmeyer, H.E.; Kim, C.H. Vitamin A metabolites induce gut-homing Foxp3+ regulatory T cells. J. Immunol. 2007, 179, 3724–3733. [Google Scholar] [CrossRef] [PubMed]
- Mucida, D.; Pino-Lagos, K.; Kim, G.; Nowak, E.; Benson, M.J.; Kronenberg, M.; Noelle, R.J.; Cheroutre, H. Retinoic acid can directly promote TGF-β-mediated Foxp3+ Treg cell conversion of naive T cells. Immunity 2009, 30, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.; Tejon, G.; Fuentes, C.; Hidalgo, Y.; Bono, M.R.; Maldonado, P.; Fernandez, R.; Wood, K.J.; Fierro, J.A.; Rosemblatt, M.; et al. Alloreactive regulatory T cells generated with retinoic acid prevent skin allograft rejection. Eur. J. Immunol. 2015, 45, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.J.; Pino-Lagos, K.; Rosemblatt, M.; Noelle, R.J. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J. Exp. Med. 2007, 204, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Golovina, T.N.; Mikheeva, T.; Brusko, T.M.; Blazar, B.R.; Bluestone, J.A.; Riley, J.L. Retinoic acid and rapamycin differentially affect and synergistically promote the ex vivo expansion of natural human T regulatory cells. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhou, X.; Wang, J.; Zheng, S.G.; Horwitz, D.A. Characterization of protective human CD4+ CD25+ Foxp3+ regulatory T cells generated with IL-2, TGF-β and retinoic acid. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Scotta, C.; Esposito, M.; Fazekasova, H.; Fanelli, G.; Edozie, F.C.; Ali, N.; Xiao, F.; Peakman, M.; Afzali, B.; Sagoo, P.; et al. Differential effects of rapamycin and retinoic acid on expansion, stability and suppressive qualities of human CD4+ CD25+ Foxp3+ T regulatory cell subpopulations. Haematologica 2013, 98, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huizinga, T.W.; Toes, R.E. De novo generation and enhanced suppression of human CD4+ CD25+ regulatory T cells by retinoic acid. J. Immunol. 2009, 183, 4119–4126. [Google Scholar] [CrossRef] [PubMed]
- Nolting, J.; Daniel, C.; Reuter, S.; Stuelten, C.; Li, P.; Sucov, H.; Kim, B.G.; Letterio, J.J.; Kretschmer, K.; Kim, H.J.; et al. Retinoic acid can enhance conversion of naive into regulatory T cells independently of secreted cytokines. J. Exp. Med. 2009, 206, 2131–2139. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Hall, J.A.; Sun, C.M.; Cai, Q.; Ghyselinck, N.; Chambon, P.; Belkaid, Y.; Mathis, D.; Benoist, C. Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi cells. Immunity 2008, 29, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Yokota-Nakatsuma, A.; Ohoka, Y.; Kagechika, H.; Kato, C.; Song, S.Y.; Iwata, M. Retinoid X receptor agonists modulate Foxp3+ regulatory T cell and Th17 cell differentiation with differential dependence on retinoic acid receptor activation. J. Immunol. 2013, 191, 3725–3733. [Google Scholar] [CrossRef] [PubMed]
- Tejon, G.; Manriquez, V.; de Calisto, J.; Flores-Santibanez, F.; Hidalgo, Y.; Crisostomo, N.; Fernandez, D.; Sauma, D.; Mora, J.R.; Bono, M.R.; et al. Vitamin A impairs the reprogramming of Tregs into IL-17-producing cells during intestinal inflammation. Biomed. Res. Int. 2015, 2015, 137893. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Kong, N.; Wang, J.; Fan, H.; Zou, H.; Horwitz, D.; Brand, D.; Liu, Z.; Zheng, S.G. Cutting edge: All-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. J. Immunol. 2010, 185, 2675–2679. [Google Scholar] [CrossRef] [PubMed]
- Mucida, D.; Park, Y.; Cheroutre, H. From the diet to the nucleus: Vitamin a and TGF-β join efforts at the mucosal interface of the intestine. Semin. Immunol. 2009, 21, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Jin, H.; Korn, T.; Liu, S.M.; Oukka, M.; Lim, B.; Kuchroo, V.K. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-β-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J. Immunol. 2008, 181, 2277–2284. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.; Sauma, D.; Morales, J.; Bono, M.R.; Rosemblatt, M.; Fierro, J.A. Transforming growth factor-β and all-trans retinoic acid generate ex vivo transgenic regulatory T cells with intestinal homing receptors. Transplant. Proc. 2009, 41, 2670–2672. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, P.; Maldonado, P.; Hidalgo, Y.; Sauma, D.; Rosemblatt, M.; Bono, M.R. Alloreactive regulatory T cells allow the generation of mixed chimerism and transplant tolerance. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Mucida, D.; Park, Y.; Kim, G.; Turovskaya, O.; Scott, I.; Kronenberg, M.; Cheroutre, H. Reciprocal Th17 and regulatory T cell differentiation mediated by retinoic acid. Science 2007, 317, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Kawazoe, Y.; Sugita, S.; Keino, H.; Yamada, Y.; Imai, A.; Horie, S.; Mochizuki, M. Retinoic acid from retinal pigment epithelium induces T regulatory cells. Exp. Eye Res. 2012, 94, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Van, Y.H.; Lee, W.H.; Ortiz, S.; Lee, M.H.; Qin, H.J.; Liu, C.P. All-trans retinoic acid inhibits type 1 diabetes by T regulatory (Treg)-dependent suppression of interferon-γ-producing T-cells without affecting Th17 cells. Diabetes 2009, 58, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.A.; Cannons, J.L.; Grainger, J.R.; Dos Santos, L.M.; Hand, T.W.; Naik, S.; Wohlfert, E.A.; Chou, D.B.; Oldenhove, G.; Robinson, M.; et al. Essential role for retinoic acid in the promotion of CD4+ T cell effector responses via retinoic acid receptor alpha. Immunity 2011, 34, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Hadis, U.; Wahl, B.; Schulz, O.; Hardtke-Wolenski, M.; Schippers, A.; Wagner, N.; Muller, W.; Sparwasser, T.; Forster, R.; Pabst, O. Intestinal tolerance requires gut homing and expansion of Foxp3+ regulatory T cells in the lamina propria. Immunity 2011, 34, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.R.; Chang, S.Y.; Chang, J.H.; Kim, J.O.; Yang, J.Y.; Kim, C.H.; Kweon, M.N. Downregulation of Th17 cells in the small intestine by disruption of gut flora in the absence of retinoic acid. J. Immunol 2010, 184, 6799–6806. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.C.; Esterhazy, D.; Sarde, A.; London, M.; Pullabhatla, V.; Osma-Garcia, I.; Al-Bader, R.; Ortiz, C.; Elgueta, R.; Arno, M.; et al. Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 2015, 42, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [PubMed]
- Elias, K.M.; Laurence, A.; Davidson, T.S.; Stephens, G.; Kanno, Y.; Shevach, E.M.; O'Shea, J.J. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 2008, 111, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Awasthi, A.; Ahuja, V. Retinoic acid-primed human dendritic cells inhibit Th9 cells and induce Th1/Th17 cell differentiation. J. Leukoc. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Fujimoto, K.; Jang, M.H.; Yang, B.G.; Jung, Y.J.; Nishiyama, M.; Sato, S.; Tsujimura, T.; Yamamoto, M.; Yokota, Y.; et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat. Immunol. 2008, 9, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kang, S.G.; HogenEsch, H.; Love, P.E.; Kim, C.H. Retinoic acid determines the precise tissue tropism of inflammatory Th17 cells in the intestine. J. Immunol. 2010, 184, 5519–5526. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Kanno, T.; Nakayamada, S.; Hirahara, K.; Sciume, G.; Muljo, S.A.; Kuchen, S.; Casellas, R.; Wei, L.; Kanno, Y.; et al. TGF-[beta] and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat. Immunol. 2012, 13, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.P.; Wilhelm, C.; Yang, Q.; Hall, J.A.; Bouladoux, N.; Boyd, A.; Nutman, T.B.; Urban, J.F., Jr.; Wang, J.; Ramalingam, T.R.; et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science 2014, 343, 432–437. [Google Scholar] [CrossRef] [PubMed]
- DePaolo, R.W.; Abadie, V.; Tang, F.; Fehlner-Peach, H.; Hall, J.A.; Wang, W.; Marietta, E.V.; Kasarda, D.D.; Waldmann, T.A.; Murray, J.A.; et al. Co-adjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature 2011, 471, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Cantorna, M.T.; Nashold, F.E.; Hayes, C.E. In vitamin A deficiency multiple mechanisms establish a regulatory T helper cell imbalance with excess Th1 and insufficient Th2 function. J. Immunol. 1994, 152, 1515–1522. [Google Scholar] [PubMed]
- Carman, J.A.; Hayes, C.E. Abnormal regulation of IFN-gamma secretion in vitamin A deficiency. J. Immunol. 1991, 147, 1247–1252. [Google Scholar] [PubMed]
- Carman, J.A.; Pond, L.; Nashold, F.; Wassom, D.L.; Hayes, C.E. Immunity to trichinella spiralis infection in vitamin A-deficient mice. J. Exp. Med. 1992, 175, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Stephensen, C.B.; Moldoveanu, Z.; Gangopadhyay, N.N. Vitamin A deficiency diminishes the salivary immunoglobulin A response and enhances the serum immunoglobulin G response to influenza A virus infection in BALB/c mice. J. Nutr. 1996, 126, 94–102. [Google Scholar] [PubMed]
- Stephensen, C.B.; Franchi, L.M.; Hernandez, H.; Campos, M.; Gilman, R.H.; Alvarez, J.O. Adverse effects of high-dose vitamin A supplements in children hospitalized with pneumonia. Pediatrics 1998, 101. [Google Scholar] [CrossRef]
- Cui, D.; Moldoveanu, Z.; Stephensen, C.B. High-level dietary vitamin A enhances T-helper type 2 cytokine production and secretory immunoglobulin A response to influenza A virus infection in BALB/c mice. J. Nutr. 2000, 130, 1132–1139. [Google Scholar] [PubMed]
- Racke, M.K.; Burnett, D.; Pak, S.H.; Albert, P.S.; Cannella, B.; Raine, C.S.; McFarlin, D.E.; Scott, D.E. Retinoid treatment of experimental allergic encephalomyelitis. IL-4 production correlates with improved disease course. J. Immunol. 1995, 154, 450–458. [Google Scholar] [PubMed]
- Stephensen, C.B.; Jiang, X.; Freytag, T. Vitamin A deficiency increases the in vivo development of IL-10-positive Th2 cells and decreases development of Th1 cells in mice. J. Nutr. 2004, 134, 2660–2666. [Google Scholar] [PubMed]
- Stephensen, C.B.; Rasooly, R.; Jiang, X.; Ceddia, M.A.; Weaver, C.T.; Chandraratna, R.A.; Bucy, R.P. Vitamin A enhances in vitro Th2 development via retinoid X receptor pathway. J. Immunol. 2002, 168, 4495–4503. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Eshima, Y.; Kagechika, H. Retinoic acids exert direct effects on T cells to suppress Th1 development and enhance Th2 development via retinoic acid receptors. Int. Immunol. 2003, 15, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Crozat, K.; Henri, S.; Tamoutounour, S.; Grenot, P.; Devilard, E.; de Bovis, B.; Alexopoulou, L.; Dalod, M.; Malissen, B. Skin-draining lymph nodes contain dermis-derived CD103− dendritic cells that constitutively produce retinoic acid and induce Foxp3+ regulatory T cells. Blood 2010, 115, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.; Greuter, M.; van der Marel, A.P.; Roozendaal, R.; Martin, S.F.; Edele, F.; Huehn, J.; Forster, R.; O'Toole, T.; Jansen, W.; et al. Lymph node stromal cells support dendritic cell-induced gut-homing of T cells. J. Immunol. 2009, 183, 6395–6402. [Google Scholar] [CrossRef] [PubMed]
- Blackman, M.; Kappler, J.; Marrack, P. The role of the T cell receptor in positive and negative selection of developing T cells. Science 1990, 248, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Wolbach, S.B.; Howe, P.R. Tissue changes following deprivation of fat-soluble A vitamin. J. Exp. Med. 1925, 42, 753–777. [Google Scholar] [CrossRef] [PubMed]
- Kiss, I.; Ruhl, R.; Szegezdi, E.; Fritzsche, B.; Toth, B.; Pongracz, J.; Perlmann, T.; Fesus, L.; Szondy, Z. Retinoid receptor-activating ligands are produced within the mouse thymus during postnatal development. Eur. J. Immunol. 2008, 38, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Mukai, M.; Nakai, Y.; Iseki, R. Retinoic acids inhibit activation-induced apoptosis in T cell hybridomas and thymocytes. J. Immunol. 1992, 149, 3302–3308. [Google Scholar] [PubMed]
- Yagi, J.; Uchida, T.; Kuroda, K.; Uchiyama, T. Influence of retinoic acid on the differentiation pathway of T cells in the thymus. Cell Immunol. 1997, 181, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Szondy, Z.; Reichert, U.; Bernardon, J.M.; Michel, S.; Toth, R.; Karaszi, E.; Fesus, L. Inhibition of activation-induced apoptosis of thymocytes by all-trans- and 9-cis-retinoic acid is mediated via retinoic acid receptor α. Biochem. J. 1998, 331, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Szondy, Z.; Reichert, U.; Bernardon, J.M.; Michel, S.; Toth, R.; Ancian, P.; Ajzner, E.; Fesus, L. Induction of apoptosis by retinoids and retinoic acid receptor γ-selective compounds in mouse thymocytes through a novel apoptosis pathway. Mol. Pharmacol. 1997, 51, 972–982. [Google Scholar] [PubMed]
- Mebius, R.E.; Rennert, P.; Weissman, I.L. Developing lymph nodes collect CD4+ CD3− LTβ+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 1997, 7, 493–504. [Google Scholar] [CrossRef]
- Walker, J.A.; Barlow, J.L.; McKenzie, A.N. Innate lymphoid cells—how did we miss them? Nat. Rev. Immunol. 2013, 13, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Honda, K.; Shinkura, R.; Adachi, S.; Nishikawa, S.; Maki, K.; Ikuta, K.; Nishikawa, S.I. IL-7 receptor α+ CD3− cells in the embryonic intestine induces the organizing center of Peyer's patches. Int. Immunol. 1999, 11, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Eberl, G.; Marmon, S.; Sunshine, M.J.; Rennert, P.D.; Choi, Y.; Littman, D.R. An essential function for the nuclear receptor RORγt in the generation of fetal lymphoid tissue inducer cells. Nat. Immunol. 2004, 5, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Van de Pavert, S.A.; Ferreira, M.; Domingues, R.G.; Ribeiro, H.; Molenaar, R.; Moreira-Santos, L.; Almeida, F.F.; Ibiza, S.; Barbosa, I.; Goverse, G.; et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature 2014, 508, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Zheng, W.; Zhao, G.; Zhang, H.; Wang, X.; Guo, Y.; Qin, C.; Shi, Y. Peripheral lymphoid volume expansion and maintenance are controlled by gut microbiota via RALDH+ dendritic cells. Immunity 2016, 44, 330–342. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bono, M.R.; Tejon, G.; Flores-Santibañez, F.; Fernandez, D.; Rosemblatt, M.; Sauma, D. Retinoic Acid as a Modulator of T Cell Immunity. Nutrients 2016, 8, 349. https://doi.org/10.3390/nu8060349
Bono MR, Tejon G, Flores-Santibañez F, Fernandez D, Rosemblatt M, Sauma D. Retinoic Acid as a Modulator of T Cell Immunity. Nutrients. 2016; 8(6):349. https://doi.org/10.3390/nu8060349
Chicago/Turabian StyleBono, Maria Rosa, Gabriela Tejon, Felipe Flores-Santibañez, Dominique Fernandez, Mario Rosemblatt, and Daniela Sauma. 2016. "Retinoic Acid as a Modulator of T Cell Immunity" Nutrients 8, no. 6: 349. https://doi.org/10.3390/nu8060349
APA StyleBono, M. R., Tejon, G., Flores-Santibañez, F., Fernandez, D., Rosemblatt, M., & Sauma, D. (2016). Retinoic Acid as a Modulator of T Cell Immunity. Nutrients, 8(6), 349. https://doi.org/10.3390/nu8060349