Dietary Advanced Glycation End Products and Risk Factors for Chronic Disease: A Systematic Review of Randomised Controlled Trials

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Eligibility Criteria
2.2. Information Sources
2.3. Selection Process
2.4. Data Management and Collection
2.5. Data Items
2.6. Outcomes and Prioritisation
2.7. Risk of Bias and Quality Assessment
2.8. Data Synthesis
3. Results
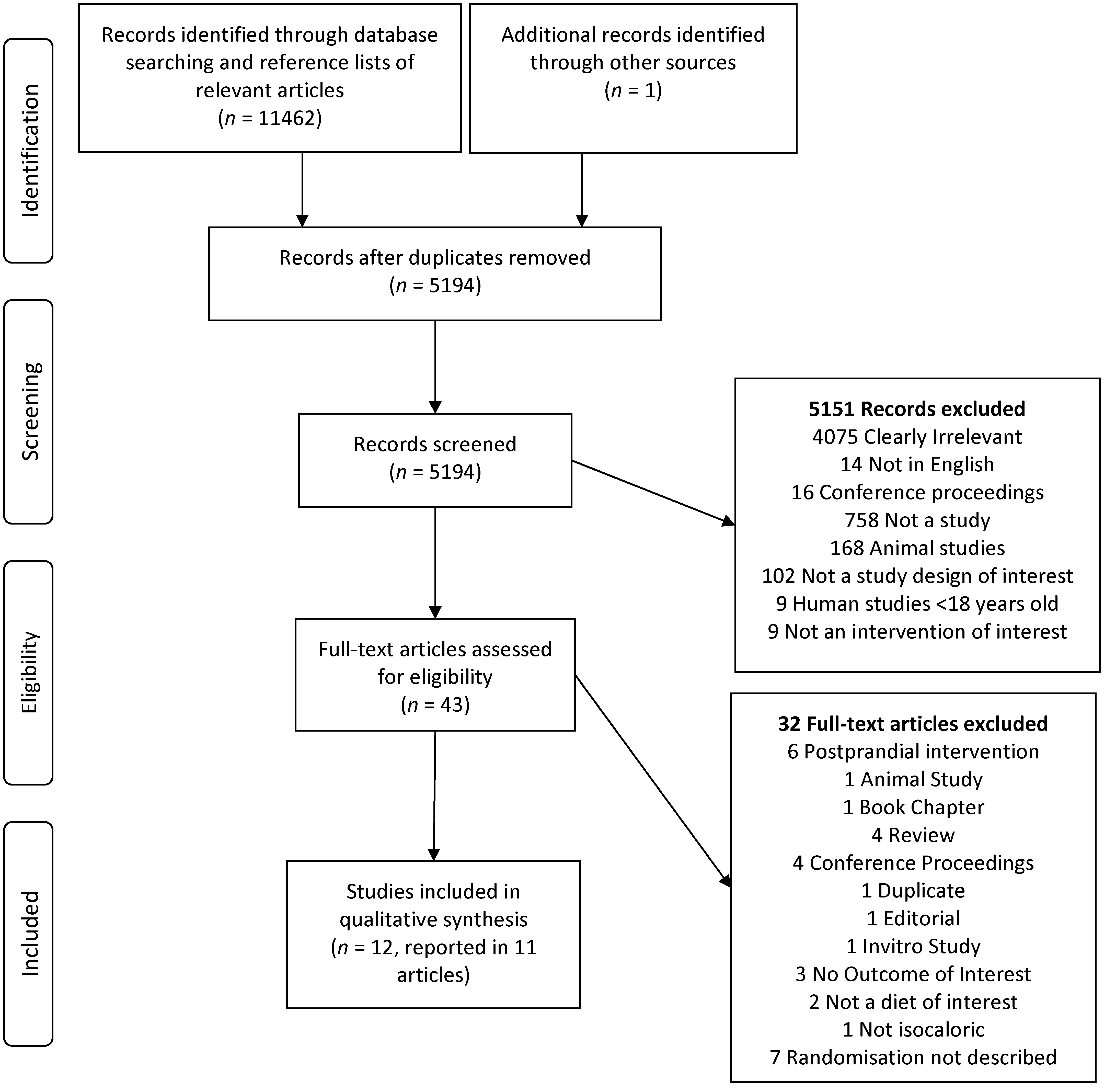
3.1. Studies Identified
3.2. Study Characteristics
3.3. Studies in Healthy Populations
3.3.1. Biomarkers of Inflammation and Oxidative Stress
3.3.2. Biomarkers of Chronic Disease Risk
3.3.3. Circulating and Excreted CML
3.4. Studies in Patients with Diabetes
3.4.1. Biomarkers of Inflammation and Oxidative Stress
3.4.2. Biomarkers of Chronic Disease Risk
3.4.3. Circulating and Excreted CML
3.5. Studies in Patients with CKD
3.5.1. Biomarkers of Inflammation and Oxidative Stress
3.5.2. Biomarkers of Chronic Disease Risk
3.5.3. Circulating and Excreted CML
3.6. Quality Assessment
4. Discussion
4.1. The Effect of a High AGE Diet
4.2. Limitations of Included Studies
4.3. Limitations of this Review
4.4. Comparison with Other Reviews
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Beaglehole, R.; Bonita, R.; Horton, R.; Adams, C.; Alleyne, G.; Asaria, P.; Baugh, V.; Bekedam, H.; Billo, N.; Casswell, S.; et al. Priority actions for the non-communicable disease crisis. Lancet. 2011, 377, 1438–1447. [Google Scholar] [CrossRef]
- Kellow, N.J.; Savige, G.S. Dietary advanced glycation end-product restriction for the attenuation of insulin resistance, oxidative stress and endothelial dysfunction: A systematic review. Eur. J. Clin. Nutr. 2013, 67, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Van Puyvelde, K.; Mets, T.; Njemini, R.; Beyer, I.; Bautmans, I. Effect of advanced glycation end product intake on inflammation and aging: A systematic review. Nutr. Rev. 2014, 72, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.X.; Hou, F.F.; Liang, M.; Wang, G.B.; Zhang, X.; Li, H.Y.; Xie, D.; Tian, J.W.; Liu, Z.Q. Restricted intake of dietary advanced glycation end products retards renal progression in the remnant kidney model. Kidney Int. 2007, 71, 901–911. [Google Scholar] [PubMed]
- Šebeková, K.; Faist, V.; Hofmann, T.; Schinzel, R.; Heidland, A. Effects of a diet rich in advanced glycation end products in the rat remnant kidney model. Am. J. Kidney Dis. 2003, 41, S48–S51. [Google Scholar] [PubMed]
- ŠEbekovÁ, K.; Hofmann, T.; Boor, P.; UlicnÁ, O.G.; Erbersdobler, H.F.; Baynes, J.W.; Thorpe, S.R.; Heidland, A.; Somoza, V. Renal effects of oral Maillard reaction product load in the form of bread crusts in healthy and subtotally nephrectomized rats. Ann. N. Y. Acad. Sci. 2005, 1043, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; He, C.; Cai, W.; Hattori, M.; Steffes, M.; Vlassara, H. Prevention of diabetic nephropathy in mice by a diet low in glycoxidation products. Diabetes Metab. Res. Rev. 2002, 18, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Somoza, V.; Lindenmeier, M.; Hofmann, T.; Frank, O.; Erbersdobler, H.F.; Baynes, J.W.; Thorpe, S.R.; Heidland, A.; Zill, H.; Bek, S.; et al. Dietary bread crust advanced glycation end products bind to the receptor for AGEs in HEK-293 kidney cells but are rapidly excreted after oral administration to healthy and subtotally nephrectomized rats. Ann. N. Y. Acad. Sci. 2005, 1043, 492–500. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. Healthy Diet Fact Sheet; World Health Organisation: Geneva, Switzerland, 2015. [Google Scholar]
- Dresden University of Technology. AGE Database. Dresden University of Technology: Dresden, Germany, 2012. [Google Scholar]
- Moher, D.; Shamseer, L.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A.; PRISMA-P Group. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst. Rev. 2015, 4, 1. [Google Scholar] [PubMed]
- National Health and Medical Research Council. NHMRC Levels of Evidence and Grades for Recommendations for Guideline Developers, National Health and Medical Research Council: Canberra, Australia, 2009.
- Higgins, J.P.; Altman, D.G.; Gøtzsche, P.C.; Jüni, P.; Moher, D.; Oxman, A.D.; Savovic, J.; Schulz, K.F.; Weeks, L.; Sterne, J.A.; et al. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ 2011, 343, d5928. [Google Scholar] [CrossRef] [PubMed]
- American Dietetic Association. Evidence Analysis Manual: Steps in the ADA Evidence Analysis Process; American Dietetic Association: Chicago, IL, USA, 2008. [Google Scholar]
- Cai, W.; Cijiang, J.; Zhu, L.; Peppa, M.; Lu, C.; Uribarri, J.; Vlassara, H. High levels of dietary advanced glycation end products transform low-density lipoprotein into a potent redox-sensitive mitogen-activated protein kinase stimulant in diabetic patients. Circulation 2004, 110, 285–291. [Google Scholar] [PubMed]
- Harcourt, B.E.; Sourris, K.C.; Coughlan, M.T.; Walker, K.Z.; Dougherty, S.L.; Andrikopoulos, S.; Morley, A.L.; Thallas-Bonke, V.; Chand, V.; Penfold, S.A.; et al. Targeted reduction of advanced glycation improves renal function in obesity. Kidney Int. 2011, 80, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Luévano-Contreras, C.; Garay-Sevilla, M.E.; Wrobel, K.; Malacara, J.M.; Wrobel, K. Dietary advanced glycation end products restriction diminishes inflammation markers and oxidative stress in patients with type 2 diabetes mellitus. J. Clin. Biochem. Nutr. 2013, 52, 22–26. [Google Scholar] [PubMed]
- Mark, A.B.; Poulsen, M.W.; Andersen, S.; Andersen, J.M.; Bak, M.J.; Ritz, C.; Holst, J.J.; Nielsen, J.; de Courten, B.; Dragsted, L.O.; et al. Consumption of a diet low in advanced glycation end products for 4 weeks improves insulin sensitivity in overweight women. Diabetes Care 2014, 37, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Peppa, M.; Uribarri, J.; Cai, W.; Lu, M.; Vlassara, H. Glycoxidation and inflammation in renal failure patients. Am. J. Kidney Dis. 2004, 43, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Gebauer, S.K.; Baer, D.J.; Sun, K.; Turner, R.; Silber, H.A.; Talegawkar, S.; Ferrucci, L.; Novotny, J.A. Dietary intake of advanced glycation end products did not affect endothelial function and inflammation in healthy adults in a randomized controlled trial. J. Nutr. 2014, 144, 1037–1042. [Google Scholar] [PubMed]
- Uribarri, J.; Cai, W.; Pyzik, R.; Goodman, S.; Chen, X.; Zhu, L.; Ramdas, M.; Striker, G.E.; Vlassara, H. Suppression of native defense mechanisms, SIRT1 and PPAR, by dietary glycoxidants precedes disease in adult humans; relevance to lifestyle-engendered chronic diseases. Amino Acids 2014, 46, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Ramdas, M.; Goodman, S.; Pyzik, R.; Xue, C.; Zhu, L.; Striker, G.E.; Vlassara, H. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: Potential role of AGER1 and SIRT1. Diabetes Care 2011, 34, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Peppa, M.; Cai, W.; Goldberg, T.; Lu, M.; Baliga, S.; Vassalotti, J.A.; Vlassara, H. Dietary glycotoxins correlate with circulating advanced glycation end product levels in renal failure patients. Am. J. Kidney Dis. 2003, 42, 532–538. [Google Scholar] [CrossRef]
- Vlassara, H.; Cai, W.; Crandall, J.; Goldberg, T.; Oberstein, R.; Dardaine, V.; Peppa, M.; Rayfield, E.J. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc. Natl. Acad. Sci. USA 2002, 99, 15596–15601. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Cai, W.; Goodman, S.; Pyzik, R.; Yong, A.; Chen, X.; Zhu, L.; Neade, T.; Beeri, M.; Silverman, J.M.; et al. Protection against loss of innate defenses in adulthood by low advanced glycation end products (AGE) intake: Role of the antiinflammatory AGE receptor-1. J. Clin. Endocrinol. Metab. 2009, 94, 4483–4491. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, T.; Cai, W.; Peppa, M.; Dardaine, V.; Baliga, B.S.; Uribarri, J.; Vlassara, H. Advanced glycoxidation end products in commonly consumed foods. J. Am. Diet. Assoc. 2004, 104, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- El Nahas, A.M.; Bello, A.K. Chronic kidney disease: The global challenge. Lancet 2005, 365, 331–340. [Google Scholar] [CrossRef]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Klöting, I.; et al. Diabetes-Associated Sustained Activation of the Transcription Factor Nuclear Factor-κB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products: Sparking the Development of Diabetic Vascular Injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Moller, D.E. Potential role of TNF-α in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol. Metab. 2000, 11, 212–217. [Google Scholar] [CrossRef]
- Basu, S. F2-isoprostanes in human health and diseases: From molecular mechanisms to clinical implications. Antioxid. Redox Signal. 2008, 10, 1405–1434. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A. New insights on oxidative stress and diabetic complications may lead to a “causal” antioxidant therapy. Diabetes Care 2003, 26, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes Care 2008, 57, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Seiquer, I.; Rubio, L.A.; Peinado, M.J.; Delgado-Andrade, C.; Navarro, M.P. Maillard reaction products modulate gut microbiota composition in adolescents. Mol. Nutr. Food Res. 2014, 58, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet—Induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Yap, F.Y.T.; Tong, D.C.K.; Andrikopoulos, S.; Gasser, A.; Thallas-Bonke, V.; Webster, D.E.; Miyazaki, J.; Kay, T.W.; Slattery, R.M.; et al. Advanced glycation end products are direct modulators of β-Cell function. Diabetes 2011, 60, 2523–2532. [Google Scholar] [PubMed]
- Hofmann, S.M.; Dong, H.-J.; Li, Z.; Cai, W.; Altomonte, J.; Thung, S.N.; Zeng, F.; Fisher, E.A.; Vlassara, H. Improved insulin sensitivity is associated with restricted intake of dietary glycoxidation products in the db/db mouse. Diabetes 2002, 51, 2082–2089. [Google Scholar] [PubMed]
- Sandu, O.; Song, K.; Cai, W.; Zheng, F.; Uribarri, J.; Vlassara, H. Insulin resistance and type 2 diabetes in high-fat–fed mice are linked to high glycotoxin intake. Diabetes 2005, 54, 2314–2319. [Google Scholar] [PubMed]
- Birlouez-Aragon, I.; Saavedra, G.; Tessier, F.J.; Galinier, A.; Ait-Ameur, L.; Lacoste, F.; Niamba, C.N.; Alt, N.; Somoza, V.; Lecerf, J.M. A diet based on high-heat-treated foods promotes risk factors for diabetes mellitus and cardiovascular diseases. Am. J. Clin. Nutr. 2010, 91, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Apple, F.S.; Christenson, R.H.; Danne, O.; Jaffe, A.S.; Mair, J.; Mockel, M.; Pagani, F.; Christenson, R.H.; Mockel, M.; Danne, O.; et al. Future biomarkers for detection of ischemia and risk stratification in acute coronary syndrome. Clin. Chem. 2005, 51, 810–824. [Google Scholar] [PubMed]
- Tsimikas, S.; Willeit, P.; Willeit, J.; Santer, P.; Mayr, M.; Xu, Q.; Mayr, A.; Witztum, J.L.; Kiechl, S. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J. Am. Coll. Cardiol. 2012, 60, 2218–2229. [Google Scholar] [CrossRef] [PubMed]
- Cerami, C.; Founds, H.; Nicholl, I.; Mitsuhashi, T.; Giordano, D.; Vanpatten, S.; Lee, A.; Al-Abed, Y.; Vlassara, H.; Bucala, R.; Cerami, A. Tobacco smoke is a source of toxic reactive glycation products. Proc. Natl. Acad. Sci. USA 1997, 94, 13915–13920. [Google Scholar] [CrossRef] [PubMed]
- Pouillart, P.; Mauprivez, H.; Ait-Ameur, L.; Cayzeele, A.; Lecerf, J.M.; Tessier, F.J.; Birlouez-Aragon, I. Strategy for the study of the health impact of dietary Maillard products in clinical studies: The example of the ICARE clinical study on healthy adults. Ann. N. Y. Acad. Sci. 2008, 1126, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Ames, J.M. Determination of Nɛ-(Carboxymethyl)lysine in foods and related systems. Ann. N. Y. Acad. Sci. 2008, 1126, 20–24. [Google Scholar] [PubMed]


{kind=link}
| Study | Intervention | Comparator | Length (Weeks) | AGE Content of Diet | Assessment of AGEs | Participants Intervention Comparator | |
|---|---|---|---|---|---|---|---|
| Harcourt et al. 2011 a [16] | High AGE diet. Food provided. Cooking methods used to generate difference in AGEs. P:F:C = 16:30:54 | Low AGE diet. Food provided. Cooking methods used to generate difference in AGEs. P:F:C = 16:30:54 | 2 | H-AGE = 14,090, L-AGE = 3302 kU AGE/day | Based on reference database not validated [26] | Healthy overweight: n = 11 (M = 100%); Age = 30 ± 9 years; BMI = 31.8 ± 4.8 kg/m2. | |
| Mark et al. 2014 b [18] | High AGE diet. Ingredients provided and participants instructed on how to prepare meals. Cooking methods used to generate difference in AGEs. P:F:C = 18.8 ± 0.4:37.3 ± 0.8:42.7 ± 0.9 | Low AGE diet. Food provided. Cooking methods used to generate difference in AGEs. P:F:C = 21.6 ± 0.4:30.6 ± 0.7:46.9 ± 0.8 | 4 | H-AGE = 24.6; L-AGE = 10.7 mg/day CML (H-AGE had 43% more AGEs) | LC-MS | Healthy overweight: n = 37 (M = 0); Age = 41.4 ± 1.4 years; BMI = 32.3 ± 0.6 kg/m2 | Healthy overweight: n = 36 (M = 0); Age = 37.9 ± 1.4 yrs; BMI = 33.2 ± 0.8 kg/m2 |
| Semba et al. 2014 b [20] | High AGE diet. Food provided. Cooking techniques and time used to produce AGEs. P:F:C = 17:29:55 * | Low AGE diet. Food provided. Cooking technique and time varied to reduce AGEs. | 6 | H-AGE = 4 times AGE content of L-AGE | Based on reference database not validated [26] | Healthy: n = 12 (M = 41.7%); Age = 60.6 ± 4.3 yrs; BMI = 26.1 ± 3.4 kg/m2 | Healthy: n = 12 (M = 41.7%); Age = 57.9 ± 6.0 yrs; BMI = 26.4 ± 4.0 kg/m2 |
| Uribarri et al. 2011 b [22] | Standard diet high in AGES. Participants prepared own food. Cooking techniques and time used to produce AGEs. | Low AGE diet. Participants prepared own foods under instruction. Cooking technique and time varied to reduce AGEs. | 16 | L-AGE = 40%–50% reduction in AGEs compared to H-AGE | Based on reference database not validated | Healthy: n = 9 (M = 22.2%); Age = 67 ± 1 years; BMI = 27.3 ± 1.4 kg/m2 | Healthy: n = 9 (M = 22.2%); Age = 67 ± 1 years; BMI = 27.3 ± 1.4 kg/m2 |
| Uribarri et al. 2014 b [21] | Standard diet high in AGES. Participants prepared own food. Cooking techniques and time used to produce AGEs. | Low AGE diet. Participants prepared own foods under instruction. Cooking technique and time varied to reduce AGEs. | 16 | H-AGE ≥ 15; L-AGE < 10 kU AGE/day | Based on reference database not validated | Healthy: n = 8 (M = 25%); Age = 63.5 ± 5 years; BMI = 29 ± 2 kg/m2 | Healthy: n = 10 (M = 30%); Age = 65 ± 2 years; BMI = 26 ± 3 kg/m2 |
| Vlassara et al. 2009 b [25] | Standard diet high in AGES. Participants prepared own food. Cooking techniques and time used to produce AGEs. | Low AGE diet. Participants prepared own foods under instruction. Cooking technique and time varied to reduce AGEs. | 16 | H-AGE > 20,000; L-AGE < 10,000 kU CML/day | Based on reference database not validated [26] | 30 Healthy participants randomised to either H-AGE or L-AGE diet. BMI = 28 ± 2 kg/m2 Further population characteristics not described. | |
| Cai et al. 2004 b [15] | High AGE diet. Food provided. Cooking methods used to generate difference in AGEs. P:F:C = 20:30:50 | Low AGE diet. Food provided. Cooking technique and time varied to reduce AGEs. P:F:C = 20:30:50 | 6 | H-AGE = 16300 ± 3700; L-AGE = 3670 ± 1200 kU CML/day | Competitive ELISA for protein foods, direct ELISA for lipid foods. Not validated | T1DM and T2DM: n = 11 (M = 54%); Age = 61 ± 7 years; BMI = 28.2 ± 3.4 kg/m2; T1DM:T2DM = 2:9 | T1DM and T2DM n = 13 (M = 38%); Age = 62 ± 5 years; BMI = 28.7 ± 5.1 kg/m2; T1DM:T2DM = 4:9 |
| Luevano-Contreras et al. 2013 b [17] | Standard diet high in AGEs. Participants prepared own food. Cooking technique and time used to produce AGEs. P:F:C = 20:30:50 | Low AGE diet. Participants prepared own foods under instruction. Cooking technique and time varied to reduce AGEs. P:F:C = 20:30:50 | 6 | H-AGE = 9910 ± 4169; L-AGE = 8956 ± 3587 kU CML/day (from baseline) | Based on reference database not validated [26] | T2DM: n = 13 (M = 15.4%); Age = 48.5 ± 6.2 years; BMI = 28.8 ± 4. kg/m2 | T2DM: n = 13 (M = 7.7%); Age = 46.0 ± 5 years; BMI = 29.8 ± 4.0 kg/m2 |
| Uribarri et al. 2011 b [22] | Standard diet high in AGES. Cooking technique and time used to produce AGEs. Participants prepared own food. | Low AGE diet. Participants prepared own foods under instruction. Cooking technique and time varied to reduce AGEs. | 16 | L-AGE = 40%–50% reduction in AGEs compared to H-AGE | Based on reference database not validated | T2DM: n = 6 (M = 22.2%); Age = 61 ± 4 years; BMI = 32.3 ± 1.6 kg/m2 | T2DM: n = 12 (M = 22.2%); Age = 61 ± 4 years; BMI = 32.3 ± 1.6 kg/m2 |
| Vlassara et al. 2002 b [24] | High AGE diet. Food provided. Cooking techniques and time used to produce AGEs. P:F:C = 20:30:50 | Low AGE diet. Food provided. Cooking technique and time varied to reduce AGEs. P:F:C = 20:30:50 | 6 | H-AGE = 16,300 ± 3700; L-AGE = 3670 ± 1200 kU CML/day | Competitive ELISA for protein foods, direct ELISAs for lipid foods. Not validated | T1DM and T2DM: n = 6 Age = 62 years; BMI = 29.5 ± 3 kg/m2 T1DM:T2DM = 4:8 | T1DM and T2DM: n = 7 Age = 62 years; BMI = 29.5 ± 3 kg/m2 |
| Vlassara et al. 2002 a [24] | High AGE diet. Food provided. Cooking methods used to generate difference in AGEs. P:F:C = 20:30:50 | Low AGE diet. Cooking technique and time varied to reduce AGEs. P:F:C = 20:30:50 | 2 | H-AGE = 16,300 ± 3700 L-AGE = 3670 ± 1200 kU CML/day | Competitive ELISA for protein foods, direct ELISAs for lipid foods. Not validated | T1DM and T2DM: n = 11 Age = 52 ± 5 years; BMI = 28 ± 3.5 kg/m2 T1DM:T2DM = 2:9 | |
| Uribarri 2003; Peppa 2004 b [19,23] | Standard diet high in AGEs. Participants prepared own food. Cooking techniques and time used to produce AGEs. | Low AGE diet. Cooking technique and time varied to reduce AGEs. Participants prepared own foods under instruction | 4 | H-AGE = 17,000 ± 3700; L-AGE = 5500 ± 900 kU CML/day | Based on reference database not validated [26] | Non diabetic peritoneal dialysis; n = 9 (M = 33.33%). Not significantly different from comparator at baseline. | Non diabetic peritoneal dialysis; n = 9 (M = 33.33%) |
| Vlassara et al. 2009 b [25] | Standard diet high in AGES. Participants prepared own food. Cooking techniques and time used to produce AGEs. | Low AGE diet. Meals prepared in the clinical research center and given to participants twice a week. Cooking technique and time varied to reduce AGEs. | 4 | H-AGE > 20,000; L-AGE < 10,000 kU CML/day | Based on reference database not validated [26] | 9 CKD (stage 3) patients randomised to either H-AGE or L-AGE diet. BMI = 23 ± 1.6 kg/m2 (intervention); 28 ± 1.9 kg/m2 (comparator). Further population characteristics not described | |
| Study | Group | n | TNFα | ES | IL-6 | ES | CRP | ES | MCP-1 | ES | 8-Isoprostane | ES |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Healthy | ||||||||||||
| Harcourt et al. 2011 [16] | L-AGE H-AGE | 11CO | NVG | NVG | H > L (plasma) | H > L (urine) | ||||||
| Semba et al. 2014 [20] | L-AGE | 12 | B: 1.48 (1.84) A: 1.53 (1.35) pg/mL | −0.03 | B: 2.11 (1.45) A: 2.62 (2.25) mg/L | −0.27 | ||||||
| H-AGE | 12 | B: 2.25 (1.84) A: 2.09 (1.35) pg/mL | +0.10 | B: 1.57 (1.45) A: 1.38 (2.25) mg/L | +0.10 | |||||||
| Uribarri et al. 2011 [22] | L-AGE | 9 | B: 10 (3.9) A: 8.4 (2.1) ng/mg (PBMC) | +0.51 | B: 135 (30) A: 90 (27) pg/mL (plasma) | +1.58 | ||||||
| H-AGE | 9 | B: 8.6 (1.8) A: 11.8 (3) ng/mg (PBMC) | −1.29 | B: 125 (54) A: 165 (69) pg/mL (plasma) | −0.65 | |||||||
| Uribarri et al. 2014 [21] | L-AGE | 10 | MD: −2.1 (1.6) * ng/mg (PBMC) | MD: 1.3 (1.4) * mg/L | B: 170 (65) A: 85 (17) MD: −48 (11) * pg/mL (serum) | +1.79 | ||||||
| H-AGE | 8 | MD: +3.2 (0.8) * ng/mg (PBMC) | MD: 0.4 (0.4) * mg/L | MD: +48 (20) pg/mL (serum) * | ||||||||
| Vlassara et al. 2009 [25] | L-AGE | 30 | B: 12 (1) * A: 8 (0.5) * ng/mg (PBMC) | B: 240 (67) * A: 100 (13) * ng/mL (plasma) | - | |||||||
| H-AGE | B: 9 (1) * A: 12 (1) * ng/mg (PBMC) | B: 122 (4) * A: 173 (38) * ng/mL (plasma) | ||||||||||
| Diabetes | ||||||||||||
| Luevano-Contreras et al. 2013 [17] | L-AGE | 13 | MD: −18.36 (17.1) * pg/mL (serum) | MD: −1.69 (5.4) * mg/L (serum) | ||||||||
| H-AGE | 13 | MD: +12.5 (14.7) * pg/mL (serum) | MD: −1.21 (5.5) * mg/L (serum) | |||||||||
| Uribarri et al. 2011 [22] | L-AGE | 12 | B: 18 (3.5) A: 14.4 (6.9) ng/mg (PBMC) | +0.66 | B: 233 (58.9) A: 141 (62.4) pg/mL (plasma) | +1.52 | ||||||
| H-AGE | 6 | B: 20 (4.9) A: 26 (4.9) ng/mg (PBMC) | −1.22 | B: 236 (61.2) A: 313 (188.6) pg/mL (plasma) | −0.55 | |||||||
| Vlassara et al. 2002 6-weeks [24] | L-AGE | 7 | MD:−20% * ng/mL (PBMC) | MD: −20% * mg/dL (serum) | ||||||||
| H-AGE | 6 | MD: +86.3% * ng/mL (PBMC) | MD: +35% * mg/dL (serum) | |||||||||
| Vlassara et al. 2002 2-weeks [24] | L-AGE | 11CO | MD: 4.1 (4.8) * mg/dL (serum) | |||||||||
| H-AGE | MD: 6 (8.6) * mg/dL (serum) | |||||||||||
| Kidney Disease | ||||||||||||
| Uribarri et al. 2003; Peppa 2004 et al. [19,23] | L-AGE | 9 | B: 44 (18) A: 31 (10.8) pg/mg (PBMC) | +0.88 | Graph only | |||||||
| H-AGE | 9 | B: 43 (21) A: 44 (21) pg/mg (PBMC) | −0.05 | Graph only | ||||||||
| Vlassara et al. 2009 [25] | L-AGE | 9 | B: 22 (4) * A: 16 (3) * ng/mg (PBMC) | B: 328 (51) * A: 154 (25) * ng/mL (plasma) | ||||||||
| H-AGE | B: 15 (4) * A: 16 (3) * ng/mg (PBMC) | B: 211 (7) * A: 167 (21) * ng/mL (plasma) | ||||||||||
| Study | Group | n | T2DM | CVD | CKD | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HOMA IR | ES | FBG | ES | HbA1c % | ES | oxLDL | ES | VCAM-1 | ES | Alb | ES | Cr | ES | |||
| Healthy | ||||||||||||||||
| Harcourt et al. 2011 [16] | L-AGE | 11CO | A: 5.1 (0.3) mmol/L | H > L | A: 72.3 (18.3) μmol/L (serum) | |||||||||||
| H-AGE | ||||||||||||||||
| Mark et al. 2014 [18] | L-AGE | 36 | B: 2.65 (1.8) A: 2.43 (1.8) | +0.12 | B: 5.4 (0.6) A: 5.5 (0.6) mmol/L | −0.17 | ||||||||||
| H-AGE | 37 | B: 2.14 (1.8) A: 2.40 (1.2) | −0.17 | B: 5.5 (0.61) A: 5.5 (0.61) mmol/L | 0 | |||||||||||
| Semba et al. 2014 [20] | L-AGE | 12 | B: 97 (10.4) A: 93 (10.4) mg/dL | +0.38 | B: 1.26 (0.8) A: 1.01 (0.9) ug/mL | +0.29 | ||||||||||
| H-AGE | 12 | B: 93 (10.4) A: 96 (10.4) mg/dL | −0.29 | B: 1.39 (0.8) A: 1.34 (0.9) ug/mL | −0.06 | |||||||||||
| Uribarri et al. 2011 [22] | L-AGE | 9 | B: 2.2 (0.9) A: 2.5 (1.8) | −0.33 | B: 86 (9) A: 88 (18) mg/dL | −0.14 | ||||||||||
| H-AGE | 9 | B: 1.3 (0.9) A: 1.5 (1.8) | −0.22 | B: 83 (12) A: 80 (12) mg/dL | +0.25 | |||||||||||
| Uribarri et al. 2014 [21] | L-AGE | 10 | MD: 0.04 (0.44) * | MD: −270 (92) * pg/mL (serum) | ||||||||||||
| H-AGE | 8 | MD: 0.14 (0.15) * | MD:+182 (46) * pg/mL (serum) | |||||||||||||
| Vlassara et al. 2009 [25] | L-AGE | 30 | B: 81 (3) * A: 78 (3) * mg/dL | B: 1001 (115) * A: 576 (49) * ng/mL (plasma) | B: 99 (12) * A: 118 (15) * mL/min (Cr clearance) | |||||||||||
| H-AGE | B: 84 (3) * A: 79 (3) * mg/dL | B: 717 (68) * A: 771 (58) * ng/mL (plasma) | B: 115 (10) * A: 100 (8) * mL/min (Cr clearance) | |||||||||||||
| Diabetes | ||||||||||||||||
| Cai et al. 2004 [15] | L-AGE | 13 | B: 122 (90.1) A: 118 (68.5) mg/dL | +0.05 | B: 7.2 (3.6) A: 7.0 (2.88) | +0.06 | A:1.5 (0.5) nmol/mg | |||||||||
| H-AGE | 11 | B: 116 (146.6) A:128 (71.6) mg/dL | −0.10 | B: 7.3 (2.0) A: 7.4 (4.3) | −0.03 | A:5.7 (2.3) nmol/mg | ||||||||||
| Luevano-Contreras et al. 2013 [17] | L-AGE | 13 | MD: −2.29 (3.7) * | MD: −18 (56.7) * mg/dL | MD: 0.19 (1.3) * | |||||||||||
| H-AGE | 13 | MD: −2.5 (6.1)* | MD: 4.55 (35.6) * mg/dL | MD: −0.11 (1.9) * | ||||||||||||
| Uribarri et al. 2011 [22] | L-AGE | 12 | B: 5.3 (1.4) A: 3.4 (2.1) | +1.06 | B: 114 (24.2) A: 111 (31.2) mg/dL | +0.11 | B: 6.4 (0.7) A: 6.6 (1.4) | −0.18 | ||||||||
| H-AGE | 6 | B: 4.5 (2.9) A: 6.2 (1.2) | −0.77 | B: 131 (90) A: 129 (63.7) mg/dL | +0.03 | B: 6.7 (1.2) A: 6.5 (1.0) | +0.18 | |||||||||
| Vlassara et al. 2002 6-weeks [24] | L-AGE | 7 | B: 7.0 (2.7) A: 5.6 (1.3) mmol/L | +0.68 | MD: −20% * ng/mL (serum) | |||||||||||
| H-AGE | 6 | B: 6.5 (2.9) A: 8.1 (2.7) mmol/L | −0.57 | |||||||||||||
| Vlassara et al. 2002 2-weeks [24] | L-AGE | 11CO | A: 7.5 (0.7) * mmol/L | MD:698 (347) * ng/mL (serum) | ||||||||||||
| H-AGE | A: 8.1 (0.4) * mmol/L | MD:1108 (429) * ng/mL (serum) | ||||||||||||||
| Kidney Disease | ||||||||||||||||
| Uribarri et al. 2003; Peppa 2004 et al. [19,23] | L-AGE | 9 | B: 3448 (483) A: 3244 (708) ng/mL (serum) | +0.34 | ||||||||||||
| H-AGE | 9 | B: 3699 (306) A: 3735 (291) ng/mL (serum) | −0.12 | |||||||||||||
| Vlassara et al. 2009 [25] | L-AGE | 9 | B: 89 (3) * A: 90 (3) * mg/dL | B: 1033 (168) * A: 733 (52) * (ng/mL) (plasma) | B: 39.5 (11) * A: 36 (10) * mL/min (Cr clearance) | |||||||||||
| H-AGE | B: 93 (2) * A: 80 (4) * mg/dL | B: 1086 (210) * A: 956 (133) * (ng/mL) (plasma) | B: 46.5 (15) * A: 43 (11) * mL/min (Cr clearance) | |||||||||||||
| Study | Group | n | Circulating CML | ES | Urinary CML | ES |
|---|---|---|---|---|---|---|
| Healthy | ||||||
| Harcourt et al. 2011 [16] | L-AGE H-AGE | 11CO | L > H | H > L | ||
| Mark et al. 2014 [18] | L-AGE H-AGE | 36 37 | H > L | |||
| Semba et al. 2014 [20] | L-AGE | 12 | B: 763 (83) A: 678 (100) ng/mL (serum) | +0.92 | B: 1.37 (5.10) A: 0.77 (6.96) μg/mg creatinine | +0.10 |
| H-AGE | 12 | B: 751 (83) A: 711 (100) ng/mL (serum) | +0.44 | B: 1.03 (5.10) A: 1.21 (6.96) μg/mg creatinine | −-0.03 | |
| Uribarri et al. 2011 [22] | L-AGE | 9 | B: 12.4 (1.5) A: 9.3 (3.0) U/mL (serum) | +1.31 | ||
| H-AGE | 9 | B: 11.7 (3.9) A: 14.0 (3.0) U/mL (serum) | -0.66 | |||
| Uribarri et al. 2014 [21] | L-AGE | 10 | B: 13.7 (3.2) A: 9.2 (2.5) MD: −3.71 (1.03) * U/mL (serum) | +1.57 | ||
| H-AGE | 8 | MD: +1.87 (1.05) * U/mL serum) | - | |||
| Vlassara et al. 2009 [25] | L-AGE | 30 | B: 14 (1) * A: 9 (1) * U/mL (serum) | - | ||
| H-AGE | B: 11 (1) * A: 13 (1) * U/mL (serum) | |||||
| Diabetes | ||||||
| Cai et al. 2004 [15] | L-AGE | 13 | B: 12.5 (7.9) A: 7.9 (4.0) U/mL (serum) | +0.73 | ||
| H-AGE | 11 | B: 13.1 (8.6) A: 18.0 (5.6) U/mL (serum) | −0.68 | |||
| Uribarri et al. 2011 [22] | L-AGE | 12 | B: 17.1 (4.5) A: 11.6 (3.8) U/mL (serum) | +1.32 | ||
| H-AGE | 6 | B: 17.8 (4.9) A: 24.2 (9.8) U/mL (serum) | −0.83 | |||
| Study | Group | n | Circulating CML | ES | Urinary CML | ES |
| Vlassara et al. 2002 6-weeks [24] | L-AGE | 7 | MD: −40% U/mL (serum) | |||
| H-AGE | 6 | MD: 28.2 % U/mL (serum) | ||||
| Vlassara et al. 2002 2-weeks [24] | L-AGE | 11CO | A: 7.7 (2.4) * U/mL (serum) A: 13 (6) * U/mL (serum) | A: 15.26 (10) × 10−3 U/24 h A: 30.4 (12) × 10−3 U/24 h | ||
| H-AGE | ||||||
| Kidney Disease | ||||||
| Uribarri et al. 2003; Peppa 2004 et al. [19,23] | L-AGE | 9 | MD: −34% (serum) | |||
| H-AGE | 9 | MD: 29% (serum) | ||||
| Vlassara et al. 2009 [25] | L-AGE | 9 | B: 25 (3) * A: 14.2 (2) * U/mL (serum) | |||
| H-AGE | B: 19 (4) * A: 17 (3) * U/mL (serum) |
| Adequate Sequence Generation | Adequate Allocation Concealment | Blinding- Outcome Assessors | Incomplete Outcome Data Addressed | Free of Selective Outcome Reporting | Free of Other Bias | Overall Risk of Bias | Quality (+, −, or Neutral) | |
|---|---|---|---|---|---|---|---|---|
| Healthy | ||||||||
| Harcourt et al. 2011 [16] | Unclear | Unclear | Unclear | + | Unclear | + | Unclear | Neutral |
| Mark et al. 2014 [18] | Unclear | Unclear | - | + | Unclear | –(weight loss occurred and supplemented with high fructose or glucose beverages) | High | Neutral |
| Semba et al. 2014 [20] | + | + | + | + | + | + | Low | Neutral |
| Uribarri et al. 2011 [22] | Unclear | Unclear | Unclear | + | Unclear | Unclear (differences in intervention groups at baseline, smoking and dietary intakes not reported) | Unclear | Neutral |
| Uribarri et al. 2014 [21] | Unclear | Unclear | Unclear | + | + | Unclear (dietary intake not reported) | Unclear | Neutral |
| Vlassara et al. 2009 [25] | Unclear | Unclear | Unclear | + | Unclear | Unclear (differences in intervention groups at baseline, smoking and dietary intakes not reported) | Unclear | - |
| Diabetes | ||||||||
| Cai et al. 2004 [15] | Unclear | Unclear | Unclear | + | Unclear | Unclear (dietary intake not reported) | Unclear | Neutral |
| Luevano- Contreras et al. 2013 [17] | + | + | + | + | Unclear | –(difference in TNFα levels between groups at baseline) | High | Neutral |
| Vlassara et al. 2002 2-weeks [24] | Unclear | Unclear | Unclear | + | - | Unclear (baseline characteristics not reported in detail) | High | Neutral |
| Vlassara et al. 2002 6-weeks [24] | Unclear | Unclear | Unclear | Unclear | Unclear | Unclear (baseline characteristics not reported) | Unclear | Neutral |
| Kidney Disease | ||||||||
| Uribarri et al. 2003/Peppa et al. 2004 [19,23] | Unclear | Unclear | Unclear | + | Unclear | Unclear (intervention group significantly increased caloric intake from baseline, smoking not reported) | Unclear | - |
| Vlassara et al. 2009 [25] | Unclear | Unclear | Unclear | + | Unclear | Unclear (differences in intervention groups at baseline, smoking and dietary intakes not reported) | Unclear | - |
| Study | Length (weeks) | Population | Inflammation | Oxidative Stress | T2DM Risk | CVD Risk | CKD Risk | cAGEs | uAGEs | Risk of Bias | Quality |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Healthy | |||||||||||
| Harcourt et al. 2011 [16] | 2 | Healthy overweight | ↑/↔ | ↑ | ↔ | N/A | ↑/↔ | ↓ | ↑ | Unclear | Neutral |
| Mark et al. 2014 [18] | 4 | Healthy overweight | N/A | N/A | ↑/↔ | N/A | N/A | N/A | ↑ | High | Neutral |
| Semba et al. 2014 [20] | 6 | Healthy | ↔ | N/A | ↔ | ↔ | N/A | ↔ | ↔ | Low | Neutral |
| Uribarri et al. 2011 [22] | 16 | Healthy | ↑ | ↑ | ↔ | N/A | N/A | ↑ | N/A | Unclear | Neutral |
| Uribarri et al. 2014 [21] | 16 | Healthy | ↑/↔ | ↑ | ↔ | ↑ | N/A | ↑ | N/A | Unclear | Neutral |
| Vlassara et al. 2009 [25] | 16 | Healthy | ↑ | ↔ | ↔ | ↔ | N/A | ↑ | N/A | Unclear | - |
| Diabetes | |||||||||||
| Cai et al. 2004 [15] | 6 | T1DM + T2DM | N/A | N/A | ↔ | ↑ | N/A | ↑ | N/A | Unclear | Neutral |
| Luevano-Contreras et al. 2013 [17] | 6 | T2DM | ↑/↔ | N/A | ↔ | N/A | N/A | ↔ | N/A | High | Neutral |
| Uribarri et al. 2011 [22] | 16 | T2DM | ↑ | ↑ | ↑/↔ | N/A | N/A | ↑ | N/A | Unclear | Neutral |
| Vlassara et al. 2002 [24] | 6 | T1DM + T2DM | ↑ | N/A | ↑ | ↑ | N/A | ↑ | ↑ | High | Neutral |
| Vlassara et al. 2002 [24] | 2 | T1DM + T2DM | ↔ | N/A | ↑ | ↑ | N/A | ↑ | N/A | Unclear | Neutral |
| Kidney Disease | |||||||||||
| Uribarri et al. 2003; Peppa 2004 [19,23] | 4 | CKD | ↑ | N/A | N/A | ↑ | N/A | ↑ | N/A | Unclear | - |
| Vlassara et al. 2009 [25] | 4 | CKD | N/A | ↑ | ↔ | ↔ | ↔ | ↑ | N/A | Unclear | - |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clarke, R.E.; Dordevic, A.L.; Tan, S.M.; Ryan, L.; Coughlan, M.T. Dietary Advanced Glycation End Products and Risk Factors for Chronic Disease: A Systematic Review of Randomised Controlled Trials. Nutrients 2016, 8, 125. https://doi.org/10.3390/nu8030125
Clarke RE, Dordevic AL, Tan SM, Ryan L, Coughlan MT. Dietary Advanced Glycation End Products and Risk Factors for Chronic Disease: A Systematic Review of Randomised Controlled Trials. Nutrients. 2016; 8(3):125. https://doi.org/10.3390/nu8030125
Chicago/Turabian StyleClarke, Rachel E., Aimee L. Dordevic, Sih Min Tan, Lisa Ryan, and Melinda T. Coughlan. 2016. "Dietary Advanced Glycation End Products and Risk Factors for Chronic Disease: A Systematic Review of Randomised Controlled Trials" Nutrients 8, no. 3: 125. https://doi.org/10.3390/nu8030125
APA StyleClarke, R. E., Dordevic, A. L., Tan, S. M., Ryan, L., & Coughlan, M. T. (2016). Dietary Advanced Glycation End Products and Risk Factors for Chronic Disease: A Systematic Review of Randomised Controlled Trials. Nutrients, 8(3), 125. https://doi.org/10.3390/nu8030125