Involvement of the Niacin Receptor GPR109a in the LocalControl of Glucose Uptake in Small Intestine of Type 2Diabetic Mice

Abstract
:1. Introduction
2. Experimental Section
2.1. Animals
2.2. Isolation of Enterocytes
2.3. Western Blot Analysis
2.4. Real-Time PCR Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Mouse β-actin (NM_007393.4) | TCCTCCTGAGCGCAAGTACTC | GTGGACAGTAGTGAGGCCAGGT |
| Mouse GPR109a (NM_030701.3) | GGCGTGGTGCAGTGAGCAGT | GGCCCACGGACAGGCTAGGT |
| Human β-actin (NC_000007.14) | GGCACCCAGCACAATGAAGATC | ATGCTTCTAGGCGGACTATGACTT |
| Human GPR109a (NM_177551.3) | TGCCGCCCTTCCTGATGGACA | TGTTCAGGGCGTGGTGGGGA |
2.5. Immunohistochemistry
2.6. Glucose Uptake by Everted Jejunal Sleeves
2.7. Glucose Uptake by Caco-2 Cells
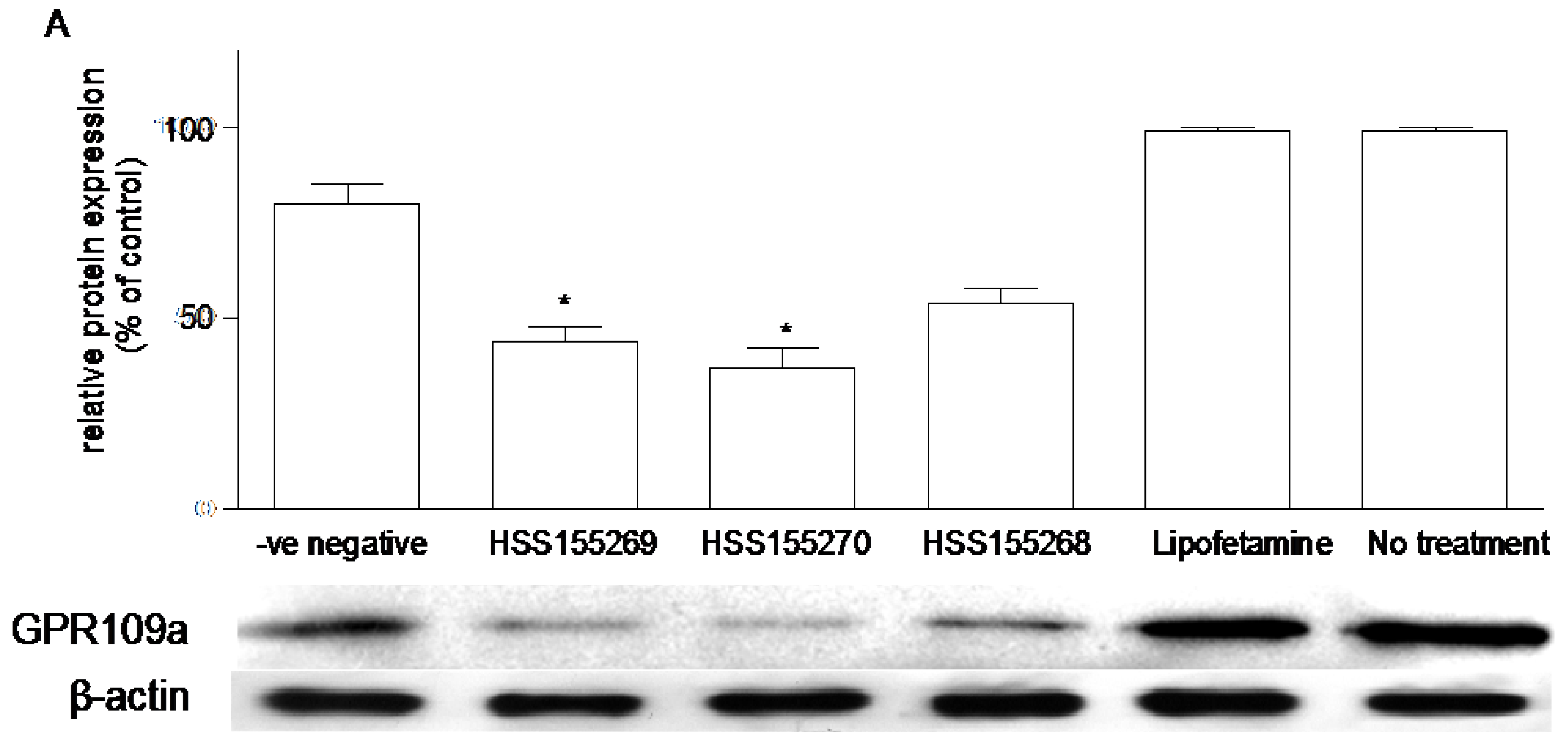
2.8. siRNA Knockdown of GPR109a in Caco-2 Cells
2.9. Statistical Analysis
3. Results
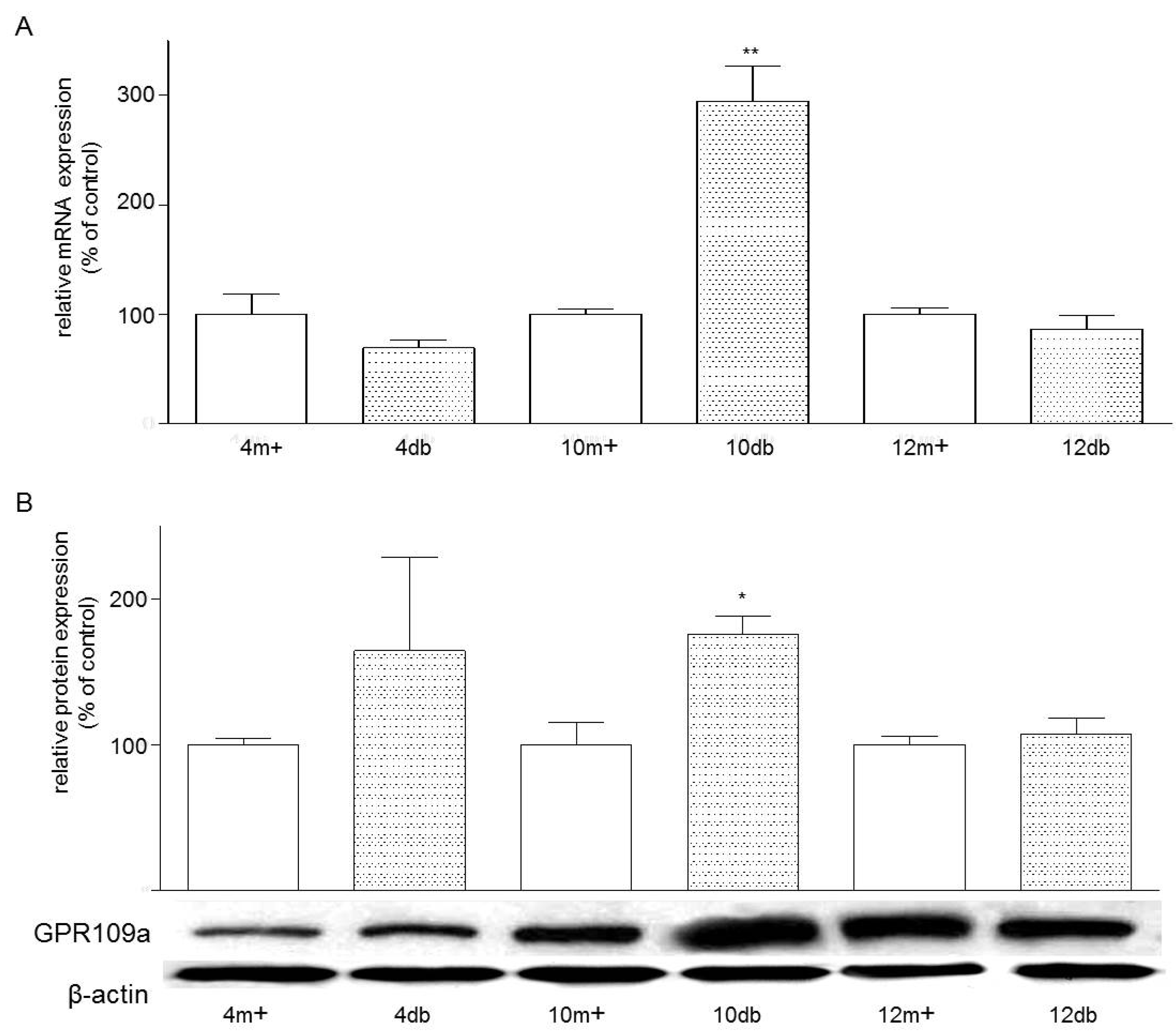
3.1. Enterocyte Expression of GPR109a
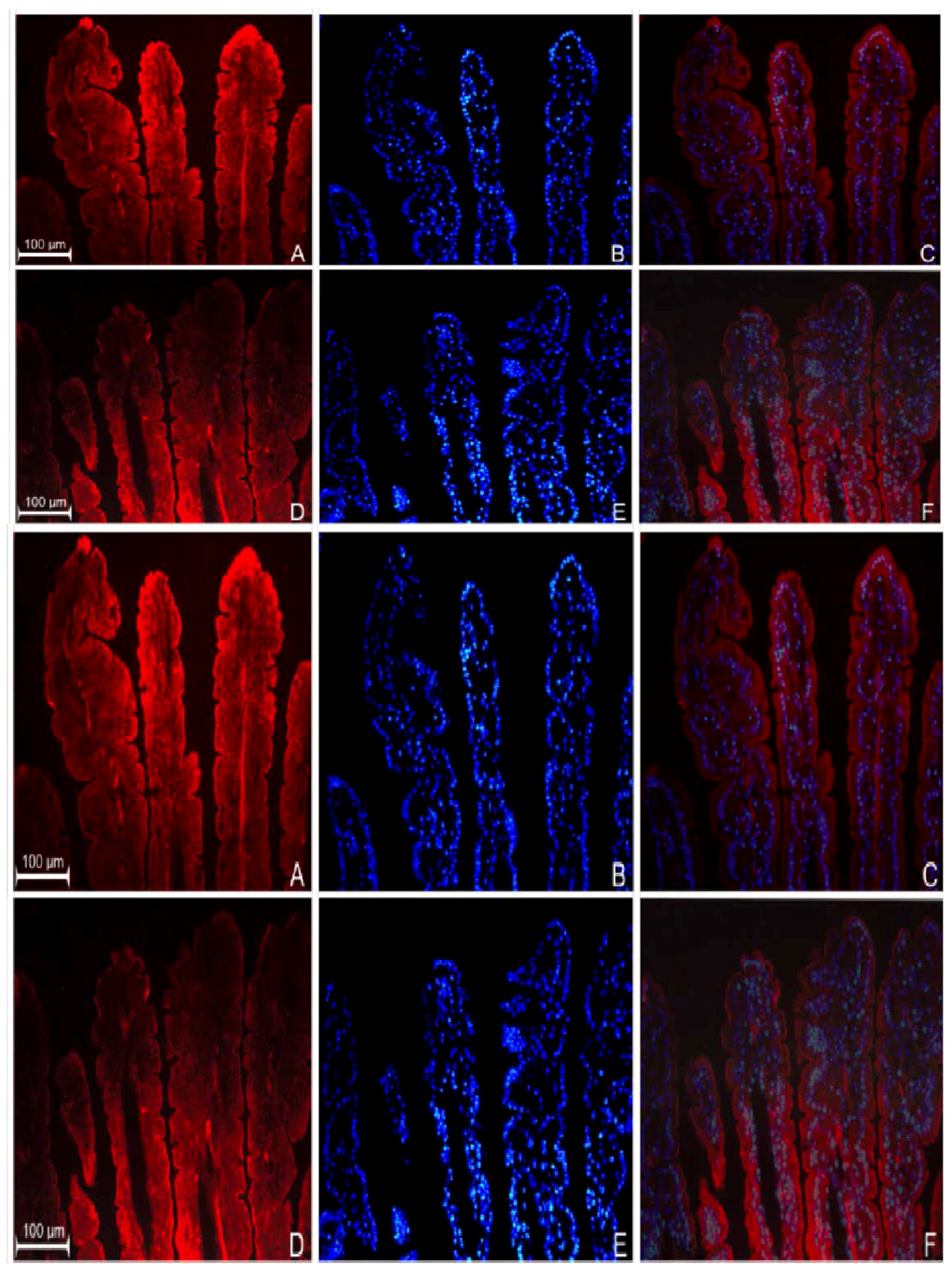
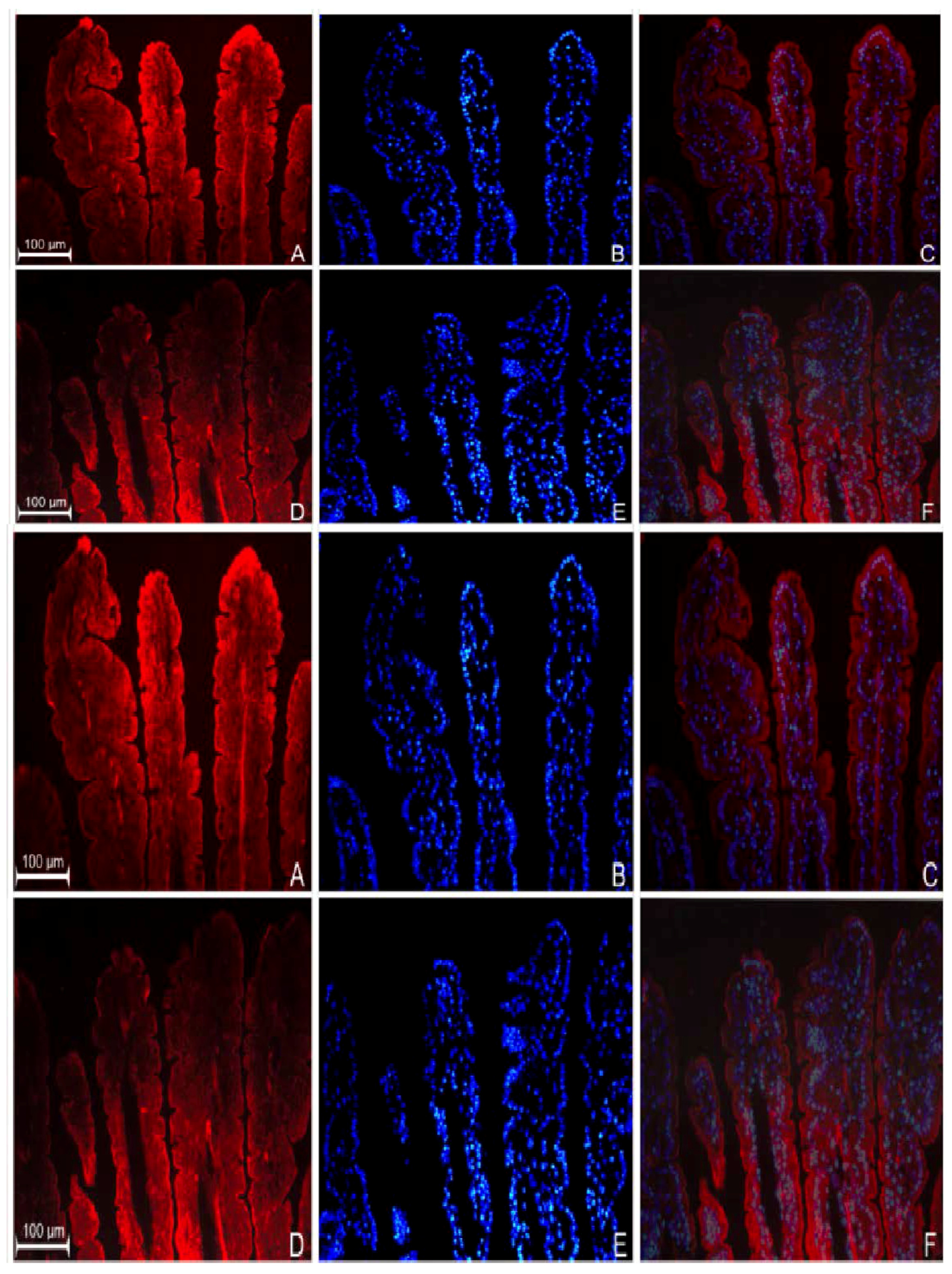
3.2. Localization of GPR109a in Jejunal Mucosa of m+/db and db/db Mice, and Caco-2 Cells
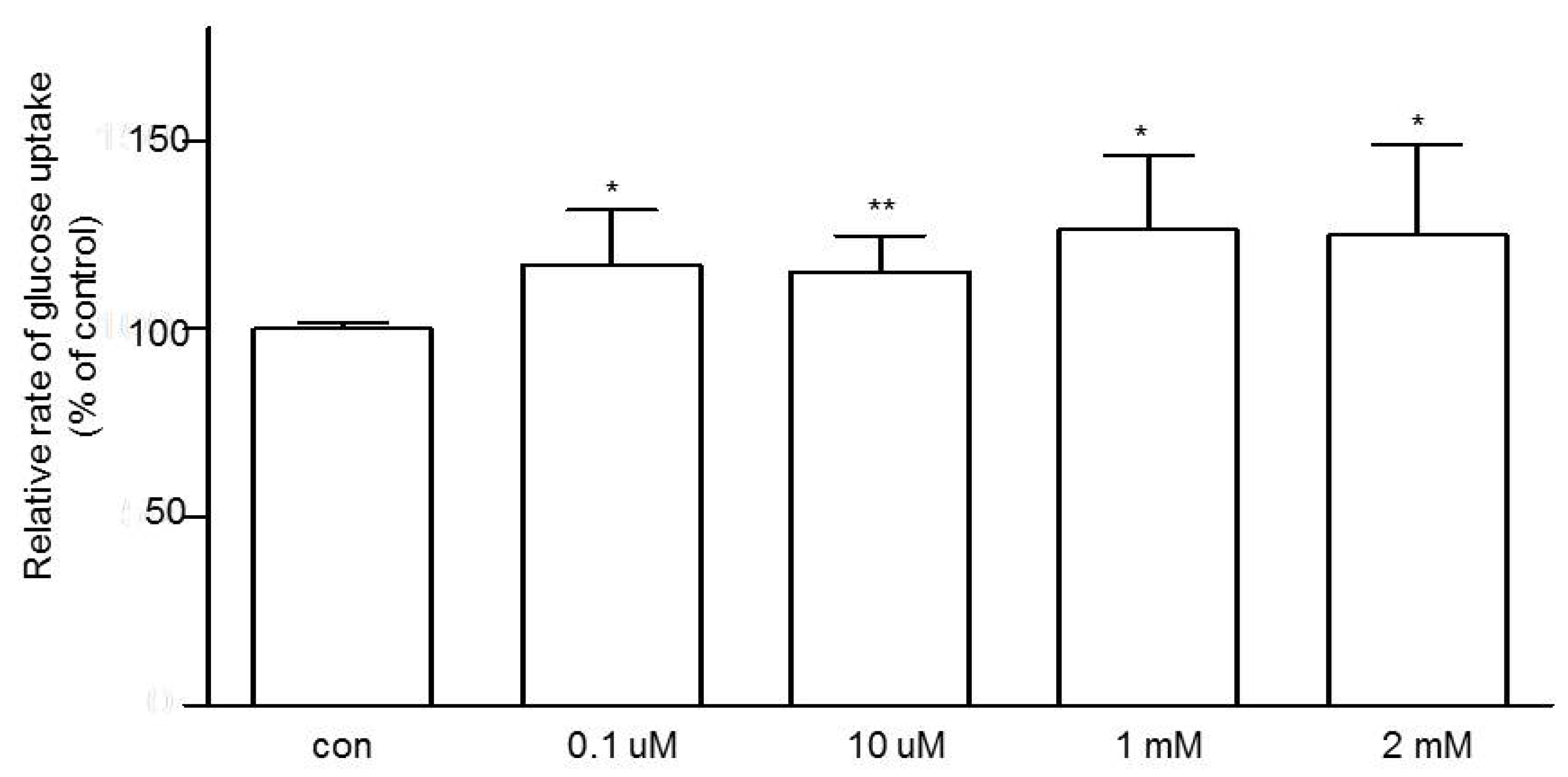
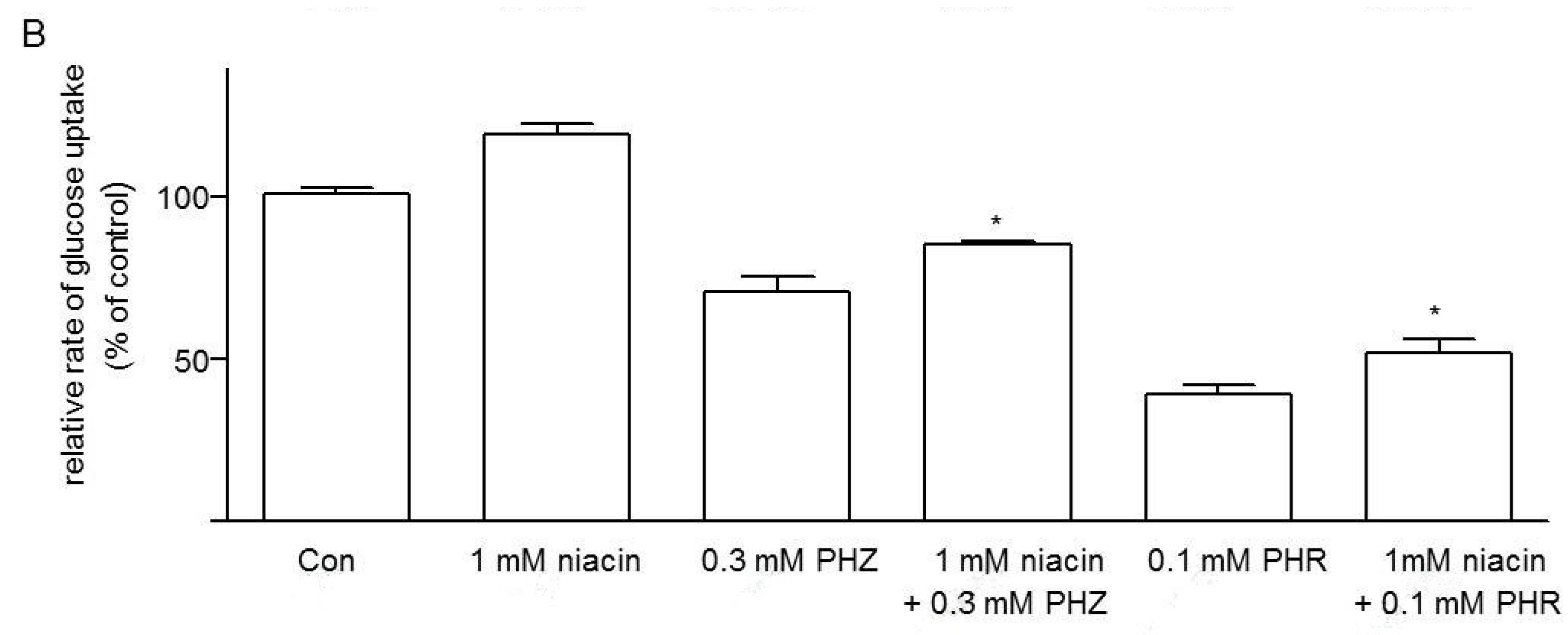
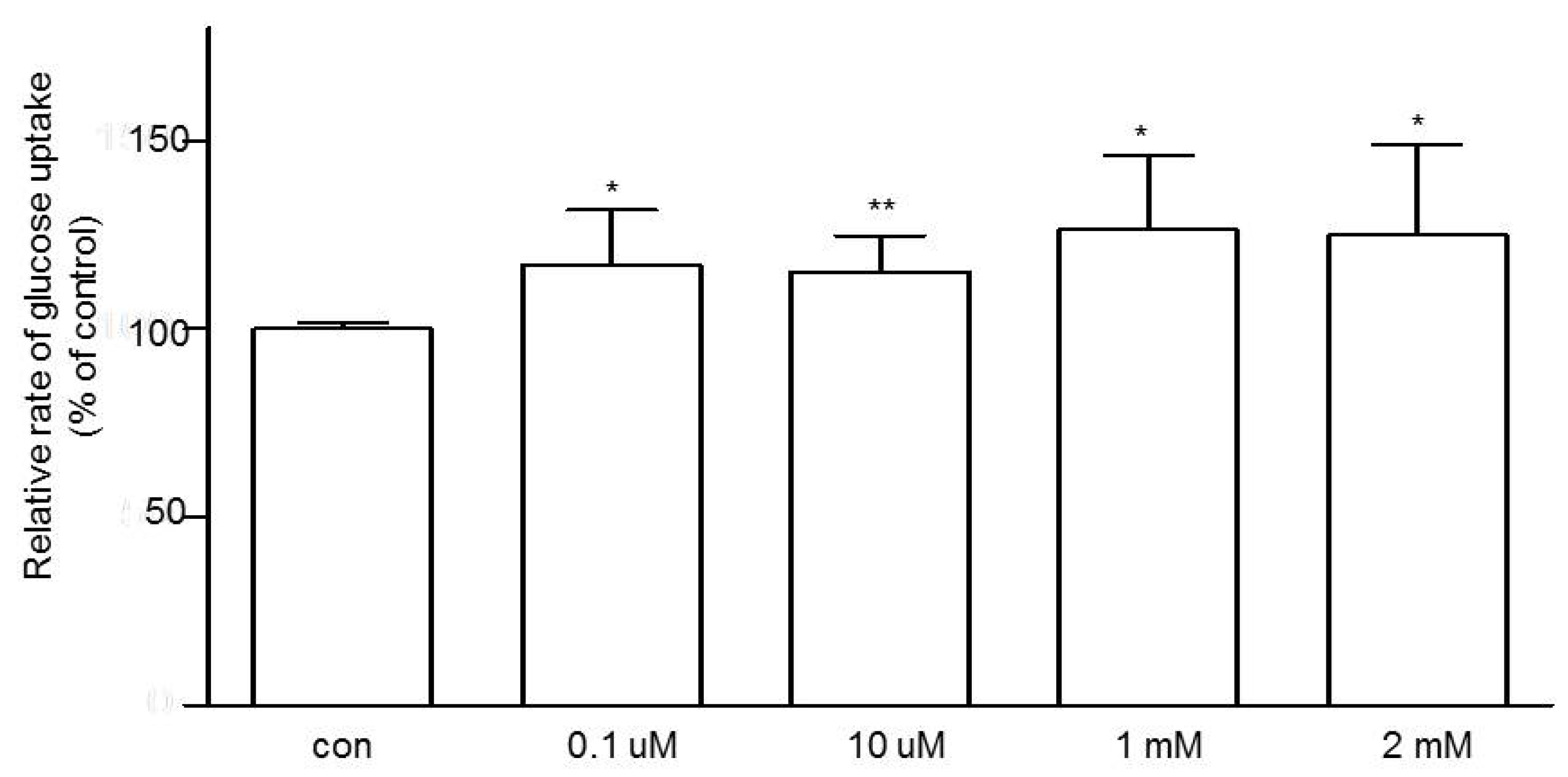
3.3. Effects of Niacin on Jejunal Glucose Uptake
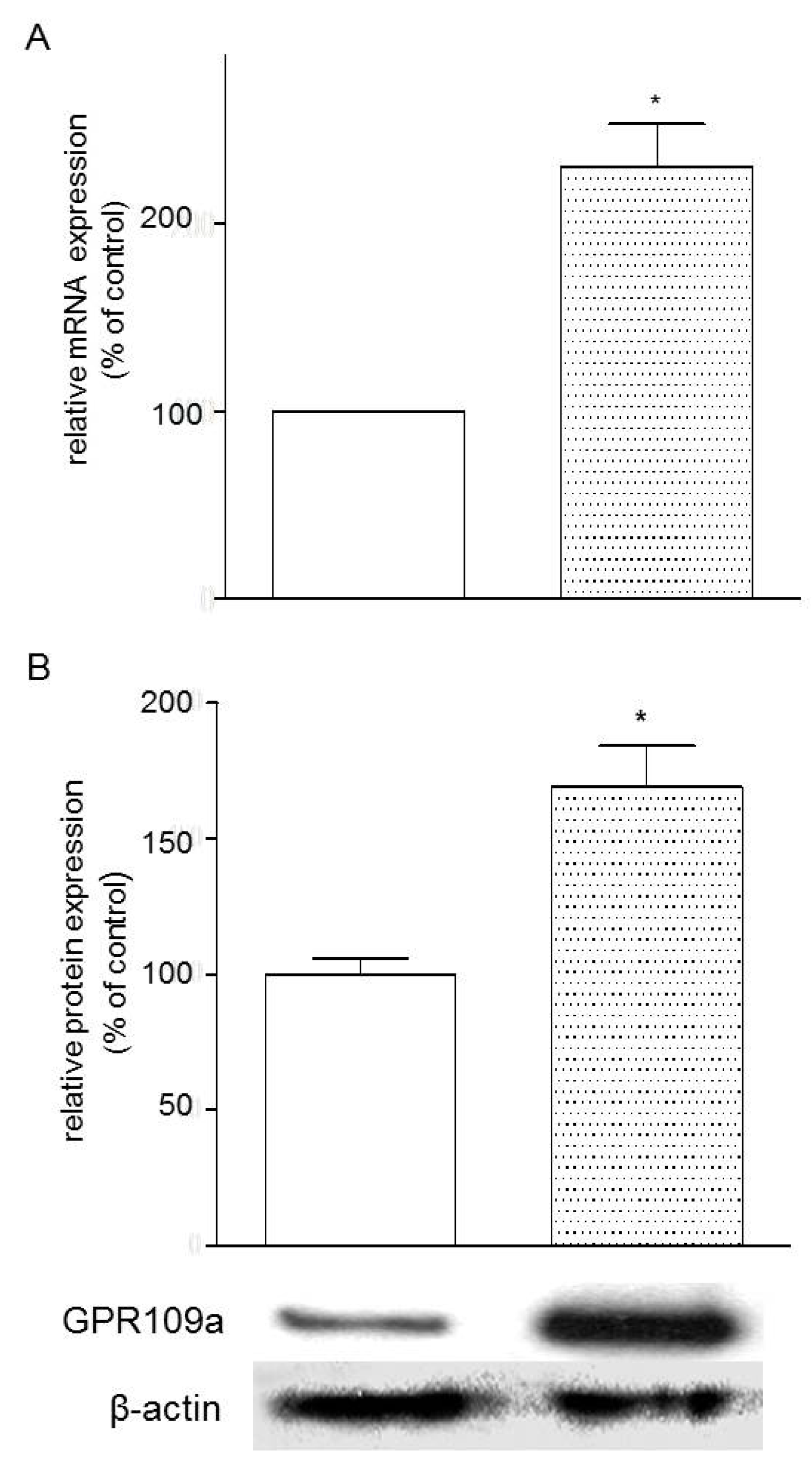
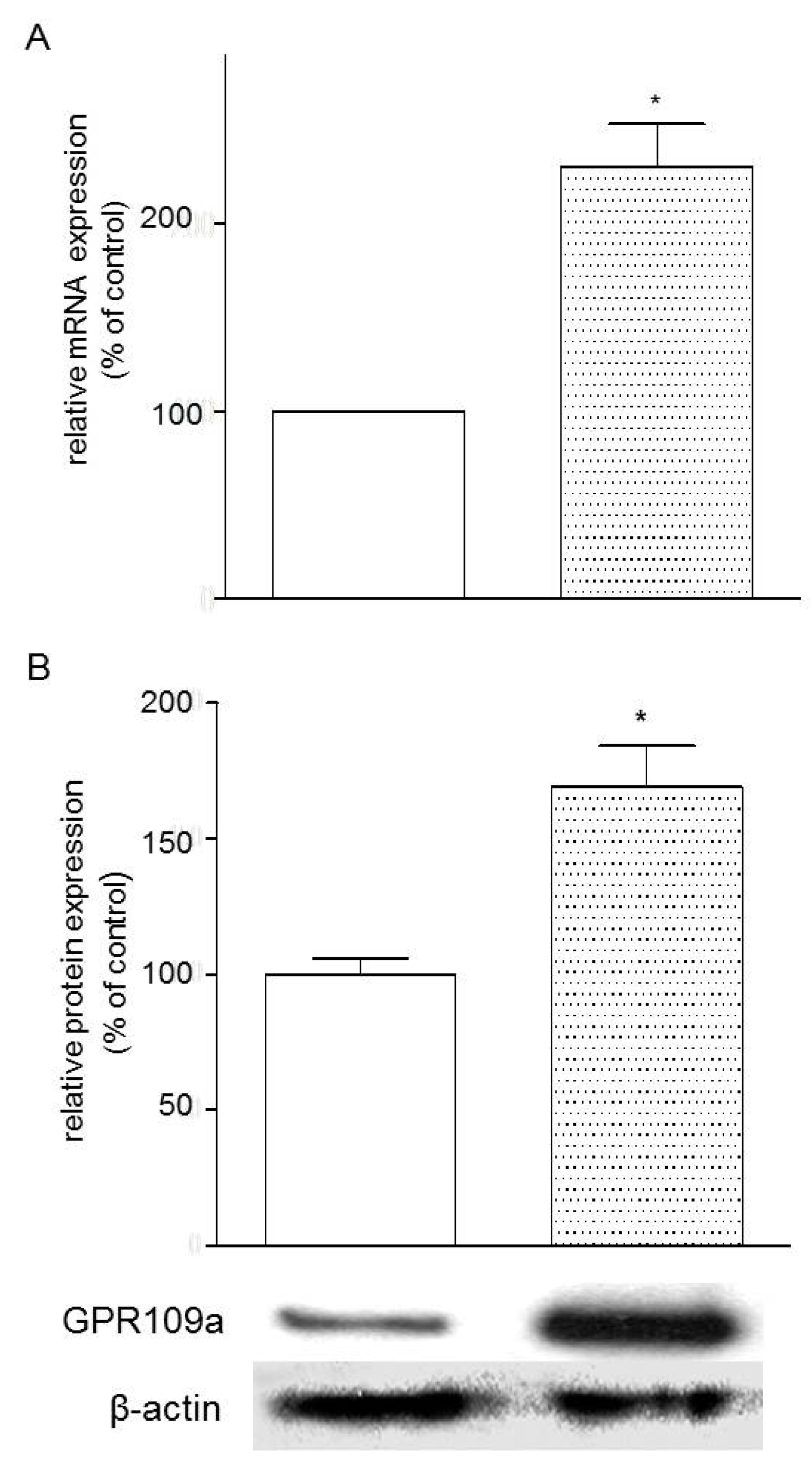
3.4. Effects of Glucose Concentrations on the Expression of GPR109a in Caco-2 Cells
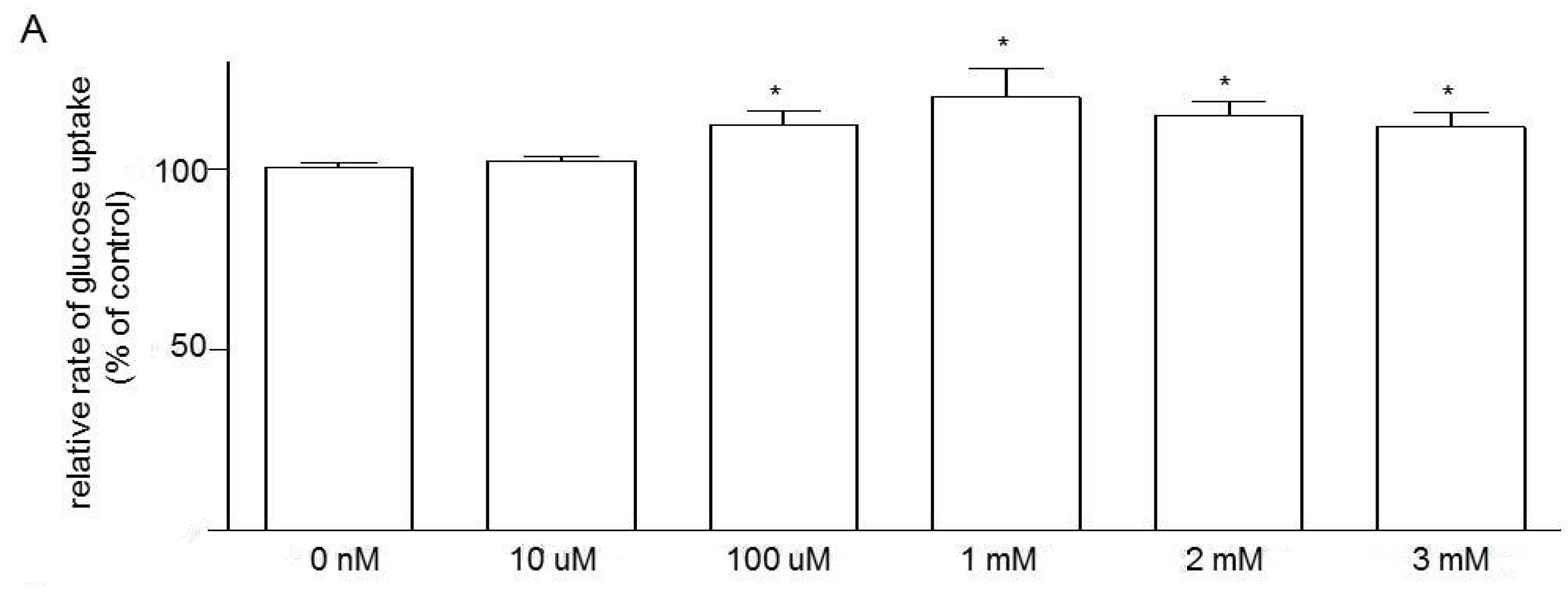
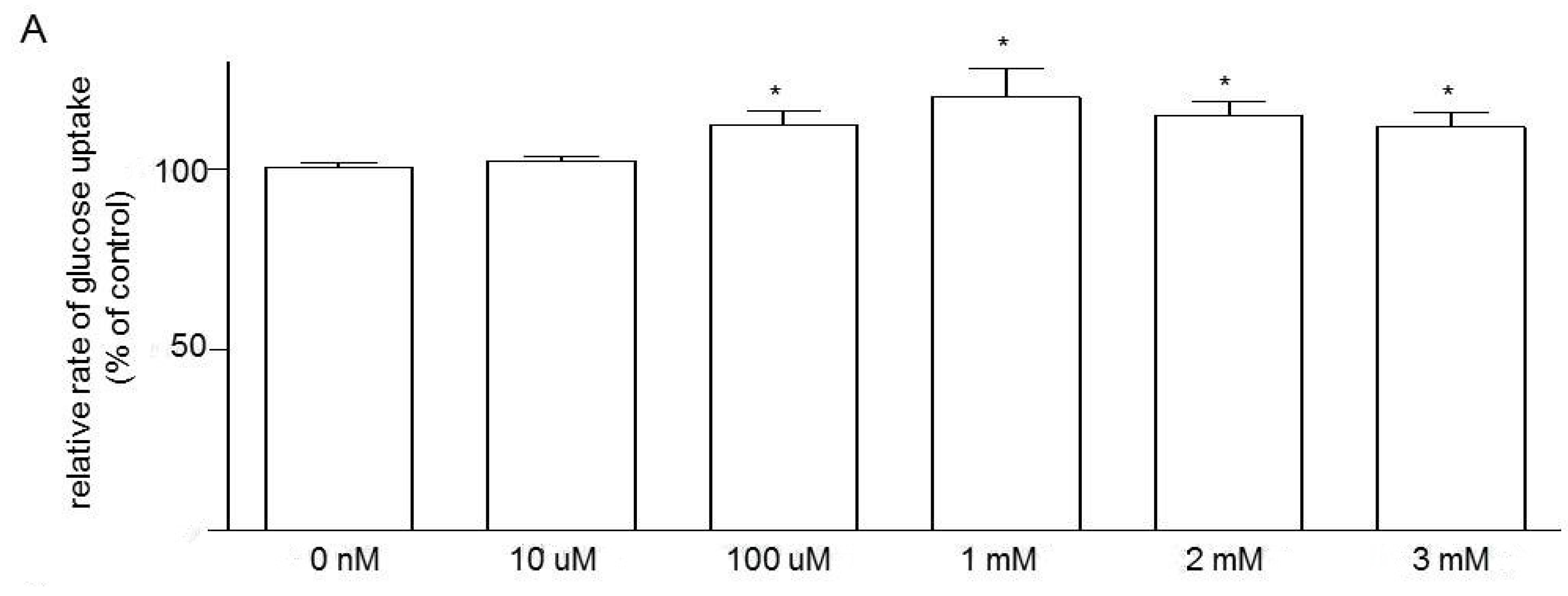
3.5. Effects of Niacin and Glucose Transporter Inhibitors on Glucose Uptake in Caco-2 Cells

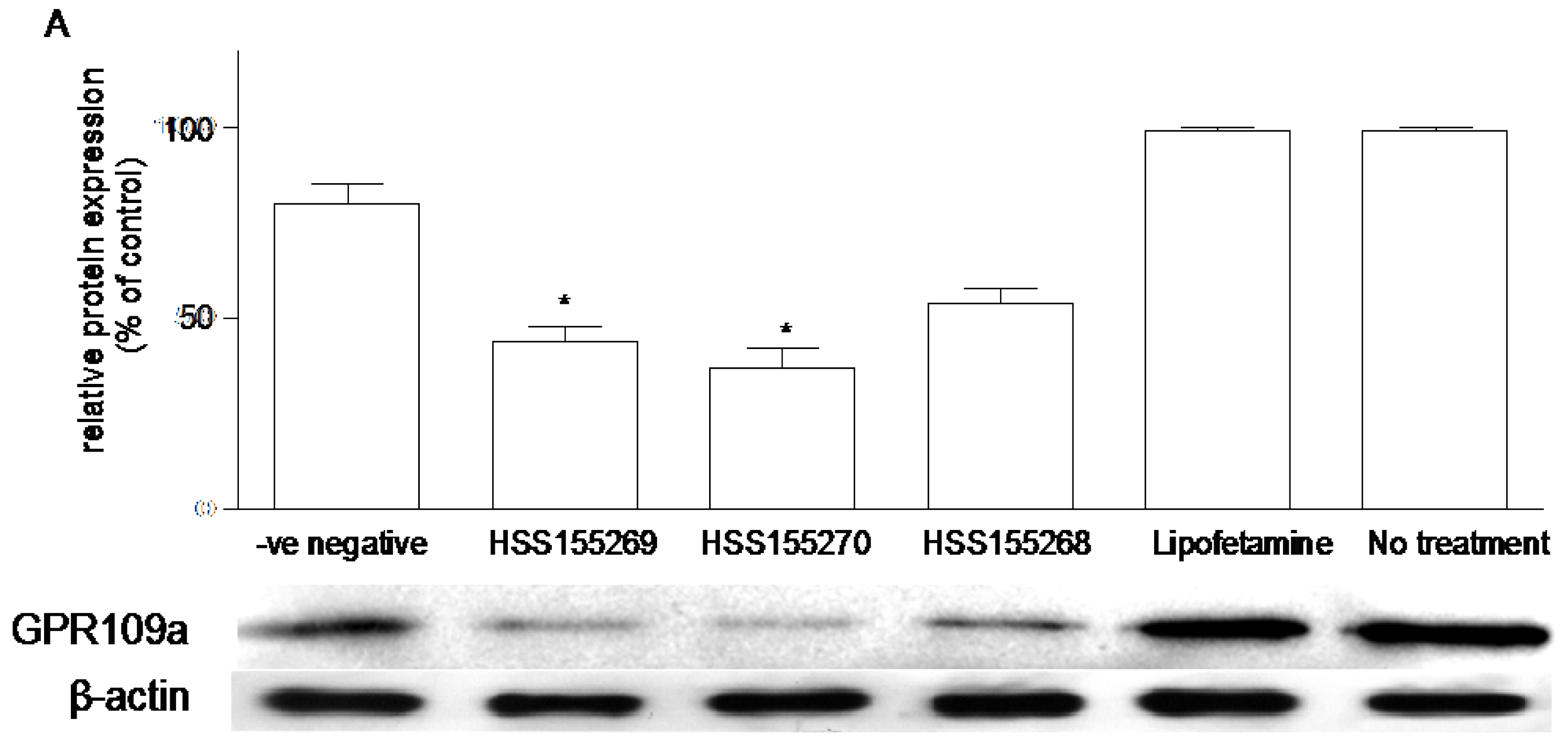
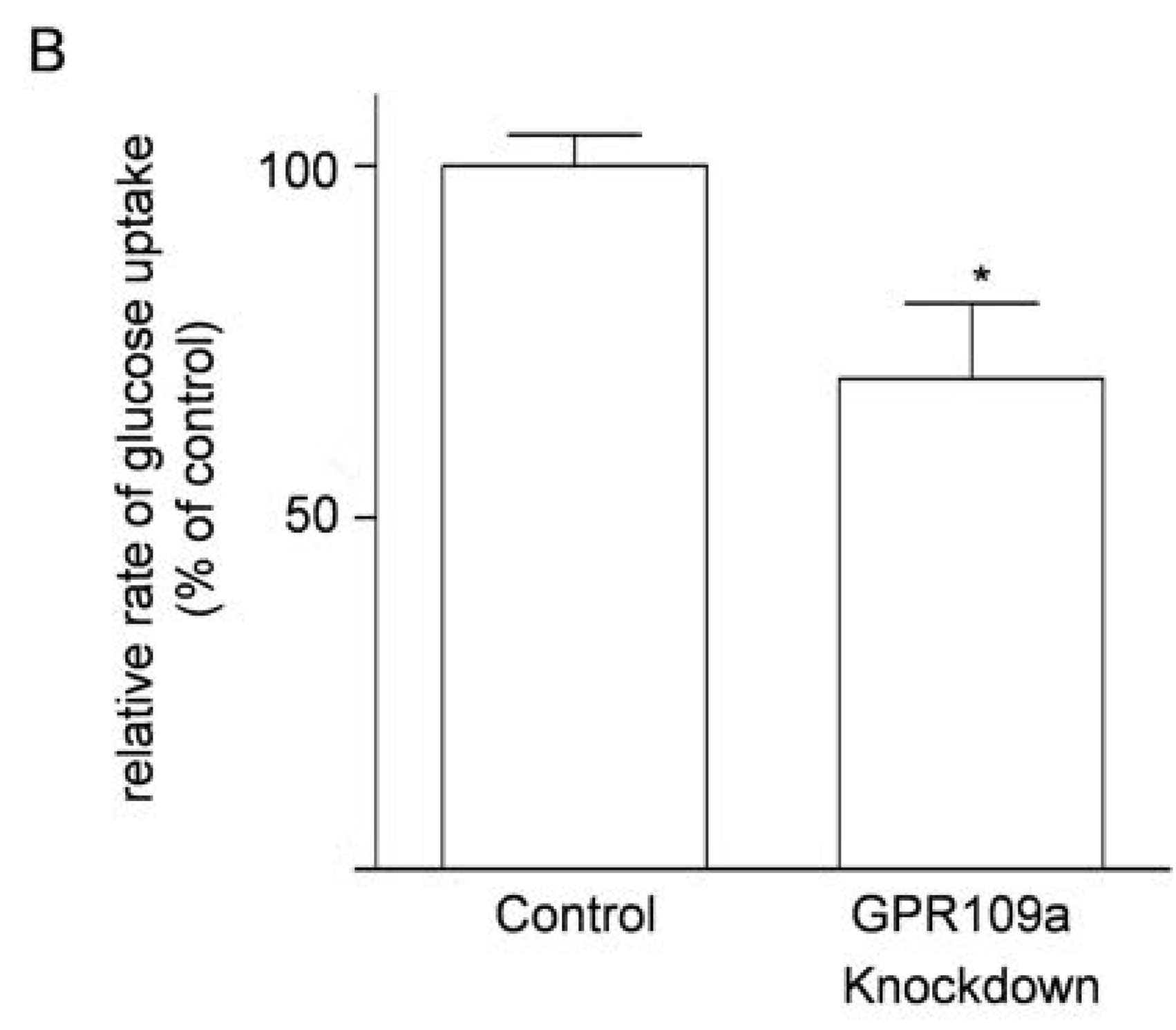
3.6. Effects of Knockdown of GPR109a on Glucose Uptake in Caco-2 Cells
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sprafka, J.M.; Burke, G.L.; Folsom, A.R.; McGovern, P.G.; Hahn, L.P. Trends in prevalence of diabetes mellitus in patients with myocardial infarction and effect of diabetes on survival. The Minnesota Heart Survey. Diabetes Care 1991, 14, 537–543. [Google Scholar] [CrossRef]
- Haffner, S.M.; Lehto, S.; Rönnemaa, T.; Pyörälä, K.; Laakso, M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N. Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef]
- Expert Panel on Detection. Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [Google Scholar]
- Guyton, J.R.; Goldberg, A.C.; Kreisberg, R.A.; Sprecher, D.L.; Superko, H.R.; O’Connor, C.M. Effectiveness of once-nightly dosing of extended release niacin alone and in combination for hypercholesterolemia. Am. J. Cardiol. 1998, 82, 737–743. [Google Scholar] [CrossRef]
- Phan, B.A.; Muñoz, L.; Shadzi, P.; Isquith, D.; Triller, M.; Brown, B.G.; Zhao, X.Q. Effects of niacin on glucose levels, coronary stenosis progression, and clinical events in subjects with normal baseline glucose levels (<100 mg/dL): A combined analysis of the Familial Atherosclerosis Treatment Study (FATS), HDL-Atherosclerosis Treatment Study (HATS), Armed Forces Regression Study (AFREGS), and Carotid Plaque Composition by MRI during lipid-lowering (CPC) study. Am. J. Cardiol. 2013, 111, 352–355. [Google Scholar]
- Hochholzer, W.; Berg, D.D.; Giugliano, R.P. The facts behind niacin. Ther. Adv. Cardiovasc. Dis. 2011, 5, 227–240. [Google Scholar] [CrossRef]
- Mcgovern, M.E. Use of nicotinic acid in patients with elevated fasting glucose, diabetes, or metabolic syndrome. Br. J. Diabetes Vasc. Dis. 2004, 4, 78. [Google Scholar] [CrossRef]
- Victoria, C.R.; Meneghelli, U.G. Kinetics of jejunal glucose transport in niacin deficient rats. Arq. Gastroenterol. 1990, 27, 191–196. [Google Scholar]
- Victoria, C.R. Jejunal electrogenic transport of glucose in rats with niacin deficiency. Arq. Gastroenterol. 1994, 31, 18–23. [Google Scholar]
- Chen, L.; So, W.Y.; Li, Y.T.; Cheng, Q.; Boucher, B.J.; Leung, P.S. Niacin-induced hyperglycemia is partially mediated via niacin receptor GPR109a in pancreatic islets. Mol. Cell. Endocrinol. 2015, 404, 56–66. [Google Scholar] [CrossRef]
- Soga, T.; Kamohara, M.; Takasaki, J.; Matsumoto, S.; Saito, T.; Ohishi, T.; Hiyama, H.; Matsuo, A.; Matsushime, H.; Furuichi, K. Molecular identification of nicotinic acid receptor. Biochem. Biophys. Res. Commun. 2003, 303, 364–369. [Google Scholar] [CrossRef]
- Tunaru, S.; Kero, J.; Schaub, A.; Wufka, C.; Blaukat, A.; Pfeffer, K.; Offermanns, S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat. Med. 2003, 9, 352–355. [Google Scholar] [CrossRef]
- Wise, A.; Foord, S.M.; Fraser, N.J.; Barnes, A.A.; Elshourbagy, N.; Eilert, M.; Ignar, D.M.; Murdock, P.R.; Steplewski, K.; Green, A.; et al. Molecular identification of high and low affinity receptors for nicotinic acid. J. Biol. Chem. 2003, 278, 9869–9874. [Google Scholar] [CrossRef]
- Offermanns, S. The nicotinic acid receptor GPR109A (HM74A or PUMA-G) as a new therapeutic target. Trends Pharmacol. Sci. 2006, 7, 384–390. [Google Scholar] [CrossRef]
- Zellner, C.; Pullinger, C.R.; Aouizerat, B.E.; Frost, P.H.; Kwok, P.Y.; Malloy, M.J.; Kane, J.P. Variations in human HM74 (GPR109B) and HM74A (GPR109A) niacin receptors. Hum. Mutat. 2005, 25, 18–21. [Google Scholar] [CrossRef]
- Cresci, G.A.; Thangaraju, M.; Mellinger, J.D.; Liu, K.; Ganapathy, V. Colonic gene expression in conventional and germ-free mice with a focus on the butyrate receptor GPR109A and the butyrate transporter SLC5A8. J. Gastrointest. Surg. 2010, 14, 449–461. [Google Scholar] [CrossRef]
- Sharp, P.A.; Boyer, S.; Srai, S.K.; Baldwin, S.A.; Debnam, E.S. Early diabetes-induced changes in rat jejunal glucose transport and the response to insulin. J. Endocrinol. 1997, 154, 19–25. [Google Scholar] [CrossRef]
- Kellett, G.L.; Helliwell, P.A. The diffusive component of intestinal glucose absorption is mediated by the glucose-induced recruitment of GLUT2 to the brush-border membrane. Biochem. J. 2000, 350, 155–162. [Google Scholar] [CrossRef]
- Kellett, G.L. The facilitated component of intestinal glucose absorption. J. Physiol. 2001, 531, 585–595. [Google Scholar] [CrossRef]
- Tobin, V.; le Gall, M.; Fioramonti, X.; Stolarczyk, E.; Blazquez, A.G.; Klein, C.; Prigent, M.; Serradas, P.; Cuif, M.H.; Magnan, C.; et al. Insulin internalizes GLUT2 in the enterocytes of healthy but not insulin-resistant mice. Diabetes 2008, 57, 55–62. [Google Scholar] [CrossRef]
- Kellett, G.L.; Brot-Laroche, E. Apical GLUT2: A major pathway of intestinal sugar absorption. Diabetes 2005, 54, 3056–3062. [Google Scholar] [CrossRef]
- Del Castillo, J.R. The use of hyperosmolar, intracellular-like solutions for the isolation of epithelial cells from guinea-pig small intestine. Biochim. Biophys. Acta 1987, 901, 201–208. [Google Scholar] [CrossRef]
- Wong, T.P.; Debnam, E.S.; Leung, P.S. Involvement of an enterocyte renin-angiotensin system in the local control of SGLT1-dependent glucose uptake across the rat small intestinal brush border membrane. J. Physiol. 2007, 584, 613–623. [Google Scholar] [CrossRef]
- Wong, T.P.; Debnam, E.S.; Leung, P.S. Diabetes mellitus and expression of the enterocyte renin-angiotensin system: Implications for control of glucose transport across the brush border membrane. Am. J. Physiol. Cell Physiol. 2009, 297, 601–610. [Google Scholar] [CrossRef]
- Ip, S.P.; Wong, T.P.; Tsai, S.J.; Leung, P.S. The recovery of some components of the renin angiotensin system in the rat pancreas after chronic exposure to hypoxic condition. J. Mol. Endocrinol. 2003, 3, 563–571. [Google Scholar] [CrossRef]
- Chu, K.Y.; Leung, P.S. Angiotensin II Type 1 receptor antagonism mediates uncoupling protein 2-driven oxidative stress and ameliorates pancreatic islet β-cell function in young Type 2 diabetic mice. Antioxid. Redox. Signal. 2007, 9, 869–878. [Google Scholar] [CrossRef]
- Lau, T.; Carlsson, P.O.; Leung, P.S. Evidence for a local angiotensin-generating system and dose-dependent inhibition of glucose-stimulated insulin release by angiotensin II in isolated pancreatic islets. Diabetologia 2004, 47, 240–248. [Google Scholar] [CrossRef]
- Debnam, E.S.; Smith, M.W.; Sharp, P.A.; Srai, S.K.; Turvey, A.; Keable, S.J. The effects of streptozotocin diabetes on sodium-glucose transporter (SGLT1) expression and function in rat jejunal and ileal villus-attached enterocytes. Pflugers 1995, 430, 151–159. [Google Scholar] [CrossRef]
- Karasov, W.H.; Pond, R.S.; Solberg, D.H.; Diamond, J.M. Regulation of proline and glucose transport in mouse intestine by dietary substrate levels. Proc. Natl. Acad. Sci. USA 1983, 80, 7674–7677. [Google Scholar] [CrossRef]
- Gilbert, T.; Rodriguez-Boulan, E. Induction of vacuolar apical compartments in the Caco-2 intestinal epithelial cell line. J. Cell Sci. 1991, 100, 451–458. [Google Scholar]
- Jumarie, C.; Malo, C. Caco-2 cells cultured in serum-free medium as a model for the study of enterocytic differentiation in vitro. J. Cell Physiol. 1991, 149, 24–33. [Google Scholar] [CrossRef]
- Gambhir, D.; Ananth, S.; Veeranan-Karmegam, R.; Elangovan, S.; Hester, S.; Jennings, E.; Offermanns, S.; Nussbaum, J.J.; Smith, S.B.; Thangaraju, M.; et al. GPR109A as an anti-inflammatory receptor in retinal pigment epithelial cells and its relevance to diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 2012, 53, 2208–2217. [Google Scholar] [CrossRef]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of GPR109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef]
- Chai, J.T.; Digby, J.E.; Choudhury, R.P. GPR109A and Vascular inflammation. Curr. Atheroscler. Rep. 2013, 15, 325. [Google Scholar] [CrossRef]
- Wong, T.P.; Ho, K.Y.; Ng, E.K.W.; Debnam, E.S.; Leung, P.S. Upregulation of ACE2-ANG-(1–7)-Mas axis in jejunal enterocytes of type 1 diabetic rats: Implications for glucose transport. Am. J. Physiol. Endocrinol. 2012, 303, E669–E681. [Google Scholar] [CrossRef]
- Bechgaard, H.; Jespersen, S. GI absorption of niacin in humans. J. Pharm. Sci. 1977, 66, 871–872. [Google Scholar] [CrossRef]
- Johnson, R.L.; Ghishan, F.K.; Kaunitz, J.D.; Merchant, J.L.; Said, H.M.; Wood, J.D. Physiology of the Gastrointestinal Tract, 5th ed.; Elsevier Inc.: San Diego, CA, USA, 2012; Volume 2. [Google Scholar]
- Taylor, W.M.; Mak, M.L.; Halperin, M.L. Effect of 3′,5′-cyclic AMP on glucose transport in rat adipocytes. Proc. Natl. Acad. Sci. USA 1976, 73, 4359–4363. [Google Scholar] [CrossRef]
- Taylor, W.M.; Halperin, M.L. Stimulation of glucose transport in rat adipocytes by insulin, adenosine, nicotinic acid and hydrogen peroxide. Role of adenosine 3′,5′-cyclic monophosphate. Biochem. J. 1979, 178, 381–389. [Google Scholar] [CrossRef]
- Stümpel, F.; Scholtka, B.; Jungermann, K. A new role for enteric glucagon-37: Acute stimulation of glucose absorption in rat small intestine. FEBS Lett. 1997, 410, 515–519. [Google Scholar] [CrossRef]
- Lee, W.Y.; Loflin, P.; Clancey, C.J.; Peng, H.; Lever, J.E. Cyclic nucleotide regulation of Na+/glucose cotransporter (SGLT1) mRNA stability. Interaction of a nucleocytoplasmic protein with a regulatory domain in the 3′-untranslated region critical for stabilization. J. Biol. Chem. 2000, 275, 33998–34008. [Google Scholar] [CrossRef]
- Rencurel, F.; Waeber, G.; Bonny, C.; Antoine, B.; Maulard, P.; Girard, J.; Leturque, A. cAMP prevents the glucose-mediated stimulation of GLUT2 gene transcription in hepatocytes. Biochem. J. 1997, 322, 441–448. [Google Scholar] [CrossRef]
- Li, G.; Deng, X.; Wu, C.; Zhou, Q.; Chen, L.; Shi, Y.; Huang, H.; Zhou, N. Distinct kinetic and spatial patterns of protein kinase C (PKC)- and epidermal growth factor receptor (EGFR)-dependent activation of extracellular signal-regulated kinases 1 and 2 by human nicotinic acid receptor GPR109A. J. Biol. Chem. 2011, 286, 31199–31212. [Google Scholar] [CrossRef]
- Cohen, M.; Kitsberg, D.; Tsytkin, S.; Shulman, M.; Aroeti, B.; Nahmias, Y. Live imaging of GLUT2 glucose dependent trafficking and its inhibition in polarized epithelial cysts. Open Biol. 2014, 4. [Google Scholar] [CrossRef]
- Thorens, B.; Dériaz, N.; Bosco, D.; DeVos, A.; Pipeleers, D.; Schuit, F.; Meda, P.; Porret, A. Protein kinase A-dependent phosphorylation of GLUT2 in pancreatic β cells. J. Biol. Chem. 1996, 271, 8075–8081. [Google Scholar]
- Wright, E.M.; Hirsch, J.R.; Loo, D.D.; Zampighi, G.A. Regulation of Na+/glucose cotransporters. J. Exp. Biol. 1997, 200, 287–293. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, T.P.; Chan, L.K.Y.; Leung, P.S. Involvement of the Niacin Receptor GPR109a in the LocalControl of Glucose Uptake in Small Intestine of Type 2Diabetic Mice. Nutrients 2015, 7, 7543-7561. https://doi.org/10.3390/nu7095352
Wong TP, Chan LKY, Leung PS. Involvement of the Niacin Receptor GPR109a in the LocalControl of Glucose Uptake in Small Intestine of Type 2Diabetic Mice. Nutrients. 2015; 7(9):7543-7561. https://doi.org/10.3390/nu7095352
Chicago/Turabian StyleWong, Tung Po, Leo Ka Yu Chan, and Po Sing Leung. 2015. "Involvement of the Niacin Receptor GPR109a in the LocalControl of Glucose Uptake in Small Intestine of Type 2Diabetic Mice" Nutrients 7, no. 9: 7543-7561. https://doi.org/10.3390/nu7095352
APA StyleWong, T. P., Chan, L. K. Y., & Leung, P. S. (2015). Involvement of the Niacin Receptor GPR109a in the LocalControl of Glucose Uptake in Small Intestine of Type 2Diabetic Mice. Nutrients, 7(9), 7543-7561. https://doi.org/10.3390/nu7095352