Intestinal Microbiota and Celiac Disease: Cause, Consequence or Co-Evolution?

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Host Immune–Microbiota Interactions
3. Host Genetics and Intestinal Microbiota
4. CD Genetics and Intestinal Microbiota
5. Epigenetics and Intestinal Microbiota: A New Emerging Field
6. Environmental Factors and Intestinal Microbiota
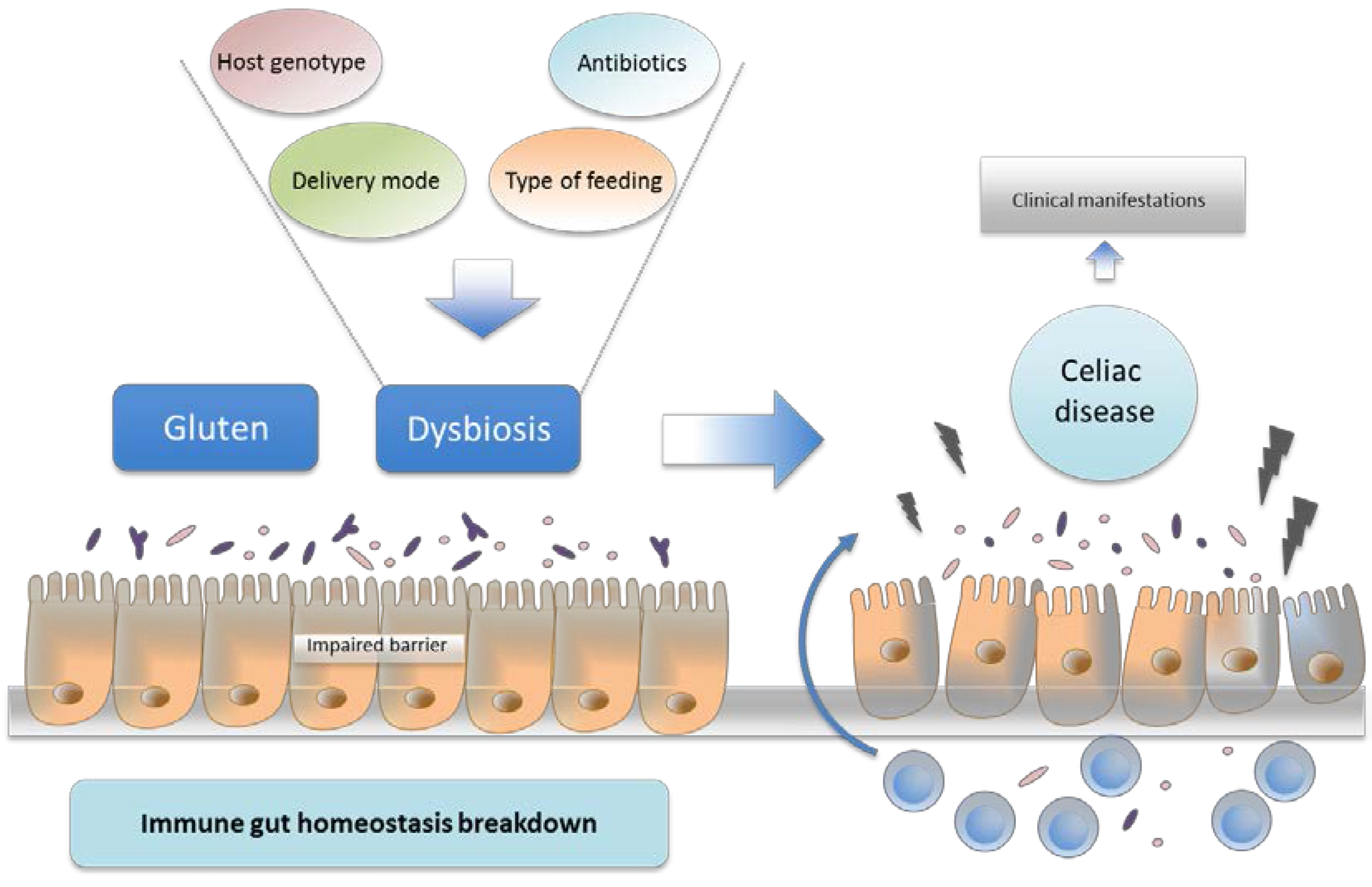
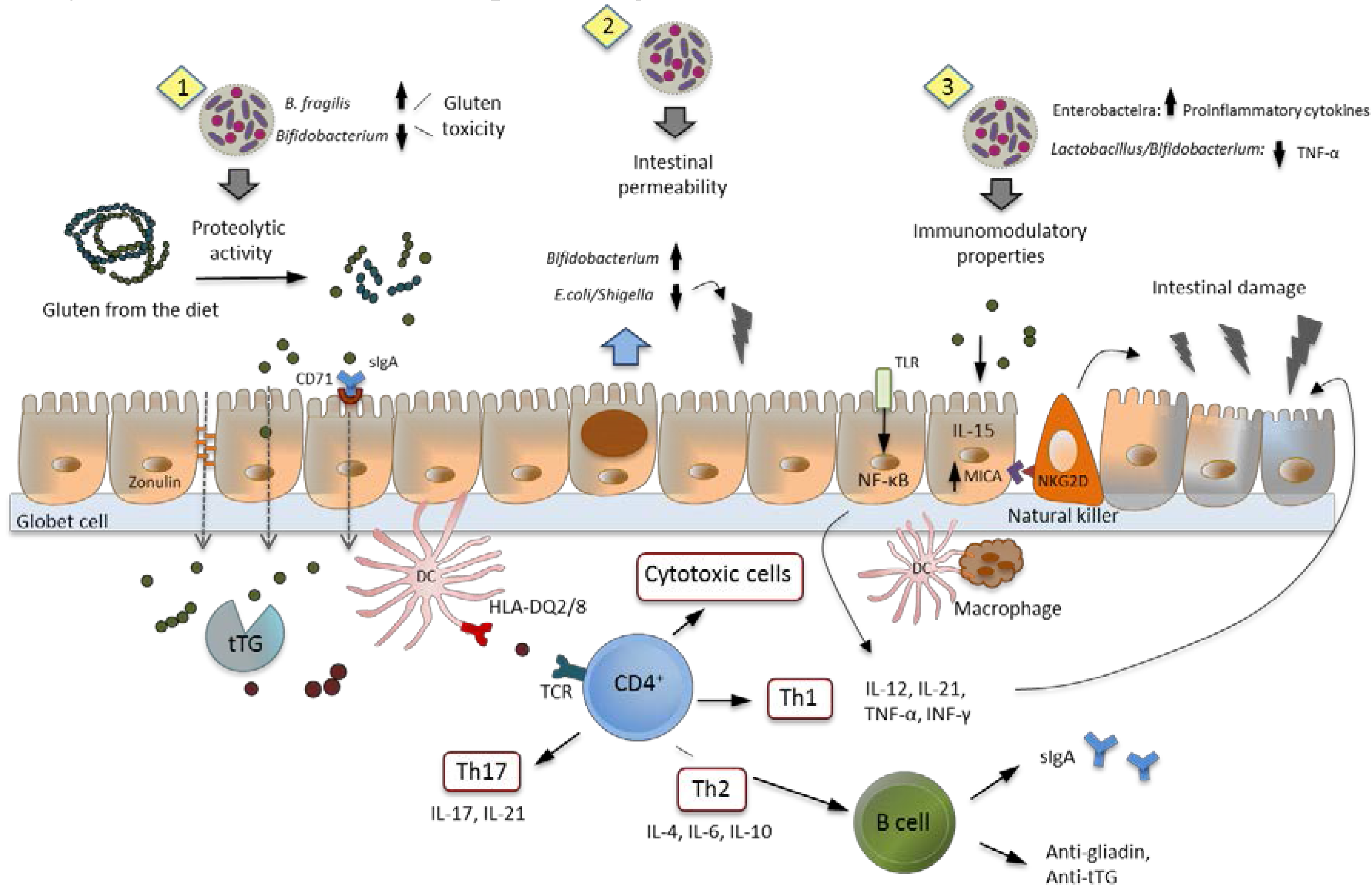
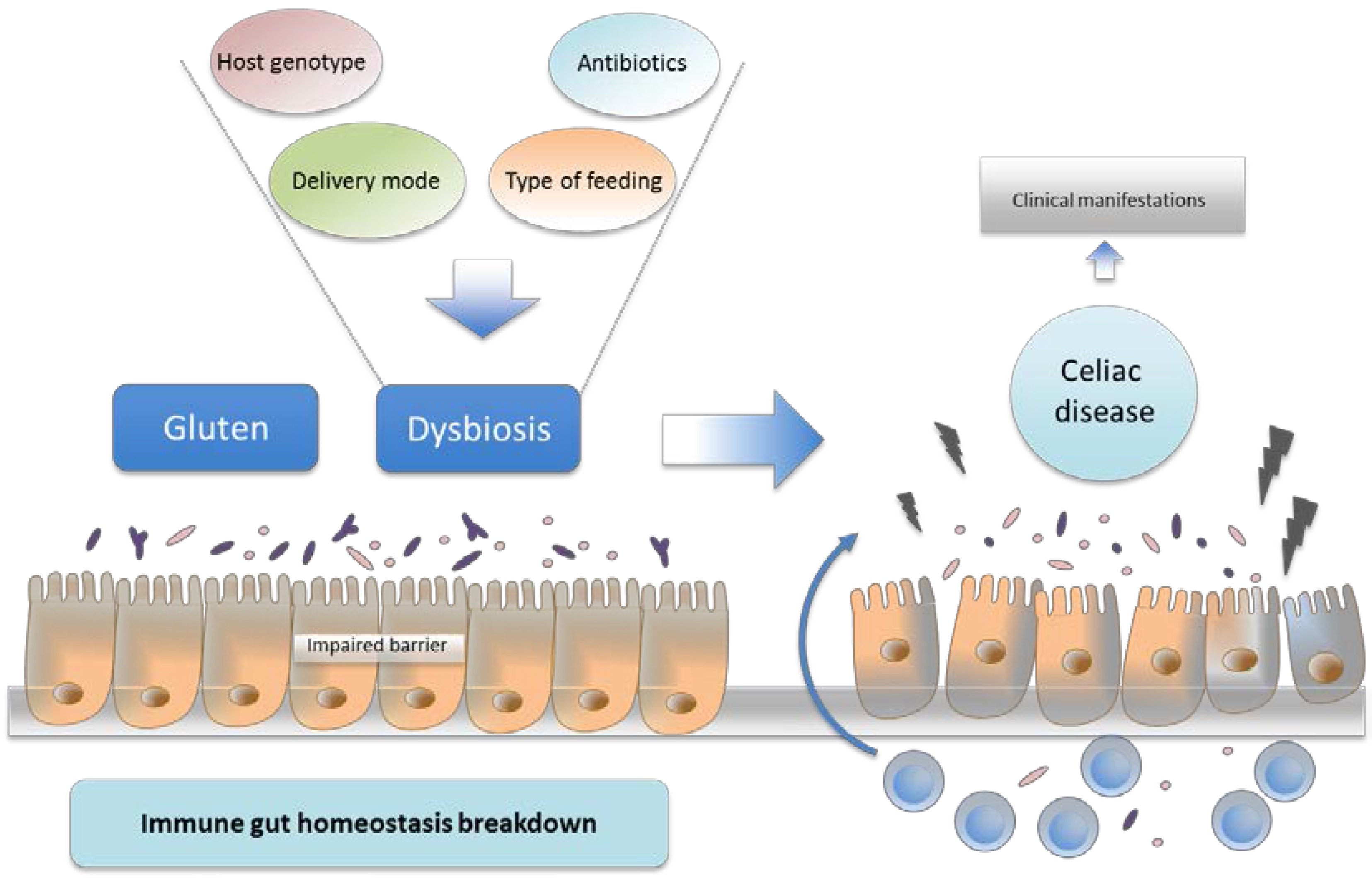
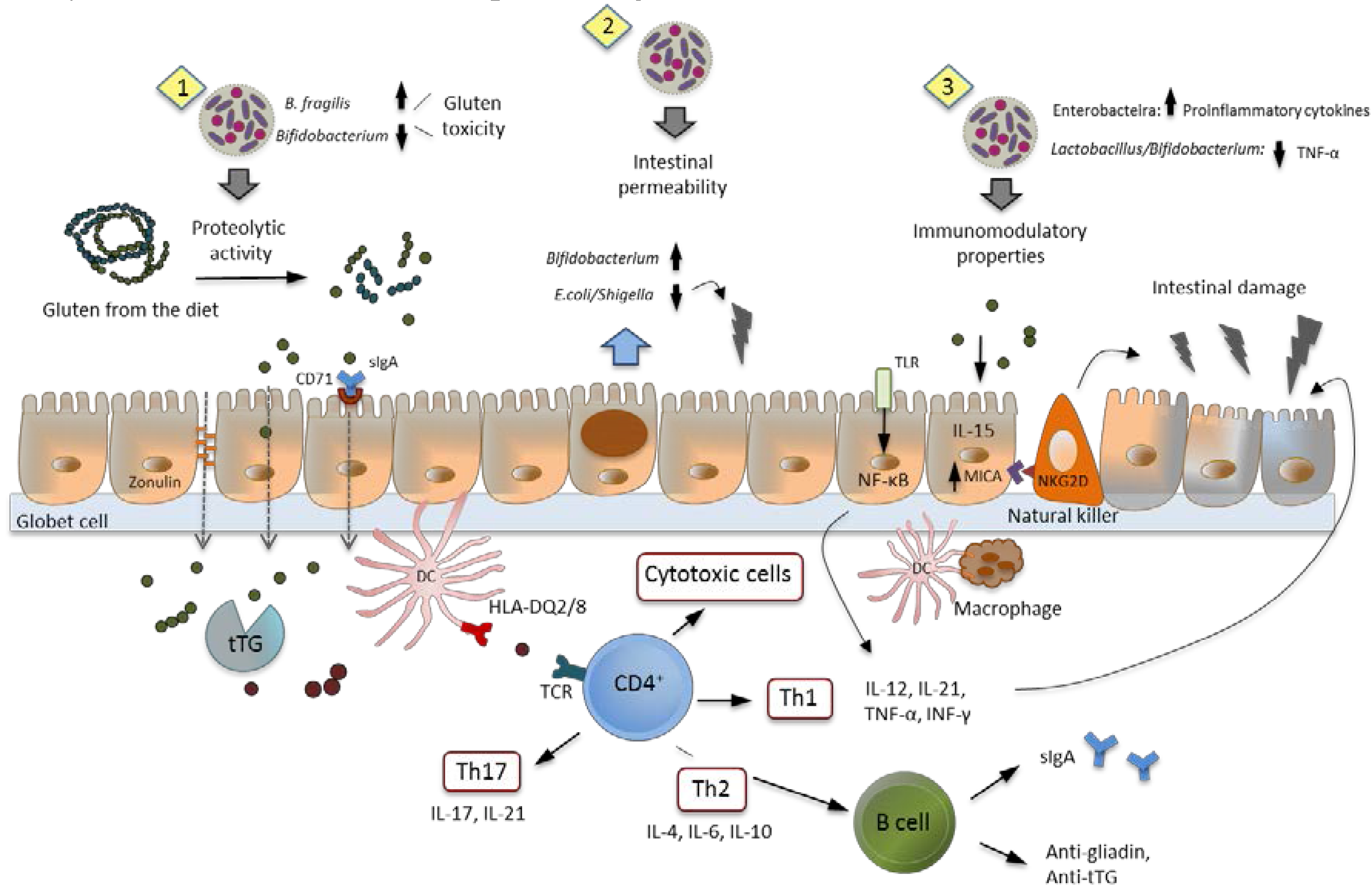
7. Intestinal Dysbiosis and Its Potential Pathogenic Role in CD
8. Role of Probiotics in CD: Human Intervention Studies
9. Concluding Remarks and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Wijmenga, C.; Gutierrez-Achury, J. Celiac disease genetics: Past, present and future challenges. J. Pediatr. Gastroenterol. Nutr. 2014, 59 (Suppl. 1), S4–S7. [Google Scholar] [CrossRef] [PubMed]
- Van Heel, D.A.; Franke, L.; Hunt, K.A.; Gwilliam, R.; Zhernakova, A.; Inouye, M.; Wapenaar, M.C.; Barnardo, M.C.N.M.; Bethel, G.; Holmes, G.K.T.; et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat. Genet. 2007, 39, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Hunt, K.A.; Zhernakova, A.; Turner, G.; Heap, G.A.R.; Franke, L.; Bruinenberg, M.; Romanos, J.; Dinesen, L.C.; Ryan, A.W.; Panesar, D.; et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat. Genet. 2008, 40, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Dubois, P.C.A.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.R.; Adány, R.; Aromaa, A.; et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010, 42, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Trynka, G.; Hunt, K.A.; Bockett, N.A.; Romanos, J.; Mistry, V.; Szperl, A.; Bakker, S.F.; Bardella, M.T.; Bhaw-Rosun, L.; Castillejo, G.; et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet. 2011, 43, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Zhernakova, A.; Withoff, S.; Wijmenga, C. Clinical implications of shared genetics and pathogenesis in autoimmune diseases. Nat. Rev. Endocrinol. 2013, 9, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Trynka, G.; Wijmenga, C.; van Heel, D.A. A genetic perspective on coeliac disease. Trends Mol. Med. 2010, 16, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Kalliomäki, M.; Satokari, R.; Lähteenoja, H.; Vähämiko, S.; Grönlund, J.; Routi, T.; Salminen, S. Expression of microbiota, toll-like receptors, and their regulators in the small intestinal mucosa in celiac disease. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Wijmenga, C.; Withoff, S. From genome-wide association studies to disease mechanisms: Celiac disease as a model for autoimmune diseases. Semin. Immunopathol. 2012, 34, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Elson, C.O.; Cong, Y. Host-microbiota interactions in inflammatory bowel disease. Gut Microbes 2012, 3, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Kalliomäki, M.; Rajala, S.; Elamo, H.; Ashorn, M.; Ruuska, T. Increased expression of CXCL16, a bacterial scavenger receptor, in the colon of children with ulcerative colitis. J. Crohns Colitis 2014, 8, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Catassi, C.; Kryszak, D.; Bhatti, B.; Sturgeon, C.; Helzlsouer, K.; Clipp, S.L.; Gelfond, D.; Puppa, E.; Sferruzza, A.; Fasano, A. Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Ann. Med. 2010, 42, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Sanz, Y.; De Pama, G.; Laparra, M. Unraveling the ties between celiac disease and intestinal microbiota. Int. Revi. Immunol. 2011, 30, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Sandberg-Bennich, S.; Dahlquist, G.; Källén, B. Coeliac disease is associated with intrauterine growth and neonatal infections. Acta Paediatr. 2002, 91, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Ivarsson, A.; Hernell, O.; Stenlund, H.; Persson, L.A. Breast-feeding protects against celiac disease. Am. J. Clin. Nutr. 2002, 75, 914–921. [Google Scholar] [PubMed]
- Sellitto, M.; Bai, G.; Serena, G.; Fricke, W.F.; Sturgeon, C.; Gajer, P.; White, J.R.; Koenig, S.S.K.; Sakamoto, J.; Boothe, D.; et al. Proof of concept of microbiome-metabolome analysis and delayed gluten exposure on celiac disease autoimmunity in genetically at-risk infants. PLoS ONE 2012, 7, e33387. [Google Scholar] [CrossRef] [PubMed]
- Mårild, K.; Stephansson, O.; Montgomery, S.; Murray, J.A.; Ludvigsson, J.F. Pregnancy outcome and risk of celiac disease in offspring: A nationwide case-control study. Gastroenterology 2012, 142, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Mårild, K.; Ye, W.; Lebwohl, B.; Green, P.H.R.; Blaser, M.J.; Card, T.; Ludvigsson, J.F. Antibiotic exposure and the development of coeliac disease: A nationwide case-control study. BMC Gastroenterol. 2013, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, H.; Li, N.; Bonnelykke, K.; Chawes, B.L.K.; Skov, T.; Paludan-Müller, G.; Stokholm, J.; Smith, B.; Krogfelt, K.A. Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. J. Allergy Clin. Immunol. 2011, 128, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Vaahtovuo, J.; Munukka, E.; Korkeamäki, M.; Luukkainen, R.; Toivanen, P. Fecal microbiota in early rheumatoid arthritis. J. Rheumatol. 2008, 35, 1500–1505. [Google Scholar] [PubMed]
- Collado, M.C.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J. Clin. Pathol. 2009, 62, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Imbalances in faecal and duodenal bifidobacterium species composition in active and non-active coeliac disease. BMC Microbiol. 2008, 8, 232. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef] [PubMed]
- Wacklin, P.; Kaukinen, K.; Tuovinen, E.; Collin, P.; Lindfors, K.; Partanen, J.; Mäki, M.; Mättö, J. The duodenal microbiota composition of adult celiac disease patients is associated with the clinical manifestation of the disease. Inflamm. Bowel Dis. 2013, 19, 934–941. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.M.; Shananhan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Ancuta, P.; Kamat, A.; Kunstman, K.J.; Kim, E.-Y.; Autissier, P.; Wurcel, A.; Zaman, T.; Stone, D.; Mefford, M.; Morgello, S.; et al. Microbial translocation is associated with increased monocyte activation and dementia in aids patients. PLoS ONE 2008, 3, e2516. [Google Scholar] [CrossRef] [PubMed]
- Hoffmanová, I.; Sánchez, D.; Hábová, V.; Anděl, M.; Tučková, L.; Tlaskalová-Hogenová, H. Serological markers of enterocyte damage and apoptosis in patients with celiac disease, autoimmune diabetes mellitus and diabetes mellitus type 2. Physiol. Res. 2014, in press. [Google Scholar]
- Human, T.; Project, M. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [Green Version]
- Kamada, N.; Seo, S.U.; Chen, G.Y.; Nuñez, C. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 312–335. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, D.H.; Erofeev, A.; Sandvik, A.; Grcic, V.; Jahnsen, F.L.; Gaustad, P.; McCoy, K.D.; Macpherson, A.J.; Meza-Zepeda, L.A.; Johansen, F.-E. Depletion of murine intestinal microbiota: Effects on gut mucosa and epithelial gene expression. PLoS ONE 2011, 6, e17996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoury, K.A.; Floch, M.H.; Hersh, T. Small intestinal mucosal cell proliferation and bacterial flora in the conventionalization of the germfree mouse. J. Exp. Med. 1969, 130, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Okkubo, T.; Tsuda, M.; Tamura, M.; Yamamura, M. Impaired superoxide production in peripheral blood neutrophils of germ-free rats. Scard. J. Immunol. 1990, 32, 727–729. [Google Scholar]
- Mitsuyama, M.; Ohara, R.; Amako, K.; Nomoto, K.; Yokokura, T. Ontogeny of macrophage function to release superoxide anion in conventional and germfree mice. Infect. Immun. 1986, 52, 236–239. [Google Scholar] [PubMed]
- Sanos, S.L.; Bui, V.L.; Mortha, A.; Oberle, K.; Heners, C.; Johner, C.; Diefenbach, A. RORgammat and commensal microflora are required for the differentiation of mucocal interleukin 22-producing NKp46+ cells. Nat. Immunol. 2009, 10, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Lee, S.M.; Li, J.; Tran, G.; Jabri, B.; Chatila, T.A.; Mazmanian, S.K. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 2011, 332, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Schippa, S.; Iebba, V.; Totino, V.; Santangelo, F.; Lepanto, M.; Alessandri, C.; Nuti, F.; Viola, F.; Di Nardo, G.; Cucchiara, S.; et al. A potential role of Escherichia coli pathobionts in the pathogenesis of pediatric inflammatory bowel disease. Can. J. Microbiol. 2012, 58, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Iebba, V.; Conte, M.P.; Lepanto, M.S.; Di Nardo, G.; Santangelo, F.; Aloi, M.; Totino, V.; Checchi, M.P.; Longhi, C.; Cucchiara, S.; et al. Microevolution in fimH gene of mucosa-associated Escherichia coli strains isolated from pediatric patients with inflammatory bowel disease. Infect. Immun. 2012, 80, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Koren, O.; Carvalho, F.A.; Ley, R.E.; Gewirtz, A.T. Aiec pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut 2014, 63, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Schippa, S.; Totino, V.; Marazzato, M.; Lepanto, M.; Santangelo, F.; Aleandri, M.; Gagliardi, A.; Longhi, C.; Pantanella, F.; Iebba, V.; et al. Escherichia coli population-based study in pediatric crohn’s disease. Adv. Microbiol. 2014, 4, 886–889. [Google Scholar] [CrossRef]
- Szebeni, B.; Veres, G.; Dezsofi, A.; Rusai, K.; Vannay, A.; Bokodi, G.; Vásárhelyi, B.; Korponay-Szabó, IR.; Tulassay, T.; Arató, A. Increased mucosal expression of Toll-like receptor (TLR)2 and TLR4 in coeliac disease. J. Pediatr. Gastroenterol. Nutr. 2007, 45, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Kalliomäki, M.; Heilig, H.G.; Palva, A.; Lähteenoja, H.; de Vos, W.M.; Salojärvi, J.; Satokari, R. Duodenal microbiota composition and mucosal homeostasis in pediatric celiac disease. BMC Gastroenterol. 2013, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Kurashima, Y.; Goto, Y.; Kiyono, H. Mucosal innate immune cells regulate both gut homeostasis and intestinal inflammation. Eur. J. Immunol. 2013, 43, 3108–3115. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.A.; Chadwick, V.S.; Murray, A. Investigations into the influence of host genetics on the predominant eubacteria in the faecal microflora of children. J. Med. Microbiol. 2005, 54, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Makino, H.; Kushiro, A.; Ishikawa, E.; Kubota, H.; Gawad, A.; Sakai, T.; Oishi, K.; Martin, R.; Ben-Amor, K.; Knol, J.; et al. Mother-to-infant transmission of intestinal bifidobacterial strains has an impact on the early development of vaginally delivered infant’s microbiota. PLoS ONE 2013, 8, e78331. [Google Scholar] [CrossRef] [PubMed]
- Makino, H.; Kushiro, A.; Ishikawa, E.; Muylaert, D.; Kubota, H.; Sakai, T.; Oishi, K.; Martin, R.; Ben Amor, K.; Oozeer, R.; et al. Transmission of intestinal bifidobacterium longum subsp. Longum strains from mother to infant, determined by multilocus sequencing typing and amplified fragment length polymorphism. Appl. Environ. Microbiol. 2011, 77, 6788–6793. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K.; et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18933–18938. [Google Scholar] [CrossRef] [PubMed]
- Khachatryan, Z.A.; Ktsoyan, Z.A.; Manukyan, G.P.; Kelly, D.; Ghazaryan, K.A.; Aminov, R.I. Predominant role of host genetics in controlling the composition of gut microbiota. PLoS ONE 2008, 3, e3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbioal ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.-P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. Nlrp6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.L.; Hofer, M.J.; Campbell, I.L.; Holmes, A.J. Community dynamics in the mouse gut microbiota: A possible role for irf9-regulated genes in community homeostasis. PLoS ONE 2010, 5, e10335. [Google Scholar] [CrossRef] [PubMed]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.A.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Bevins, C.L.; Salzman, N.H. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 2011, 9, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Silverberg, M.S.; Weersma, R.K.; Gevers, D.; Dijkstra, G.; Huang, H.; Tyler, A.D.; van Sommeren, S.; Imhann, F.; Stempak, J.M.; et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med. 2014, 6, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petnicki-Ocwieja, T.; Hrncir, T.; Liu, Y.-J.; Biswas, A.; Hudcovic, T.; Tlaskalova-Hogenova, H.; Kobayashi, K.S. NOD2 is required for the regulation of commensal microbiota in the intestine. Proc. Natl. Acad. Sci. USA 2009, 106, 15813–15818. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; Robertson, C.E.; Hamm, C.M.; Kpadeh, Z.; Zhang, T.; Chen, H.; Zhu, W.; Sartor, R.B.; Boedeker, E.C.; Harpaz, N.; et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm. Bowel Dis. 2011, 17, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; Kitani, A.; Fuss, I.; Asano, N.; Watanabe, T. The molecular basis of NOD2 susceptibility mutations in crohn’s disease. Mucosal Immunol. 2008, 1 (Suppl. 1), S5–S9. [Google Scholar] [CrossRef] [PubMed]
- Staubach, F.; Künzel, S.; Baines, A.C.; Yee, A.; McGee, B.M.; Bäckhed, F.; Baines, J.F.; Johnsen, J.M. Expression of the blood-group-related glycosyltransferase B4galnt2 influences the intestinal microbiota in mice. ISME J. 2012, 6, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Schippa, S.; Iebba, V.; Santangelo, F.; Gagliardi, A.; De Biase, R.V.; Stamato, A.; Bertasi, S.; Lucarelli, M.; Conte, M.P.; Quattrucci, S. Cystic fibrosis transmembrane conductance regulator (CFTR) allelic variants relate to shifts in faecal microbiota of cystic fibrosis patients. PLoS ONE 2013, 8, e61176. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Lundin, E.E.; Andreasson, A.N.; Greco, D.; Rafter, J.; Talley, N.J.; Agreus, L.; Andersson, A.F.; Engstrand, L.; D’Amato, M. The impact of crohn’s disease genes on healthy human gut microbiota: A pilot study. Gut 2013, 62, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Palma, G.D.; Capilla, A.; Nova, E.; Castillejo, G.; Varea, V.; Pozo, T.; Garrote, J.A.; Polanco, I.; López, A.; Ribes-Koninckx, C.; et al. Influence of milk-feeding type and genetic risk of developing coeliac disease on intestinal microbiota of infants: The proficel study. PLoS ONE 2012, 7, e30791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivares, M.; Neef, A.; Castillejo, G.; Palma, G.D.; Varea, V.; Capilla, A.; Palau, F.; Nova, E.; Marcos, A.; Polanco, I.; et al. The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut 2014, 64, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Toivanen, P.; Vaahtovuo, J.; Eerola, E. Influence of major histocompatibility complex on bacterial composition of fecal flora. Infect. Immun. 2001, 69, 2372–2377. [Google Scholar] [CrossRef] [PubMed]
- Bolnick, D.I.; Snowberg, L.K.; Caporaso, J.G.; Lauber, C.; Knight, R.; Stutz, W.E. Major histocompatibility complex class iib polymorphism influences gut microbiota composition and diversity. Mol. Ecol. 2014, 23, 4831–4845. [Google Scholar] [CrossRef] [PubMed]
- Fagarasan, S.; Honjo, T. Intestinal iga synthesis: Regulation of front-line body defences. Nat. Rev. Immunol. 2003, 3, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Cebula, A.; Seweryn, M.; Rempala, G.A.; Pabla, S.S.; McIndoe, R.A.; Denning, T.L.; Bry, L.; Kraj, P.; Kisielow, P.; Ignatowicz, L. Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature 2013, 497, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, G.; Polich, G.; Sen, M.M.; Singh, M.; Epstein, B.E.; Lytle, A.K.; Rouse, M.S.; Patel, R.; David, C.S. Evaluating the role of HLA-DQ polymorphisms on immune response to bacterial superantigens using transgenic mice. Tissue Antigens 2008, 71, 135–145. [Google Scholar] [CrossRef] [PubMed]
- McGovern, D.P.B.; Jones, M.R.; Taylor, K.D.; Marciante, K.; Yan, X.; Dubinsky, M.; Ippoliti, A.; Vasiliauskas, E.; Berel, D.; Derkowski, C.; et al. Fucosyltransferase 2 (FUT2) non-secretor status is associated with Crohn’s disease. Hum. Mol. Genet. 2010, 19, 3468–3476. [Google Scholar] [CrossRef] [PubMed]
- Parmar, A.S.; Alakulppi, N.; Paavola-Sakki, P.; Kurppa, K.; Halme, L.; Färkkilä, M.; Turunen, U.; Lappalainen, M.; Kontula, K.; Kaukinen, K.; et al. Association study of FUT2 (rs601338) with celiac disease and inflammatory bowel disease in the finnish population. Tissue Antigens 2012, 80, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Weiss, F.U.; Schurmann, C.; Guenther, A.; Ernst, F.; Teumer, A.; Mayerle, J.; Simon, P.; Völzke, H.; Radke, D.; Greinacher, A.; et al. Fucosyltransferase 2 (FUT2) non-secretor status and blood group b are associated with elevated serum lipase activity in asymptomatic subjects, and an increased risk for chronic pancreatitis: A genetic association study. Gut 2015, 64, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Ludvigsson, J.F.; Montgomery, S.M.; Ekbom, A. Risk of pancreatitis in 14,000 individuals with celiac disease. Clin. Gastroenterol. Hepatol. 2007, 5, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Wacklin, P.; Tuimala, J.; Nikkilä, J.; Tims, S.; Mäkivuokko, H.; Alakulppi, N.; Laine, P.; Rajilic-Stojanovic, M.; Paulin, L.; de Vos, W.M.; et al. Faecal microbiota composition in adults is associated with the FUT2 gene determining the secretor status. PLoS ONE 2014, 9, e94863. [Google Scholar] [CrossRef] [PubMed]
- Rausch, P.; Rehman, A.; Künzel, S.; Häsler, R.; Ott, S.J.; Schreiber, S.; Rosenstiel, P.; Franke, A.; Baines, J.F. Colonic mucosa-associated microbiota is influenced by an interaction of crohn disease and FUT2 (secretor) genotype. Proc. Natl. Acad. Sci. USA 2011, 108, 19030–19035. [Google Scholar] [CrossRef] [PubMed]
- Wacklin, P.; Mäkivuokko, H.; Alakulppi, N.; Nikkilä, J.; Tenkanen, H.; Räbinä, J.; Partanen, J.; Aranko, K.; Mättö, J. Secretor genotype (FUT2 gene) is strongly associated with the composition of bifidobacteria in the human intestine. PLoS ONE 2011, 6, e20113. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Lund, R.; Laiho, A.; Lundelin, K.; Ley, R.E.; Isolauri, E.; Salminen, S. Gut microbiota as an epigenetic regulator: Pilot study based on whole-genome methylation analysis. mBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Remely, M.; Aumueller, E.; Merold, C.; Dworzak, S.; Hippe, B.; Zanner, J.; Pointner, A.; Brath, H.; Haslberger, A.G. Effects of short chain fatty acid producing bacteria on epigenetic regulation of FFAR3 in type 2 diabetes and obesity. Gene 2014, 537, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Gonsky, R.; Deem, R.L.; Landers, C.J.; Derkowski, C.A.; Berel, D.; McGovern, D.P.B.; Targan, S.R. Distinct ifng methylation in a subset of ulcerative colitis patients based on reactivity to microbial antigens. Inflamm. Bowel Dis. 2011, 17, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Masotti, A. Interplays between gut microbiota and gene expression regulation by mirnas. Front. Cell. Infect. Microbiol. 2012, 2, 137. [Google Scholar] [CrossRef] [PubMed]
- Dalmasso, G.; Nguyen, H.T.T.; Yan, Y.; Laroui, H.; Charania, M.A.; Ayyadurai, S.; Sitaraman, S.V.; Merlin, D. Microbiota modulate host gene expression via micrornas. PLoS ONE 2011, 6, e19293. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Shirdel, E.A.; Waldron, L.; Zhang, R.-H.; Jurisica, I.; Comelli, E.M. The murine caecal microrna signature depends on the presence of the endogenous microbiota. Int. J. Biol. Sci. 2012, 8, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Alenghat, T.; Artis, D. Epigenomic regulation of host-microbiota interactions. Trends Immunol. 2014, 35, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y, M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from europe and rural africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, J.; Lange, B.; Frick, J.-S.; Sauer, H.; Zimmermann, K.; Schwiertz, A.; Rusch, K.; Klosterhalfen, S.; Enck, P. A vegan or vegetarian diet substantially alters the human colonic faecal microbiota. Eur. J. Clin. Nutr. 2012, 66, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.-Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Guaraldi, F.; Salvatori, G. Effect of breast and formula feeding on gut microbiota shaping in newborns. Front. Cell. Infect. Microbiol. 2012, 2, 94. [Google Scholar] [CrossRef] [PubMed]
- Akobeng, A.K.; Ramanan, A.V.; Buchan, I.; Heller, R.F. Effect of breast feeding on risk of coeliac disease: A systematic review and meta-analysis of observational studies. Arch. Dis. Child. 2006, 91, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Aronsson, C.A.; Lee, H.-S.; Liu, E.; Uusitalo, U.; Hummel, S.; Yang, J.; Hummel, M.; Rewers, M.; She, J.-X.; Simell, O.; et al. Age at gluten introduction and risk of celiac disease. Pediatrics 2015, 135, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Størdal, K.; White, R.A.; Eggesbø, M. Early feeding and risk of celiac disease in a prospective birth cohort. Pediatrics 2013, 132, e1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Olivares, M.; Albrecht, S.; De Palma, G.; Ferrer, M.D.; Castillejo, G.; Schols, H.A.; Sanz, Y. Human milk composition differs in healthy mothers and mothers with celiac disease. Eur. J. Nutr. 2015, 54, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, T.; Ozeke, T.; Meral, A. Gliadin-specific IgA antibodies in breast milk. J. Int. Med. Res. 2000, 28, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [PubMed]
- Decker, E.; Engelmann, G.; Findeisen, A.; Gerner, P.; Laass, M.; Ney, D.; Posovszky, C.; Hoy, L.; Hornef, M.W. Cesarean delivery is associated with celiac disease but not inflammatory bowel disease in children. Pediatrics 2010, 125, e1433–e1440. [Google Scholar] [CrossRef] [PubMed]
- De Palma, G.; Nadal, I.; Collado, M.C.; Sanz, Y. Effects of a gluten-free diet on gut microbiota and immune function in healthy adult human subjects. Br. J. Nutr. 2009, 102, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, S.; Blumberg, R.S. Life at the beginning: Perturbation of the microbiota by antibiotics in early life and its role in health and disease. Nat. Immunol. 2014, 15, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Di Cagno, R.; De Angelis, M.; De Pasquale, I.; Ndagijimana, M.; Vernocchi, P.; Ricciuti, P.; Gagliardi, F.; Laghi, L.; Crecchio, C.; Guerzoni, M.E.; et al. Duodenal and faecal microbiota of celiac children: Molecular, phenotype and metabolome characterization. BMC Microbiol. 2011, 11, 219. [Google Scholar] [CrossRef] [PubMed]
- Nistal, E.; Caminero, A.; Vivas, S.; Ruiz de Morales, J.M.; Sáenz de Miera, L.E.; Rodríguez-Aparicio, L.B.; Casqueiro, J. Differences in faecal bacteria populations and faecal bacteria metabolism in healthy adults and celiac disease patients. Biochimie 2012, 94, 1724–1729. [Google Scholar] [CrossRef] [PubMed]
- Schippa, S.; Iebba, V.; Barbato, M.; Di Nardo, G.; Totino, V.; Checchi, M.P.; Longhi, C.; Maiella, G.; Cucchiara, S.; Conte, M.P. A distinctive ‘microbial signature’ in celiac pediatric patients. BMC Microbiol. 2010, 10, 175. [Google Scholar] [CrossRef] [PubMed]
- Ou, G.; Hedberg, M.; Hörstedt, P.; Baranov, V.; Forsberg, G.; Drobni, M.; Sandström, O.; Wai, S.N.; Johansson, I.; Hammarström, M.-L.; et al. Proximal small intestinal microbiota and identification of rod-shaped bacteria associated with childhood celiac disease. Am. J. Gastroenterol. 2009, 104, 3058–3067. [Google Scholar] [CrossRef] [PubMed]
- Wacklin, P.; Laurikka, P.; Lindfors, K.; Collin, P.; Salmi, T.; Lähdeaho, M.-L.; Saavalainen, P.; Mäki, M.; Mättö, J.; Kurppa, K.; et al. Altered duodenal microbiota composition in celiac disease patients suffering from persistent symptoms on a long-term gluten-free diet. Am. J. Gastroenterol. 2014, 109, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Meji, T.G.; Budding, A.E.; Grasman, M.E.; Kneepkens, C.M.; Savelkoul, P.H.; Mearin, M.L. Composition and diversity of the duodenal mucosa-associated microbiome in children with untreated coeliac disease. Scand. J. Gastroenterol. 2013, 48, 530–536. [Google Scholar]
- Sánchez, E.; Nadal, I.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Reduced diversity and increased virulence-gene carriage in intestinal enterobacteria of coeliac children. BMC Gastroenterol. 2008, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, E.; Laparra, J.M.; Sanz, Y. Discerning the role of bacteroides fragilis in celiac disease pathogenesis. Appl. Environ. Microbiol. 2012, 78, 6507–6515. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A.; Shea-Donohue, T. Mechanisms of disease: The role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat. Clin. Pract. Gastroenterol. Hepatol. 2005, 2, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Intestinal staphylococcus spp. and virulent features associated with coeliac disease. J. Clin. Pathol. 2012, 65, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Laparra, J.M.; Sanz, Y. Bifidobacteria inhibit the inflammatory response induced by gliadins in intestinal epithelial cells via modifications of toxic peptide generation during digestion. J. Cell. Biochem. 2010, 109, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Cinova, J.; De Palma, G.; Stepankova, R.; Kofronova, O.; Kverka, M.; Sanz, Y.; Tuckova, L. Role of intestinal bacteria in gliadin-induced changes in intestinal mucosa: Study in germ-free rats. PLoS ONE 2011, 6, e16169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, A.; Linsalata, M.; Notarnicola, M.; Tutino, V.; Russo, F. Lactobacillus gg restoration of the gliadin induced epithelial barrier disruption: The role of cellular polyamines. BMC Microbiol. 2014, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Olivares, M.; Sanz, Y. Intestinal microbiota and Celiac Disease. In Advances in the Understanding of Gluten Related Pathology and the Evolution of Gluten-Free Foods; OmniaScience: Barcelona, Spain, 2015; in press. [Google Scholar]
- Sjöberg, V.; Sandström, O.; Hedberg, M.; Hammarström, S.; Hernell, O.; Hammarström, M.-L. Intestinal T-cell responses in celiac disease—Impact of celiac disease associated bacteria. PLoS ONE 2013, 8, e53414. [Google Scholar] [CrossRef] [PubMed]
- D’Arienzo, R.; Stefanile, R.; Maurano, F.; Mazzarella, G.; Ricca, E.; Troncone, R.; Auricchio, S.; Rossi, M. Immunomodulatory effects of lactobacillus casei administration in a mouse model of gliadin-sensitive enteropathy. Scand. J. Immunol. 2011, 74, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Laparra, J.M.; Olivares, M.; Gallina, O.; Sanz, Y. Bifidobacterium longum cect 7347 modulates immune responses in a gliadin-induced enteropathy animal model. PLoS ONE 2012, 7, e30744. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.; De Palma, G.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Bifidobacterium strains suppress in vitro the pro-inflammatory milieu triggered by the large intestinal microbiota of coeliac patients. J. Inflamm. (Lond.) 2008, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- De Palma, G.; Kamanova, J.; Cinova, J.; Olivares, M.; Drasarova, H.; Tuckova, L.; Sanz, Y. Modulation of phenotypic and functional maturation of dendritic cells by intestinal bacteria and gliadin: Relevance for celiac disease. J. Leukoc. Biol. 2012, 92, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Smecuol, E.; Hwang, H.J.; Sugai, E.; Corso, L.; Cherñavsky, A.C.; Bellavite, F.P.; González, A.; Vodánovich, F.; Moreno, M.L.; Vázquez, H.; et al. Exploratory, randomized, double-blind, placebo-controlled study on the effects of bifidobacterium infantis natren life start strain super strain in active celiac disease. J. Clin. Gastroenterol. 2013, 47, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Olivares, M.; Castillejo, G.; Varea, V.; Sanz, Y. Double-blind, randomised, placebo-controlled intervention trial to evaluate the effects of bifidobacterium longum cect 7347 in children with newly diagnosed coeliac disease. Br. J. Nutr. 2014, 112, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Klemenak, M.; Dolinšek, J.; Langerholc, T.; Di Gioia, D.; Mičetić-Turk, D. Administration of Bifidobacterium breve Decreases the Production of TNF-α in Children with Celiac Disease. Dig. Dis. Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cenit, M.C.; Olivares, M.; Codoñer-Franch, P.; Sanz, Y. Intestinal Microbiota and Celiac Disease: Cause, Consequence or Co-Evolution? Nutrients 2015, 7, 6900-6923. https://doi.org/10.3390/nu7085314
Cenit MC, Olivares M, Codoñer-Franch P, Sanz Y. Intestinal Microbiota and Celiac Disease: Cause, Consequence or Co-Evolution? Nutrients. 2015; 7(8):6900-6923. https://doi.org/10.3390/nu7085314
Chicago/Turabian StyleCenit, María Carmen, Marta Olivares, Pilar Codoñer-Franch, and Yolanda Sanz. 2015. "Intestinal Microbiota and Celiac Disease: Cause, Consequence or Co-Evolution?" Nutrients 7, no. 8: 6900-6923. https://doi.org/10.3390/nu7085314