Per-Arnt-Sim Kinase (PASK): An Emerging Regulator of Mammalian Glucose and Lipid Metabolism

Abstract
:1. Introduction
1.1. PAS Kinase Structure and Function
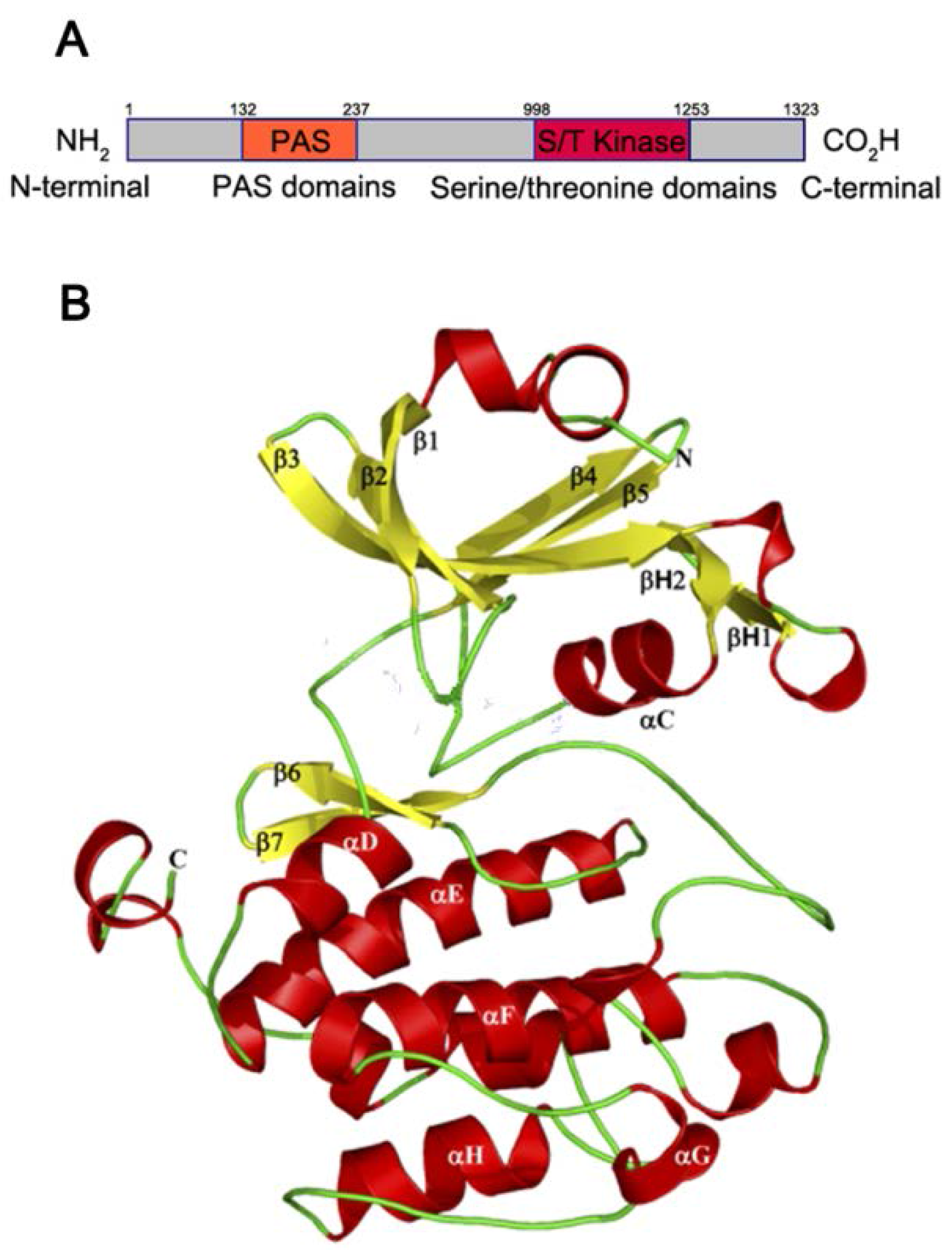
1.2. PAS Kinase Activation

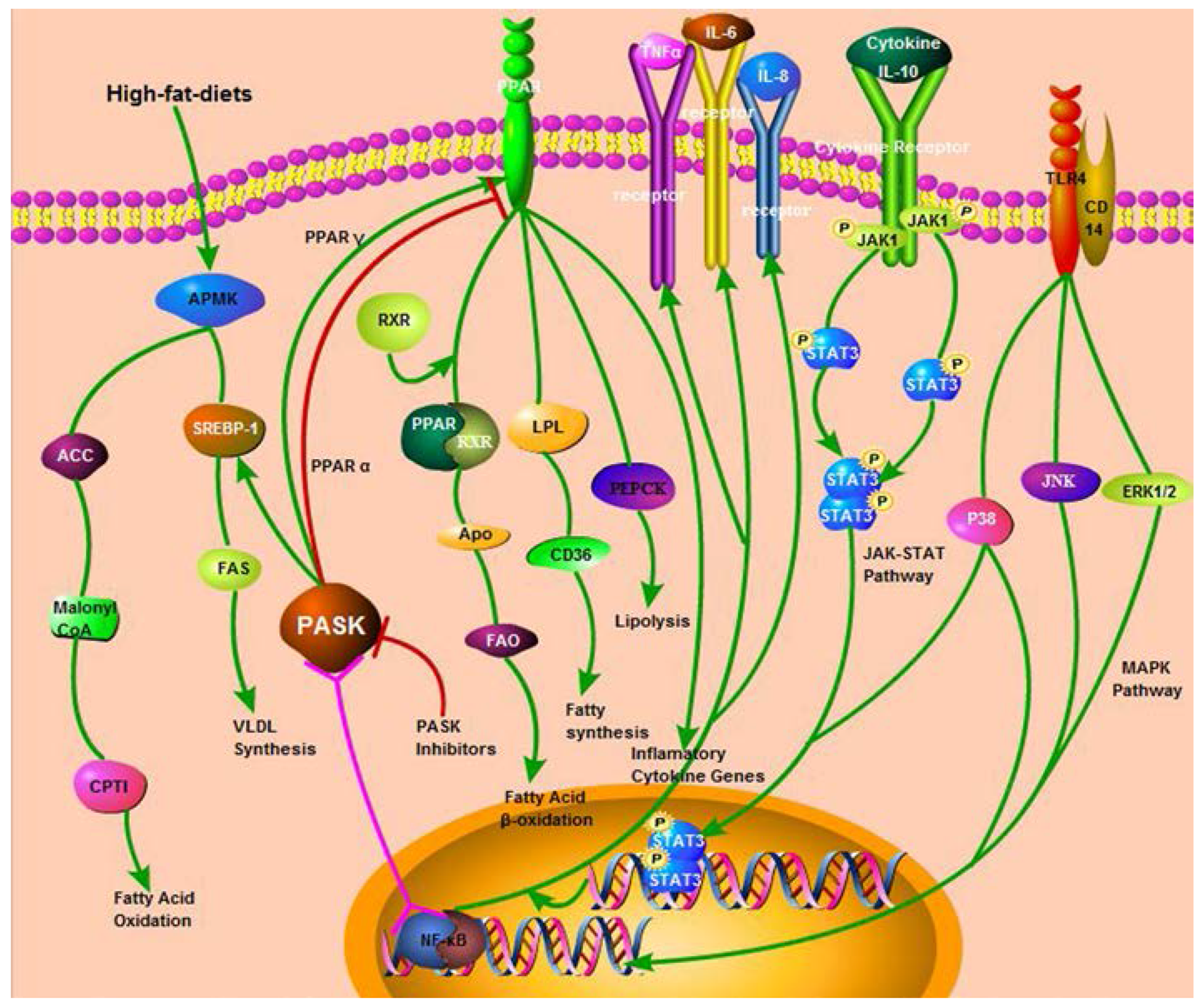
2. The Function and Mechanism of PASK in Mammalian Glucose and Lipid Metabolism
2.1. The Regulation of PASK in Mammalian Glucose Metabolism

{kind=link}
{kind=link}
{kind=link}
| Name | Expression Level | Biological Function | Reference |
|---|---|---|---|
| Insulin | Up-regulated | Promotes the absorption of glucose from the blood to skeletal muscles and fat tissue; causes fat to be stored. | [25,26,27] |
| Glucagon | Down-regulated | Elevates the concentration of glucose in the blood by promoting gluconeogenesis and glycogenolysis. | [17] |
| Glycogen synthase (GS) | Down-regulated | A key enzyme in glycogenesis; involves in converting glucose to glycogen. | [26,28] |
| GSK-3β | Down-regulated | Phosphorylates and inactivates its downstream target GS; active in a number of central intracellular signaling pathways (cellular proliferation, migration, inflammation, immune responses, glucose homeostasis, and apoptosis) | [29] |
2.1.1. PASK and Pancreatic α Cells
2.1.2. PASK and Pancreatic β Cells
2.1.3. PASK and Glycogen Synthase
2.2. The Regulation of PASK in Mammalian Lipid Metabolism
| Name | Expression Level | Biological Function | Reference |
|---|---|---|---|
| AMPK | Up-regulated | Stimulates hepatic fatty acid oxidation and ketogenesis; inhibits cholesterol synthesis, lipogenesis, and triglyceride synthesis; inhibits adipocyte lipolysis and lipogenesis; stimulates skeletal muscle fatty acid oxidation and muscle glucose uptake, modulates insulin secretion. | [9,40] |
| SREBP-1c | Down-regulated | Promotes cholesterol biosynthesis and uptake; stimulates fatty acid biosynthesis. | [37] |
| FAS | Down-regulated | An enzyme complex responsible for fatty acid biosynthesis; catalyzes the synthesis of palmitate from acetyl-CoA and malonyl-CoA. | [6,33] |
| CD36/FAT | Down-regulated | Functions in long-chain fatty acid uptake and signaling; promotes sterile inflammation. | [6,33] |
| SCD1 | Down-regulated | A key enzyme in fatty acid metabolism, catalyzes a rate-limiting step in the synthesis of unsaturated fatty acids. | [6,33] |
| PPARγ | Down-regulated | Regulates fatty acid storage and glucose metabolism; stimulates lipid uptake and adipogenesis. | [6,33] |
2.2.1. PASK and AMPK/SREBP-1
2.2.2. PASK and other Enzymes or Receptors Involved in Lipid Metabolism
3. Future Directions
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ford, E.S. Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome: A summary of the evidence. Diabetes Care 2005, 28, 1769–1778. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Jung, D.J.; Cho, K.H.; Park, J.W.; Yoon, K.W.; Do, J.Y. The Association between Metabolic Syndrome or Chronic Kidney Disease and Hearing Thresholds in Koreans: The Korean National Health and Nutrition Examination Survey 2009–2012. PLoS ONE 2015, 10, e0120372. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [PubMed]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Grose, J.H.; Rutter, J. The role of PAS kinase in PASsing the glucose signal. Sensors (Basel) 2010, 10, 5668–5682. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.-X.; Rutter, J. The role of PAS kinase in regulating energy metabolism. IUBMB Life 2008, 60, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Rutter, J.; Michnoff, C.H.; Harper, S.M.; Gardner, K.H.; McKnight, S.L. PAS kinase: An evolutionarily conserved PAS domain-regulated serine/threonine kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 8991–8996. [Google Scholar] [CrossRef] [PubMed]
- Beausoleil, S.A.; Jedrychowski, M.; Schwartz, D.; Elias, J.E.; Villen, J.; Li, J.; Cohn, M.A.; Cantley, L.C.; Gygi, S.P. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12130–12135. [Google Scholar] [CrossRef] [PubMed]
- Hurtado-Carneiro, V.; Roncero, I.; Blazquez, E.; Alvarez, E.; Sanz, C. PAS kinase as a nutrient sensor in neuroblastoma and hypothalamic cells required for the normal expression and activity of other cellular nutrient and energy sensors. Mol. Neurobiol. 2013, 48, 904–920. [Google Scholar] [CrossRef] [PubMed]
- Katschinski, D.M.; Marti, H.H.; Wagner, K.F.; Shibata, J.; Eckhardt, K.; Martin, F.; Depping, R.; Paasch, U.; Gassmann, M.; Ledermann, B.; et al. Targeted disruption of the mouse PAS domain serine/threonine kinase PASKIN. Mol. Cell. Biol. 2003, 23, 6780–6789. [Google Scholar] [CrossRef] [PubMed]
- Fritz, W.A.; Lin, T.M.; Peterson, R.E. The aryl hydrocarbon receptor (AhR) inhibits vanadate-induced vascular endothelial growth factor (VEGF) production in TRAMP prostates. Carcinogenesis 2008, 29, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, K.; Troger, J.; Reissmann, J.; Katschinski, D.M.; Wagner, K.F.; Stengel, P.; Paasch, U.; Hunziker, P.; Borter, E.; Barth, S.; et al. Male germ cell expression of the PAS domain kinase PASKIN and its novel target eukaryotic translation elongation factor eEF1A1. Cell. Physiol. Biochem. 2007, 20, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Huh, W.K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O’Shea, E.K. Global analysis of protein localization in budding yeast. Nature 2003, 425, 686–691. [Google Scholar] [CrossRef] [PubMed]
- DeMille, D.; Grose, J.H. PAS kinase: A nutrient sensing regulator of glucose homeostasis. IUBMB Life 2013, 65, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Cardon, C.M.; Rutter, J. PAS kinase: Integrating nutrient sensing with nutrient partitioning. Semin. Cell Dev. Biol. 2012, 23, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Cardon, C.M.; Beck, T.; Hall, M.N.; Rutter, J. PAS kinase promotes cell survival and growth through activation of Rho1. Sci. Signal. 2012, 5, ra9. [Google Scholar] [CrossRef] [PubMed]
- Schlafli, P.; Borter, E.; Spielmann, P.; Wenger, R.H. The PAS-domain kinase PASKIN: A new sensor in energy homeostasis. Cell. Mol. Life Sci. 2009, 66, 876–883. [Google Scholar] [CrossRef] [PubMed]
- DeMille, D.; Bikman, B.T.; Mathis, A.D.; Prince, J.T.; Mackay, J.T.; Sowa, S.W.; Hall, T.D.; Grose, J.H. A comprehensive protein-protein interactome for yeast PAS kinase 1 reveals direct inhibition of respiration through the phosphorylation of Cbf1. Mol. Biol. Cell 2014, 25, 2199–2215. [Google Scholar] [CrossRef] [PubMed]
- DeMille, D.; Badal, B.D.; Evans, J.B.; Mathis, A.D.; Anderson, J.F.; Grose, J.H. PAS kinase is activated by direct SNF1-dependent phosphorylation and mediates inhibition of TORC1 through the phosphorylation and activation of Pbp1. Mol. Biol. Cell 2015, 26, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Amezcua, C.A.; Harper, S.M.; Rutter, J.; Gardner, K.H. Structure and interactions of PAS kinase N-terminal PAS domain: Model for intramolecular kinase regulation. Structure 2002, 10, 1349–1361. [Google Scholar] [CrossRef]
- Moglich, A.; Ayers, R.A.; Moffat, K. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure 2009, 17, 1282–1294. [Google Scholar] [CrossRef] [PubMed]
- Kikani, C.K.; Antonysamy, S.A.; Bonanno, J.B.; Romero, R.; Zhang, F.F.; Russell, M.; Gheyi, T.; Iizuka, M.; Emtage, S.; Sauder, J.M.; et al. Structural bases of PAS domain-regulated kinase (PASK) activation in the absence of activation loop phosphorylation. J. Biol. Chem. 2010, 285, 41034–41043. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [PubMed]
- Rutter, J.; Probst, B.L.; McKnight, S.L. Coordinate regulation of sugar flux and translation by PAS kinase. Cell 2002, 111, 17–28. [Google Scholar] [CrossRef]
- Semplici, F.; Vaxillaire, M.; Fogarty, S.; Semache, M.; Bonnefond, A.; Fontes, G.; Philippe, J.; Meur, G.; Diraison, F.; Sessions, R.B.; et al. Human mutation within Per-Arnt-Sim (PAS) domain-containing protein kinase (PASK) causes basal insulin hypersecretion. J. Biol. Chem. 2011, 286, 44005–44014. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Xavier, G.; Rutter, J.; Rutter, G.A. Involvement of Per-Arnt-Sim (PAS) kinase in the stimulation of preproinsulin and pancreatic duodenum homeobox 1 gene expression by glucose. Proc. Natl. Acad. Sci. USA 2004, 101, 8319–8324. [Google Scholar] [CrossRef] [PubMed]
- Fontes, G.; Semache, M.; Hagman, D.K.; Tremblay, C.; Shah, R.; Rhodes, C.J.; Rutter, J.; Poitout, V. Involvement of Per-Arnt-Sim Kinase and extracellular-regulated kinases-1/2 in palmitate inhibition of insulin gene expression in pancreatic β-cells. Diabetes 2009, 58, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.A.; Skurat, A.V.; Probst, B.; de Paoli-Roach, A.; Roach, P.J.; Rutter, J. Control of mammalian glycogen synthase by PAS kinase. Proc. Natl. Acad. Sci. USA 2005, 102, 16596–16601. [Google Scholar] [CrossRef] [PubMed]
- Semache, M.; Zarrouki, B.; Fontes, G.; Fogarty, S.; Kikani, C.; Chawki, M.B.; Rutter, J.; Poitout, V. Per-Arnt-Sim kinase regulates pancreatic duodenal homeobox-1 protein stability via phosphorylation of glycogen synthase kinase 3β in pancreatic βcells. J. Biol. Chem. 2013, 288, 24825–24833. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.; Mouralidarane, A.; Ray, S.; Soeda, J.; Oben, J. Recent advancements in drug treatment of obesity. Clin. Med. 2012, 12, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Xavier, G.; Farhan, H.; Kim, H.; Caxaria, S.; Johnson, P.; Hughes, S.; Bugliani, M.; Marselli, L.; Marchetti, P.; Birzele, F.; et al. Per-arnt-sim (PAS) domain-containing protein kinase is downregulated in human islets in type 2 diabetes and regulates glucagon secretion. Diabetologia 2011, 54, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Hayes, H.L.; Moss, L.G.; Schisler, J.C.; Haldeman, J.M.; Zhang, Z.; Rosenberg, P.B.; Newgard, C.B.; Hohmeier, H.E. Pdx-1 activates islet α- and β-cell proliferation via a mechanism regulated by transient receptor potential cation channels 3 and 6 and extracellular signal-regulated kinases 1 and 2. Mol. Cell. Biol. 2013, 33, 4017–4029. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.X.; Cardon, C.M.; Swiatek, W.; Cooksey, R.C.; Smith, T.L.; Wilde, J.; Boudina, S.; Abel, E.D.; McClain, D.A.; Rutter, J. PAS kinase is required for normal cellular energy balance. Proc. Natl. Acad. Sci. USA 2007, 104, 15466–15471. [Google Scholar] [CrossRef] [PubMed]
- Eto, K.; Yamashita, T.; Matsui, J.; Terauchi, Y.; Noda, M.; Kadowaki, T. Genetic manipulations of fatty acid metabolism in β-cells are associated with dysregulated insulin secretion. Diabetes 2002, 51, S414–S420. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, P.E.; Rorsman, P. Per-arnt-sim (PAS) domain kinase (PASK) as a regulator of glucagon secretion. Diabetologia 2011, 54, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Skurat, A.V.; Dietrich, A.D. Phosphorylation of Ser640 in muscle glycogen synthase by DYRK family protein kinases. J. Biol. Chem. 2004, 279, 2490–2498. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Romero, D.; Swiatek, W.I.; Dorweiler, I.; Kikani, C.K.; Sabic, H.; Zweifel, B.S.; McKearn, J.; Blitzer, J.T.; Nickols, G.A.; et al. PAS kinase drives lipogenesis through SREBP-1 maturation. Cell Rep. 2014, 8, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Soliz, J.; Soulage, C.; Borter, E.; van Patot, M.T.; Gassmann, M. Ventilatory responses to acute and chronic hypoxia are altered in female but not male Paskin-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R649–R658. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.W.; Treebak, J.T.; Wojtaszewski, J.F.; Sakamoto, K. Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes 2011, 60, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.J.; Kwon, S.W.; Jung, B.H.; Oh, S.H.; Lee, B.H. Role of the AMPK/SREBP-1 pathway in the development of orotic acid-induced fatty liver. J. Lipid Res. 2011, 52, 1617–1625. [Google Scholar] [CrossRef] [PubMed]
- Rybakowska, I.; Slominska, E.M.; Romaszko, P.; Lipinski, M.; Zukowska, P.; Smolenski, R.T. Activity of AMP-regulated protein kinase and AMP-deaminase in the heart of mice fed high-fat diet. Nucleosides Nucleotides and Nucleic Acids 2014, 33, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Xu, M.; Fang, Q. Role of AMPKα in skeletal muscle glycometabolism regulation and adaptation in relation to sepsis. Biomed. Res. Int. 2014, 2014, 390760. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Hawley, S.A. AMP-activated protein kinase: The energy charge hypothesis revisited. Bioessays 2001, 23, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Leek, B.T.; Mudaliar, S.R.; Henry, R.; Mathieu-Costello, O.; Richardson, R.S. Effect of acute exercise on citrate synthase activity in untrained and trained human skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R441–R447. [Google Scholar] [PubMed]
- Li, W.; Li, Y.; Wang, Q.; Yang, Y. Crude extracts from Lycium barbarum suppress SREBP-1c expression and prevent diet-induced fatty liver through AMPK activation. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhai, Y.; Mu, Y.; Gong, H.; Uppal, H.; Toma, D.; Ren, S.; Evans, R.M.; Xie, W. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J. Biol. Chem. 2006, 281, 15013–15020. [Google Scholar] [CrossRef] [PubMed]
- Tagle Arrospide, M. Non-alcoholic fatty liver. Rev. Gastroenterol. Peru. 2003, 23, 49–57. [Google Scholar] [PubMed]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Marzocchi, R. Metabolic syndrome and NASH. Clin. Liver Dis. 2007, 11, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Chinetti, G.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors (PPARs): Nuclear receptors with functions in the vascular wall. Z. Kardiol. 2001, 90, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Jiao, M.; Ren, F.; Zhou, L.; Duan, Z.; Zhao, C. Roles of peroxisome proliferator-activated receptor-alpha in acute liver failure and its pathogenetic mechanism in mice. Zhonghua Yi Xue Za Zhi 2014, 94, 2059–2063. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, D.-d.; Zhang, J.-g.; Wang, Y.-z.; Liu, Y.; Liu, G.-l.; Li, X.-y. Per-Arnt-Sim Kinase (PASK): An Emerging Regulator of Mammalian Glucose and Lipid Metabolism. Nutrients 2015, 7, 7437-7450. https://doi.org/10.3390/nu7095347
Zhang D-d, Zhang J-g, Wang Y-z, Liu Y, Liu G-l, Li X-y. Per-Arnt-Sim Kinase (PASK): An Emerging Regulator of Mammalian Glucose and Lipid Metabolism. Nutrients. 2015; 7(9):7437-7450. https://doi.org/10.3390/nu7095347
Chicago/Turabian StyleZhang, Dan-dan, Ji-gang Zhang, Yu-zhu Wang, Ying Liu, Gao-lin Liu, and Xiao-yu Li. 2015. "Per-Arnt-Sim Kinase (PASK): An Emerging Regulator of Mammalian Glucose and Lipid Metabolism" Nutrients 7, no. 9: 7437-7450. https://doi.org/10.3390/nu7095347
APA StyleZhang, D.-d., Zhang, J.-g., Wang, Y.-z., Liu, Y., Liu, G.-l., & Li, X.-y. (2015). Per-Arnt-Sim Kinase (PASK): An Emerging Regulator of Mammalian Glucose and Lipid Metabolism. Nutrients, 7(9), 7437-7450. https://doi.org/10.3390/nu7095347