Mechanistic Effects of Calcitriol in Cancer Biology



{kind=link}
Abstract
:1. Vitamin D. General Concepts
2. Vitamin D and Cancer
2.1. Biochemical Basis of Cancer
2.2. Deficiency of Vitamin D in Cancer
3. Mechanistic Effects of Calcitriol in Cancer Prevention and Treatment
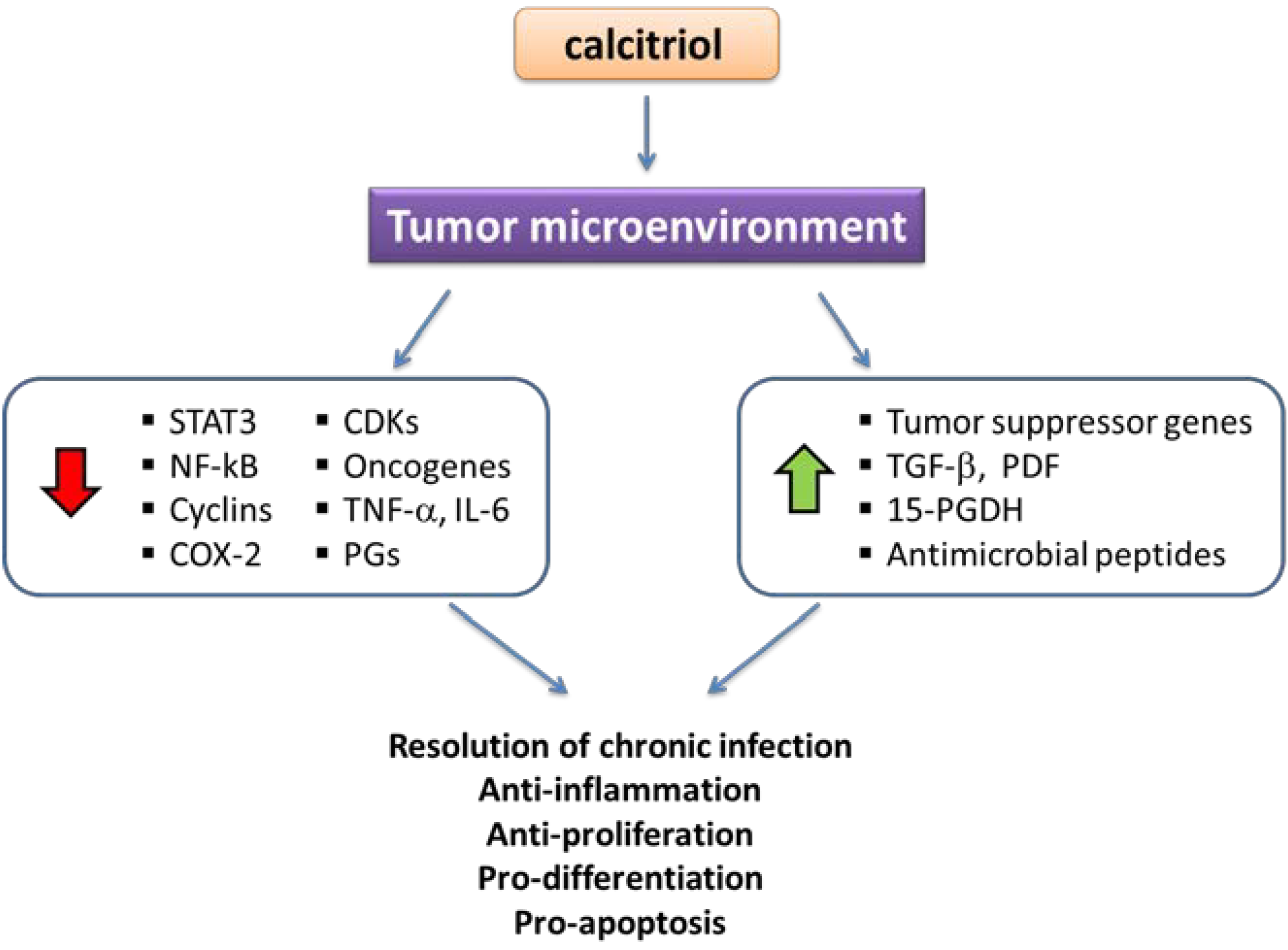
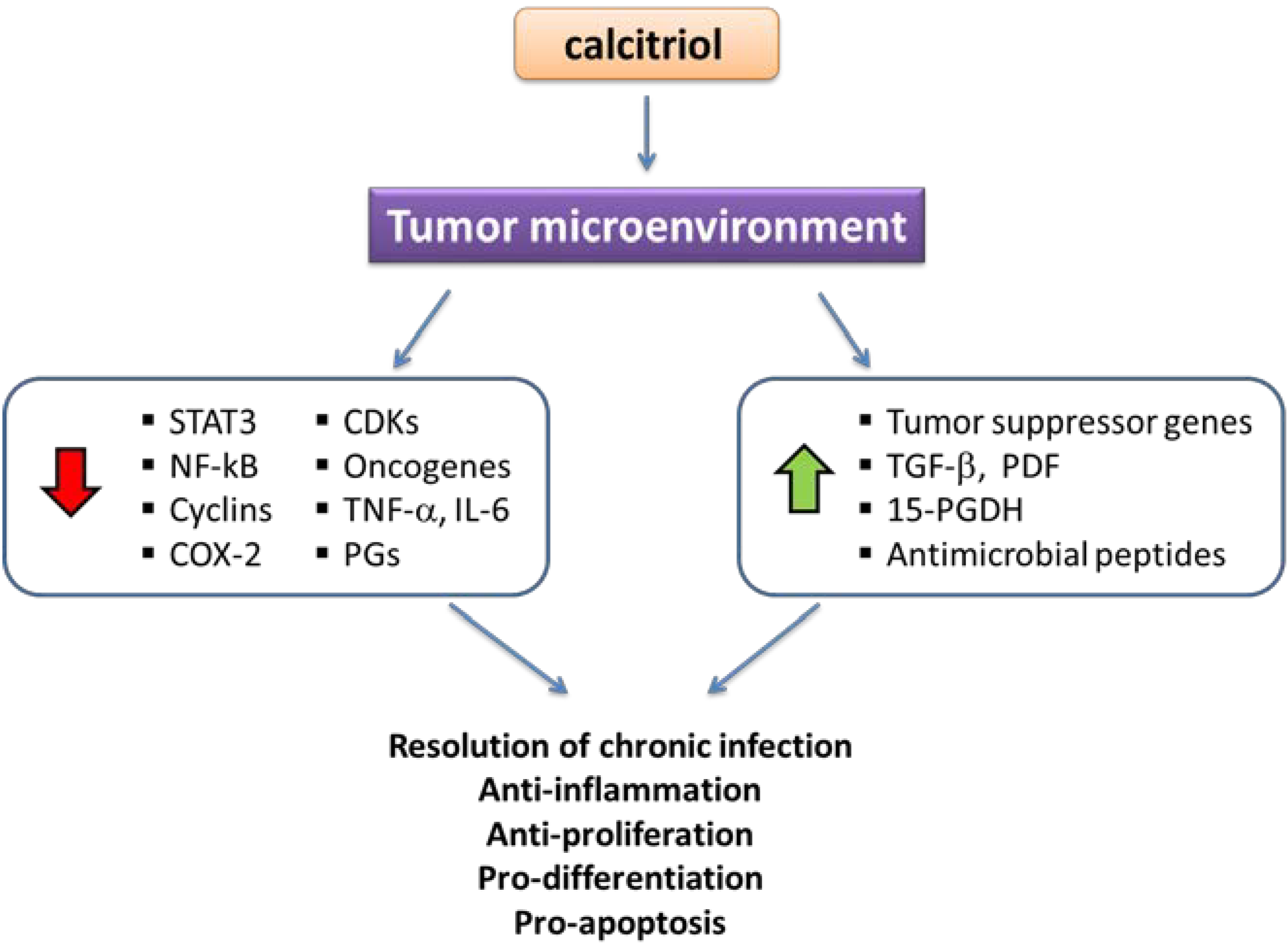
3.1. Calcitriol and Anti-Proliferative Effects
3.2. Calcitriol Pro-Differentiating Effects
3.3. Apoptosis
3.4. Regulation of Oncogenes
3.5. Regulation of Angiogenesis
3.6. Epigenetic Regulation of the VDR
3.7. Calcitriol Anti-Inflammatory Effects
3.7.1. Inflammation-Dependent Carcinogenesis
3.7.2. Calcitriol Effects upon Inflammation-Dependent Carcinogenesis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Holick, M.F. McCollum Award Lecture, 1994: Vitamin D—New horizons for the 21st century. Am. J. Clin. Nutr. 1994, 60, 619–630. [Google Scholar] [PubMed]
- Lips, P. Vitamin D physiology. Prog. Biophys. Mol. Biol. 2006, 92, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.Q.; Chen, T.C.; Matsuoka, L.Y.; Wortsman, J.; Holick, M.F. Kinetic and thermodynamic studies of the conversion of previtamin D3 to vitamin D3 in human skin. J. Biol. Chem. 1993, 268, 14888–14892. [Google Scholar] [PubMed]
- Takeyama, K.; Kitanaka, S.; Sato, T.; Kobori, M.; Yanagisawa, J.; Kato, S. 25-Hydroxy vitamin D3 1α-hydroxylase and vitamin D synthesis. Science 1997, 277, 1827–1830. [Google Scholar] [CrossRef] [PubMed]
- Jones, G. Expanding role for vitamin D in chronic kidney disease: Importance of blood 25-OH-D levels and extra-renal 1α-hydroxylase in the classical and nonclassical actions of 1α,25-dihydroxyvitamin D3. Semin. Dial. 2007, 20, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J. Regulation of vitamin D action. Nephrol. Dial. Transplant. 1999, 14, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D: Evolutionary, physiological and health perspectives. Curr. Drug Targets 2011, 12, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Toner, C.D.; Davis, C.D.; Milner, J.A. The vitamin D and cancer conundrum: Aiming at a moving target. J. Am. Diet. Assoc. 2010, 110, 1492–1500. [Google Scholar] [CrossRef] [PubMed]
- Barsony, J.; Renyi, I.; McKoy, W. Subcellular distribution of normal and mutant vitamin D receptors in living cells. Studies with a novel fluorescent ligand. J. Biol. Chem. 1997, 272, 5774–5782. [Google Scholar] [CrossRef] [PubMed]
- Tagami, T.; Lutz, W.H.; Kumar, R.; Jameson, J.L. The interaction of the vitamin D receptor with nuclear receptor corepressors and coactivators. Biochem. Biophys. Res. Commun. 1998, 253, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Wranicz, J.; Szostak-Wegierek, D. Health outcomes of vitamin D. Part II. Role in prevention of diseases. Rocz. Panstw. Zakl. Hig. 2014, 65, 273–279. [Google Scholar] [PubMed]
- Sylvia, V.L.; Schwartz, Z.; Ellis, E.B.; Helm, S.H.; Gomez, R.; Dean, D.D.; Boyan, B.D. Nongenomic regulation of protein kinase C isoforms by the vitamin D metabolites 1α,25-(OH)2 D3 and 24R,25-(OH)2 D3. J. Cell. Physiol. 1996, 167, 380–393. [Google Scholar] [CrossRef]
- Santillan, G.E.; Boland, R.L. Studies suggesting the participation of protein kinase A in 1,25(OH)2-vitamin D3-dependent protein phosphorylation in cardiac muscle. J. Mol. Cell. Cardiol. 1998, 30, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, G.; Boland, R.; de Boland, A.R. Modulation by 1,25(OH)2-vitamin D3 of the adenylyl cyclase/cyclic AMP pathway in rat and chick myoblasts. Biochim. Biophys. Acta 1995, 1269, 91–97. [Google Scholar] [CrossRef]
- Barrera, D.; Avila, E.; Hernandez, G.; Mendez, I.; Gonzalez, L.; Halhali, A.; Larrea, F.; Morales, A.; Diaz, L. Calcitriol affects hCG gene transcription in cultured human syncytiotrophoblasts. Reprod. Biol. Endocrinol. RBE 2008, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Wali, R.K.; Baum, C.L.; Sitrin, M.D.; Brasitus, T.A. 1,25(OH)2 vitamin D3 stimulates membrane phosphoinositide turnover, activates protein kinase C, and increases cytosolic calcium in rat colonic epithelium. J. Clin. Investig. 1990, 85, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Morelli, S.; de Boland, A.R.; Boland, R.L. Generation of inositol phosphates, diacylglycerol and calcium fluxes in myoblasts treated with 1,25-dihydroxyvitamin D3. Biochem. J. 1993, 289, 675–679. [Google Scholar] [PubMed]
- Bellido, T.; Morelli, S.; Fernandez, L.M.; Boland, R. Evidence for the participation of protein kinase C and 3′,5′-cyclic AMP-dependent protein kinase in the stimulation of muscle cell proliferation by 1,25-dihydroxy-vitamin D3. Mol. Cell. Endocrinol. 1993, 90, 231–238. [Google Scholar] [CrossRef]
- Fleet, J.C. Rapid, membrane-initiated actions of 1,25 dihydroxyvitamin D: What are they and what do they mean? J. Nutr. 2004, 134, 3215–3218. [Google Scholar] [PubMed]
- Morelli, S.; Buitrago, C.; Boland, R.; de Boland, A.R. The stimulation of MAP kinase by 1,25(OH)2-vitamin D3 in skeletal muscle cells is mediated by protein kinase C and calcium. Mol. Cell. Endocrinol. 2001, 173, 41–52. [Google Scholar] [CrossRef]
- Vazquez, G.; de Boland, A.R. Stimulation of dihydropyridine-sensitive Ca2+ influx in cultured myoblasts by 1,25(OH)2-vitamin D3. Biochem. Mol. Biol. Int. 1993, 31, 677–684. [Google Scholar] [PubMed]
- Karlsson, S.; Olausson, J.; Lundh, D.; Sogard, P.; Mandal, A.; Holmstrom, K.O.; Stahel, A.; Bengtsson, J.; Larsson, D. Vitamin D and prostate cancer: The role of membrane initiated signaling pathways in prostate cancer progression. J. Steroid Biochem. Mol. Biol. 2010, 121, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Irazoqui, A.P.; Boland, R.L.; Buitrago, C.G. Actions of 1,25(OH)2-vitamin D3 on the cellular cycle depend on VDR and p38 MAPK in skeletal muscle cells. J. Mol. Endocrinol. 2014, 53, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Buitrago, C.; Pardo, V.G.; Boland, R. Role of VDR in 1α,25-dihydroxy vitamin D3-dependent non-genomic activation of MAPKs, Src and Akt in skeletal muscle cells. J. Steroid Biochem. Mol. Biol. 2013, 136, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, Y.; Ozono, K.; Uchida, M.; Yoshimura, M.; Shinki, T.; Suda, T.; Yamamoto, O. Functional assessment of two vitamin D-responsive elements in the rat 25-hydroxyvitamin D3 24-hydroxylase gene. J. Biol. Chem. 1996, 271, 30381–30385. [Google Scholar] [CrossRef] [PubMed]
- Zierold, C.; Darwish, H.M.; DeLuca, H.F. Two vitamin D response elements function in the rat 1,25-dihydroxyvitamin D 24-hydroxylase promoter. J. Biol. Chem. 1995, 270, 1675–1678. [Google Scholar] [PubMed]
- Dwivedi, P.P.; Omdahl, J.L.; Kola, I.; Hume, D.A.; May, B.K. Regulation of rat cytochrome P450C24 (CYP24) gene expression. Evidence for functional cooperation of Ras-activated Ets transcription factors with the vitamin D receptor in 1,25-dihydroxyvitamin D3-mediated induction. J. Biol. Chem. 2000, 275, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, P.P.; Hii, C.S.; Ferrante, A.; Tan, J.; Der, C.J.; Omdahl, J.L.; Morris, H.A.; May, B.K. Role of MAP kinases in the 1,25-dihydroxyvitamin D3-induced transactivation of the rat cytochrome P450C24 (CYP24) promoter. Specific functions for ERK1/ERK2 and ERK5. J. Biol. Chem. 2002, 277, 29643–29653. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Boltz, M.A.; Armbrecht, H.J. Effects of 1,25-dihydroxy vitamin D3 and phorbol ester on 25-hydroxyvitamin D3 24-hydroxylase cytochrome P450 messenger ribonucleic acid levels in primary cultures of rat renal cells. Endocrinology 1993, 132, 1782–1788. [Google Scholar] [PubMed]
- Armbrecht, H.J.; Hodam, T.L.; Boltz, M.A.; Chen, M.L. Phorbol ester markedly increases the sensitivity of intestinal epithelial cells to 1,25-dihydroxyvitamin D3. FEBS Lett. 1993, 327, 13–16. [Google Scholar] [CrossRef]
- Koyama, H.; Inaba, M.; Nishizawa, Y.; Ohno, S.; Morii, H. Protein kinase C is involved in 24-hydroxylase gene expression induced by 1,25(OH)2D3 in rat intestinal epithelial cells. J. Cell. Biochem. 1994, 55, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Nutchey, B.K.; Kaplan, J.S.; Dwivedi, P.P.; Omdahl, J.L.; Ferrante, A.; May, B.K.; Hii, C.S. Molecular action of 1,25-dihydroxy vitamin D3 and phorbol ester on the activation of the rat cytochrome P450C24 (CYP24) promoter: Role of MAP kinase activities and identification of an important transcription factor binding site. Biochem. J. 2005, 389, 753–762. [Google Scholar] [PubMed]
- Murayama, A.; Kim, M.S.; Yanagisawa, J.; Takeyama, K.; Kato, S. Transrepression by a liganded nuclear receptor via a bHLH activator through co-regulator switching. EMBO J. 2004, 23, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Avila, E.; Diaz, L.; Barrera, D.; Halhali, A.; Mendez, I.; Gonzalez, L.; Zuegel, U.; Steinmeyer, A.; Larrea, F. Regulation of Vitamin D hydroxylases gene expression by 1,25-dihydroxyvitamin D3 and cyclic AMP in cultured human syncytiotrophoblasts. J. Steroid Biochem. Mol. Biol. 2007, 103, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Birge, S.J.; Alpers, D.H. Stimulation of intestinal mucosal proliferation by vitamin D. Gastroenterology 1973, 64, 977–982. [Google Scholar] [PubMed]
- Wang, X.; Pesakhov, S.; Weng, A.; Kafka, M.; Gocek, E.; Nguyen, M.; Harrison, J.S.; Danilenko, M.; Studzinski, G.P. ERK 5/MAPK pathway has a major role in 1α,25-(OH)2 vitamin D3-induced terminal differentiation of myeloid leukemia cells. J. Steroid Biochem. Mol. Biol. 2014, 144, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Dhup, S.; Dadhich, R.K.; Porporato, P.E.; Sonveaux, P. Multiple biological activities of lactic acid in cancer: Influences on tumor growth, angiogenesis and metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ngo, D.C.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Introduction to the molecular basis of cancer metabolism and the Warburg effect. Mol. Biol. Rep. 2015, 42, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Huang, P. Mitochondrial defects in cancer. Mol. Cancer 2002, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Li, H.; Chan, A.T.; Hollis, B.W.; Lee, I.M.; Stampfer, M.J.; Wu, K.; Giovannucci, E.; Ma, J. Circulating levels of vitamin D and colon and rectal cancer: The Physicians’ Health Study and a meta-analysis of prospective studies. Cancer Prev. Res. (Phila) 2011, 4, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.F.; Comstock, G.W.; Garland, F.C.; Helsing, K.J.; Shaw, E.K.; Gorham, E.D. Serum 25-hydroxyvitamin D and colon cancer: Eight-year prospective study. Lancet 1989, 2, 1176–1178. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Scarmo, S.; Afanasyeva, Y.; Lenner, P.; Koenig, K.L.; Horst, R.L.; Clendenen, T.V.; Arslan, A.A.; Chen, Y.; Hallmans, G.; Lundin, E.; et al. Circulating levels of 25-hydroxyvitamin D and risk of breast cancer: A nested case-control study. Breast Cancer Res. BCR 2013, 15, R15. [Google Scholar] [CrossRef] [PubMed]
- Bertone-Johnson, E.R.; Chen, W.Y.; Holick, M.F.; Hollis, B.W.; Colditz, G.A.; Willett, W.C.; Hankinson, S.E. Plasma 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D and risk of breast cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1991–1997. [Google Scholar] [CrossRef] [PubMed]
- Villasenor, A.; Ballard-Barbash, R.; Ambs, A.; Bernstein, L.; Baumgartner, K.; Baumgartner, R.; Ulrich, C.M.; Hollis, B.W.; McTiernan, A.; Neuhouser, M.L. Associations of serum 25-hydroxyvitamin D with overall and breast cancer-specific mortality in a multiethnic cohort of breast cancer survivors. Cancer Causes Control CCC 2013, 24, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Prentice, R.L.; Pettinger, M.B.; Jackson, R.D.; Wactawski-Wende, J.; Lacroix, A.Z.; Anderson, G.L.; Chlebowski, R.T.; Manson, J.E.; Van Horn, L.; Vitolins, M.Z.; et al. Health risks and benefits from calcium and vitamin D supplementation: Women’s Health Initiative clinical trial and cohort study. Osteoporos. Int. 2013, 24, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.F.; Gorham, E.D.; Mohr, S.B.; Grant, W.B.; Giovannucci, E.L.; Lipkin, M.; Newmark, H.; Holick, M.F.; Garland, F.C. Vitamin D and prevention of breast cancer: Pooled analysis. J. Steroid Biochem. Mol. Biol. 2007, 103, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; von Versen-Hoynck, F. Vitamin D—Roles in women’s reproductive health? Reprod. Biol. Endocrinol. RBE 2011, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, C.; Turati, F.; La Vecchia, C. Hepatocellular carcinoma epidemiology. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Ichida, T.; Matsuzawa, J.; Sugimura, K.; Asakura, H. p16(INK4) is inactivated by extensive CpG methylation in human hepatocellular carcinoma. Gastroenterology 1999, 116, 394–400. [Google Scholar] [CrossRef]
- Murata, H.; Tsuji, S.; Tsujii, M.; Sakaguchi, Y.; Fu, H.Y.; Kawano, S.; Hori, M. Promoter hypermethylation silences cyclooxygenase-2 (Cox-2) and regulates growth of human hepatocellular carcinoma cells. Lab. Investig. 2004, 84, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Finkelmeier, F.; Kronenberger, B.; Koberle, V.; Bojunga, J.; Zeuzem, S.; Trojan, J.; Piiper, A.; Waidmann, O. Severe 25-hydroxyvitamin D deficiency identifies a poor prognosis in patients with hepatocellular carcinoma—A prospective cohort study. Aliment. Pharmacol. Ther. 2014, 39, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Guanabens, N.; Pares, A. Management of osteoporosis in liver disease. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Fedirko, V.; Duarte-Salles, T.; Bamia, C.; Trichopoulou, A.; Aleksandrova, K.; Trichopoulos, D.; Trepo, E.; Tjonneland, A.; Olsen, A.; Overvad, K.; et al. Prediagnostic circulating vitamin D levels and risk of hepatocellular carcinoma in European populations: A nested case-control study. Hepatology 2014, 60, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Lappe, J.M.; Travers-Gustafson, D.; Davies, K.M.; Recker, R.R.; Heaney, R.P. Vitamin D and calcium supplementation reduces cancer risk: Results of a randomized trial. Am. J. Clin. Nutr. 2007, 85, 1586–1591. [Google Scholar] [PubMed]
- Ding, N.; Yu, R.T.; Subramaniam, N.; Sherman, M.H.; Wilson, C.; Rao, R.; Leblanc, M.; Coulter, S.; He, M.; Scott, C.; et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell 2013, 153, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.M.; Miki, D.; Ochi, H.; Nischalke, H.D.; Bojunga, J.; Bibert, S.; Morikawa, K.; Gouttenoire, J.; Cerny, A.; Dufour, J.F.; et al. Genetic analyses reveal a role for vitamin D insufficiency in HCV-associated hepatocellular carcinoma development. PLoS ONE 2013, 8, e64053. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.F.; Gorham, E.D.; Mohr, S.B.; Garland, F.C. Vitamin D for cancer prevention: Global perspective. Ann. Epidemiol. 2009, 19, 468–483. [Google Scholar] [CrossRef] [PubMed]
- Hanchette, C.L.; Schwartz, G.G. Geographic patterns of prostate cancer mortality. Evidence for a protective effect of ultraviolet radiation. Cancer 1992, 70, 2861–2869. [Google Scholar] [CrossRef]
- Grant, W.B. An estimate of premature cancer mortality in the U.S. due to inadequate doses of solar ultraviolet-B radiation. Cancer 2002, 94, 1867–1875. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, C.J.; Fryer, A.A.; French, M.E.; Liu, S.; Saxby, M.F.; Jones, P.W.; Strange, R.C. Exposure to ultraviolet radiation: Association with susceptibility and age at presentation with prostate cancer. Lancet 2001, 358, 641–642. [Google Scholar] [CrossRef]
- John, E.M.; Schwartz, G.G.; Dreon, D.M.; Koo, J. Vitamin D and breast cancer risk: The NHANES I Epidemiologic follow-up study, 1971–1975 to 1992. National Health and Nutrition Examination Survey. Cancer Epidemiol. Biomark. Prev. 1999, 8, 399–406. [Google Scholar]
- Radermacher, J.; Diesel, B.; Seifert, M.; Tilgen, W.; Reichrath, J.; Fischer, U.; Meese, E. Expression analysis of CYP27B1 in tumor biopsies and cell cultures. Anticancer Res. 2006, 26, 2683–2686. [Google Scholar] [PubMed]
- Cross, H.S. Extrarenal vitamin D hydroxylase expression and activity in normal and malignant cells: Modification of expression by epigenetic mechanisms and dietary substances. Nutr. Rev. 2007, 65, S108–S112. [Google Scholar] [CrossRef] [PubMed]
- Zehnder, D.; Bland, R.; Williams, M.C.; McNinch, R.W.; Howie, A.J.; Stewart, P.M.; Hewison, M. Extrarenal expression of 25-hydroxyvitamin D3-1α-hydroxylase. J. Clin. Endocrinol. Metab. 2001, 86, 888–894. [Google Scholar] [PubMed]
- Colston, K.; Colston, M.J.; Feldman, D. 1,25-dihydroxyvitamin D3 and malignant melanoma: The presence of receptors and inhibition of cell growth in culture. Endocrinology 1981, 108, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Biggs, J.R.; Kraft, A.S. Inhibitors of cyclin-dependent kinase and cancer. J. Mol. Med. (Berl) 1995, 73, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Hager, G.; Kornfehl, J.; Knerer, B.; Weigel, G.; Formanek, M. Molecular analysis of p21 promoter activity isolated from squamous carcinoma cell lines of the head and neck under the influence of 1,25(OH)2 vitamin D3 and its analogs. Acta Otolaryngol. 2004, 124, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Saramaki, A.; Banwell, C.M.; Campbell, M.J.; Carlberg, C. Regulation of the human p21(waf1/cip1) gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res. 2006, 34, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Kawa, S.; Nikaido, T.; Aoki, Y.; Zhai, Y.; Kumagai, T.; Furihata, K.; Fujii, S.; Kiyosawa, K. Vitamin D analogues up-regulate p21 and p27 during growth inhibition of pancreatic cancer cell lines. Br. J. Cancer 1997, 76, 884–889. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; Elstner, E.; Holden, S.; Uskokovic, M.; Koeffler, H.P. Inhibition of proliferation of prostate cancer cells by a 19-nor-hexafluoride vitamin D3 analogue involves the induction of p21waf1, p27kip1 and E-cadherin. J. Mol. Endocrinol. 1997, 19, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Artaza, J.N.; Sirad, F.; Ferrini, M.G.; Norris, K.C. 1,25(OH)2vitamin D3 inhibits cell proliferation by promoting cell cycle arrest without inducing apoptosis and modifies cell morphology of mesenchymal multipotent cells. J. Steroid Biochem. Mol. Biol. 2010, 119, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Verlinden, L.; Verstuyf, A.; Convents, R.; Marcelis, S.; Van Camp, M.; Bouillon, R. Action of 1,25(OH)2D3 on the cell cycle genes, cyclin D1, p21 and p27 in MCF-7 cells. Mol. Cell. Endocrinol. 1998, 142, 57–65. [Google Scholar] [CrossRef]
- Liu, M.; Lee, M.H.; Cohen, M.; Bommakanti, M.; Freedman, L.P. Transcriptional activation of the Cdk inhibitor p21 by vitamin D3 leads to the induced differentiation of the myelomonocytic cell line U937. Genes Dev. 1996, 10, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, P.A.; Modzelewski, R.A.; Shurin, Z.R.; Rueger, R.M.; Trump, D.L.; Johnson, C.S. 1,25-Dihydroxycholecalciferol (1,25-D3) inhibits the growth of squamous cell carcinoma and down-modulates p21(Waf1/Cip1) in vitro and in vivo. Cancer Res. 1999, 59, 2644–2649. [Google Scholar] [PubMed]
- Dunlap, N.; Schwartz, G.G.; Eads, D.; Cramer, S.D.; Sherk, A.B.; John, V.; Koumenis, C. 1α,25-dihydroxyvitamin D3 (calcitriol) and its analogue, 19-nor-1α,25(OH)2D2, potentiate the effects of ionising radiation on human prostate cancer cells. Br. J. Cancer 2003, 89, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, P.A.; McGuire, T.F.; Yu, W.D.; Zuhowski, E.G.; Schellens, J.H.; Egorin, M.J.; Trump, D.L.; Johnson, C.S. Cisplatin potentiates 1,25-dihydroxyvitamin D3-induced apoptosis in association with increased mitogen-activated protein kinase kinase kinase 1 (MEKK-1) expression. Mol. Cancer Ther. 2002, 1, 821–829. [Google Scholar] [PubMed]
- Ma, Y.; Yu, W.D.; Hershberger, P.A.; Flynn, G.; Kong, R.X.; Trump, D.L.; Johnson, C.S. 1α,25-Dihydroxyvitamin D3 potentiates cisplatin antitumor activity by p73 induction in a squamous cell carcinoma model. Mol. Cancer Ther. 2008, 7, 3047–3055. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Johnson, C.S.; Rueger, R.M.; Trump, D.L. Calcitriol (1,25-dihydroxycholecalciferol) potentiates activity of mitoxantrone/dexamethasone in an androgen independent prostate cancer model. J. Urol. 2002, 168, 756–761. [Google Scholar] [CrossRef]
- Hershberger, P.A.; Yu, W.D.; Modzelewski, R.A.; Rueger, R.M.; Johnson, C.S.; Trump, D.L. Calcitriol (1,25-dihydroxycholecalciferol) enhances paclitaxel antitumor activity in vitro and in vivo and accelerates paclitaxel-induced apoptosis. Clin. Cancer Res. 2001, 7, 1043–1051. [Google Scholar] [PubMed]
- Segovia-Mendoza, M.; Diaz, L.; Gonzalez-Gonzalez, M.E.; Martinez-Reza, I.; Garcia-Quiroz, J.; Prado-Garcia, H.; Ibarra-Sanchez, M.J.; Esparza-Lopez, J.; Larrea, F.; Garcia-Becerra, R. Calcitriol and its analogues enhance the antiproliferative activity of gefitinib in breast cancer cells. J. Steroid Biochem. Mol. Biol. 2015, 148, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; Gombart, A.F.; Kwok, S.H.; Park, S.; Koeffler, H.P. The anti-proliferative effects of 1α,25(OH)2D3 on breast and prostate cancer cells are associated with induction of BRCA1 gene expression. Oncogene 2000, 19, 5091–5097. [Google Scholar] [CrossRef] [PubMed]
- Baudet, C.; Chevalier, G.; Chassevent, A.; Canova, C.; Filmon, R.; Larra, F.; Brachet, P.; Wion, D. 1,25-Dihydroxyvitamin D3 induces programmed cell death in a rat glioma cell line. J. Neurosci. Res. 1996, 46, 540–550. [Google Scholar] [CrossRef]
- Audo, I.; Darjatmoko, S.R.; Schlamp, C.L.; Lokken, J.M.; Lindstrom, M.J.; Albert, D.M.; Nickells, R.W. Vitamin D analogues increase p53, p21, and apoptosis in a xenograft model of human retinoblastoma. Investig. Ophthalmol. Visual Sci. 2003, 44, 4192–4199. [Google Scholar] [CrossRef]
- Fan, F.S.; Yu, W.C. 1,25-Dihydroxyvitamin D3 suppresses cell growth, DNA synthesis, and phosphorylation of retinoblastoma protein in a breast cancer cell line. Cancer Investig. 1995, 13, 280–286. [Google Scholar] [CrossRef]
- Jensen, S.S.; Madsen, M.W.; Lukas, J.; Binderup, L.; Bartek, J. Inhibitory effects of 1α,25-dihydroxyvitamin D3 on the G1-S phase-controlling machinery. Mol. Endocrinol. 2001, 15, 1370–1380. [Google Scholar] [PubMed]
- Palmer, H.G.; Gonzalez-Sancho, J.M.; Espada, J.; Berciano, M.T.; Puig, I.; Baulida, J.; Quintanilla, M.; Cano, A.; de Herreros, A.G.; Lafarga, M.; et al. Vitamin D3 promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J. Cell Biol. 2001, 154, 369–387. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E. The epidemiology of vitamin D and colorectal cancer: Recent findings. Curr. Opin. Gastroenterol. 2006, 22, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Larriba, M.J.; Munoz, A. Vitamin D and colon cancer. Endocr. Relat. Cancer 2012, 19, R51–R71. [Google Scholar] [CrossRef] [PubMed]
- Santos-Martinez, N.; Diaz, L.; Ordaz-Rosado, D.; Garcia-Quiroz, J.; Barrera, D.; Avila, E.; Halhali, A.; Medina-Franco, H.; Ibarra-Sanchez, M.J.; Esparza-Lopez, J.; et al. Calcitriol restores antiestrogen responsiveness in estrogen receptor negative breast cancer cells: A potential new therapeutic approach. BMC Cancer 2014, 14, 230. [Google Scholar] [CrossRef] [PubMed]
- Towsend, K.; Trevino, V.; Falciani, F.; Stewart, P.M.; Hewison, M.; Campbell, M.J. Identification of VDR-responsive gene signatures in breast cancer cells. Oncology 2006, 71, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Blutt, S.E.; McDonnell, T.J.; Polek, T.C.; Weigel, N.L. Calcitriol-induced apoptosis in LNCaP cells is blocked by overexpression of Bcl-2. Endocrinology 2000, 141, 10–17. [Google Scholar] [PubMed]
- Guzey, M.; Kitada, S.; Reed, J.C. Apoptosis induction by 1α,25-dihydroxyvitamin D3 in prostate cancer. Mol. Cancer Ther. 2002, 1, 667–677. [Google Scholar] [PubMed]
- Wagner, N.; Wagner, K.D.; Schley, G.; Badiali, L.; Theres, H.; Scholz, H. 1,25-dihydroxyvitamin D3-induced apoptosis of retinoblastoma cells is associated with reciprocal changes of Bcl-2 and bax. Exp. Eye Res. 2003, 77, 1–9. [Google Scholar] [CrossRef]
- Kizildag, S.; Ates, H. Treatment of K562 cells with 1,25-dihydroxyvitamin D3 induces distinct alterations in the expression of apoptosis-related genes BCL2, BAX, BCLXL, and p21. Ann. Hematol. 2009, 89, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.D.; Ma, Y.; Flynn, G.; Muindi, J.R.; Kong, R.X.; Trump, D.L.; Johnson, C.S. Calcitriol enhances gemcitabine anti-tumor activity in vitro and in vivo by promoting apoptosis in a human pancreatic carcinoma model system. Cell Cycle 2010, 9, 3022–3029. [Google Scholar] [CrossRef] [PubMed]
- McGuire, T.F.; Trump, D.L.; Johnson, C.S. Vitamin D3-induced apoptosis of murine squamous cell carcinoma cells. Selective induction of caspase-dependent MEK cleavage and up-regulation of MEKK-1. J. Biol. Chem. 2001, 276, 26365–26373. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, I.N. Calcium signaling in cancer and vitamin D. J. Steroid Biochem. Mol. Biol. 2005, 97, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Weitsman, G.E.; Koren, R.; Zuck, E.; Rotem, C.; Liberman, U.A.; Ravid, A. Vitamin D sensitizes breast cancer cells to the action of H2O2: Mitochondria as a convergence point in the death pathway. Free Radic. Biol. Med. 2005, 39, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Ravid, A.; Koren, R. The role of reactive oxygen species in the anticancer activity of vitamin D. Recent Results Cancer Res. 2003, 164, 357–367. [Google Scholar] [PubMed]
- Lee, L.R.; Teng, P.N.; Nguyen, H.; Hood, B.L.; Kavandi, L.; Wang, G.; Turbov, J.M.; Thaete, L.G.; Hamilton, C.A.; Maxwell, G.L.; et al. Progesterone enhances calcitriol antitumor activity by upregulating vitamin D receptor expression and promoting apoptosis in endometrial cancer cells. Cancer Prev. Res. (Phila) 2013, 6, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Ikeda, N.; Uemura, H.; Ishiguro, H.; Hori, M.; Hosaka, M.; Kyo, S.; Miyamoto, K.; Takeda, E.; Kubota, Y. Combination treatment with 1α,25-dihydroxyvitamin D3 and 9-cis-retinoic acid directly inhibits human telomerase reverse transcriptase transcription in prostate cancer cells. Mol. Cancer Ther. 2003, 2, 739–746. [Google Scholar] [PubMed]
- Zhang, X.; Nicosia, S.V.; Bai, W. Vitamin D receptor is a novel drug target for ovarian cancer treatment. Curr. Cancer Drug Targets 2006, 6, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Yamada, O.; Ozaki, K.; Nakatake, M.; Akiyama, M.; Kawauchi, K.; Matsuoka, R. Multistep regulation of telomerase during differentiation of HL60 cells. J. Leukoc. Biol. 2008, 83, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Kasiappan, R.; Shen, Z.; Tse, A.K.; Jinwal, U.; Tang, J.; Lungchukiet, P.; Sun, Y.; Kruk, P.; Nicosia, S.V.; Zhang, X.; et al. 1,25-Dihydroxyvitamin D3 suppresses telomerase expression and human cancer growth through microRNA-498. J. Biol. Chem. 2012, 287, 41297–41309. [Google Scholar] [CrossRef] [PubMed]
- Saunders, D.E.; Christensen, C.; Wappler, N.L.; Schultz, J.F.; Lawrence, W.D.; Malviya, V.K.; Malone, J.M.; Deppe, G. Inhibition of c-myc in breast and ovarian carcinoma cells by 1,25-dihydroxyvitamin D3, retinoic acid and dexamethasone. Anticancer Drugs 1993, 4, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Rohan, J.N.; Weigel, N.L. 1α,25-dihydroxyvitamin D3 reduces c-Myc expression, inhibiting proliferation and causing G1 accumulation in C4–2 prostate cancer cells. Endocrinology 2009, 150, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Shachaf, C.M.; Felsher, D.W. Tumor dormancy and MYC inactivation: Pushing cancer to the brink of normalcy. Cancer Res. 2005, 65, 4471–4474. [Google Scholar] [CrossRef] [PubMed]
- Dalhoff, K.; Dancey, J.; Astrup, L.; Skovsgaard, T.; Hamberg, K.J.; Lofts, F.J.; Rosmorduc, O.; Erlinger, S.; Bach Hansen, J.; Steward, W.P.; et al. A phase II study of the vitamin D analogue Seocalcitol in patients with inoperable hepatocellular carcinoma. Br. J. Cancer 2003, 89, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.B.; Goetsch, P.D.; Pike, J.W. VDR/RXR and TCF4/β-catenin cistromes in colonic cells of colorectal tumor origin: Impact on c-FOS and c-MYC gene expression. Mol. Endocrinol. 2011, 26, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Candeliere, G.A.; Jurutka, P.W.; Haussler, M.R.; St-Arnaud, R. A composite element binding the vitamin D receptor, retinoid X receptor α, and a member of the CTF/NF-1 family of transcription factors mediates the vitamin D responsiveness of the c-fos promoter. Mol. Cell. Biol. 1996, 16, 584–592. [Google Scholar] [PubMed]
- Okano, K.; Usa, T.; Ohtsuru, A.; Tsukazaki, T.; Miyazaki, Y.; Yonekura, A.; Namba, H.; Shindoh, H.; Yamashita, S. Effect of 22-Oxa-1,25-dihydroxyvitamin D3 on human thyroid cancer cell growth. Endocr. J. 1999, 46, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Camacho, J. Ether à go-go potassium channels and cancer. Cancer Lett. 2006, 233, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Avila, E.; Garcia-Becerra, R.; Rodriguez-Rasgado, J.A.; Diaz, L.; Ordaz-Rosado, D.; Zugel, U.; Steinmeyer, A.; Barrera, D.; Halhali, A.; Larrea, F.; et al. Calcitriol down-regulates human ether à go-go 1 potassium channel expression in cervical cancer cells. Anticancer Res. 2010, 30, 2667–2672. [Google Scholar] [PubMed]
- Garcia-Becerra, R.; Diaz, L.; Camacho, J.; Barrera, D.; Ordaz-Rosado, D.; Morales, A.; Ortiz, C.S.; Avila, E.; Bargallo, E.; Arrecillas, M.; et al. Calcitriol inhibits Ether-à go-go potassium channel expression and cell proliferation in human breast cancer cells. Exp. Cell Res. 2010, 316, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Quiroz, J.; Garcia-Becerra, R.; Santos-Martinez, N.; Barrera, D.; Ordaz-Rosado, D.; Avila, E.; Halhali, A.; Villanueva, O.; Ibarra-Sanchez, M.J.; Esparza-Lopez, J.; et al. In vivo dual targeting of the oncogenic Ether-à go-go-1 potassium channel by calcitriol and astemizole results in enhanced antineoplastic effects in breast tumors. BMC Cancer 2014, 14, 745. [Google Scholar] [CrossRef] [PubMed]
- Cazares-Ordonez, V.; Gonzalez-Duarte, R.J.; Diaz, L.; Ishizawa, M.; Uno, S.; Ortiz, V.; Ordonez-Sanchez, M.L.; Makishima, M.; Larrea, F.; Avila, E. A cis-acting element in the promoter of human Ether-à go-go-1 potassium channel gene mediates repression by calcitriol in human cervical cancer cells. Biochem. Cell Biol. 2015, 93, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Bocci, G.; Francia, G.; Man, S.; Lawler, J.; Kerbel, R.S. Thrombospondin 1, a mediator of the antiangiogenic effects of low-dose metronomic chemotherapy. Proc. Natl. Acad. Sci. USA 2003, 100, 12917–12922. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Vascular endothelial growth factor (VEGF) inhibition—A critical review. Anticancer Agents Med. Chem. 2007, 7, 223–245. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.J.; Johnson, C.S.; Modzelewski, R.A.; Trump, D.L. Antiproliferative effects of 1α,25-dihydroxyvitamin D3 and vitamin D analogs on tumor-derived endothelial cells. Endocrinology 2002, 143, 2508–2514. [Google Scholar] [PubMed]
- Chung, I.; Han, G.; Seshadri, M.; Gillard, B.M.; Yu, W.D.; Foster, B.A.; Trump, D.L.; Johnson, C.S. Role of vitamin D receptor in the antiproliferative effects of calcitriol in tumor-derived endothelial cells and tumor angiogenesis in vivo. Cancer Res. 2009, 69, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Mantell, D.J.; Owens, P.E.; Bundred, N.J.; Mawer, E.B.; Canfield, A.E. 1α,25-dihydroxyvitamin D3 inhibits angiogenesis in vitro and in vivo. Circ. Res. 2000, 87, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Majewski, S.; Skopinska, M.; Marczak, M.; Szmurlo, A.; Bollag, W.; Jablonska, S. Vitamin D3 is a potent inhibitor of tumor cell-induced angiogenesis. J. Investig. Dermatol. Symp. Proc. 1996, 1, 97–101. [Google Scholar] [PubMed]
- Iseki, K.; Tatsuta, M.; Uehara, H.; Iishi, H.; Yano, H.; Sakai, N.; Ishiguro, S. Inhibition of angiogenesis as a mechanism for inhibition by lα-hydroxyvitamin D3 and 1,25-dihydroxyvitamin D3 of colon carcinogenesis induced by azoxymethane in Wistar rats. Int. J. Cancer 1999, 81, 730–733. [Google Scholar] [CrossRef]
- Fernandez-Garcia, N.I.; Palmer, H.G.; Garcia, M.; Gonzalez-Martin, A.; del Rio, M.; Barettino, D.; Volpert, O.; Munoz, A.; Jimenez, B. 1α,25-Dihydroxyvitamin D3 regulates the expression of Id1 and Id2 genes and the angiogenic phenotype of human colon carcinoma cells. Oncogene 2005, 24, 6533–6544. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.J.; Teegarden, D. 1α,25-dihydroxycholecalciferol increases the expression of vascular endothelial growth factor in C3H10T1/2 mouse embryo fibroblasts. J. Nutr. 2004, 134, 2244–2250. [Google Scholar] [PubMed]
- Lin, R.; Amizuka, N.; Sasaki, T.; Aarts, M.M.; Ozawa, H.; Goltzman, D.; Henderson, J.E.; White, J.H. 1Α,25-dihydroxyvitamin D3 promotes vascularization of the chondro-osseous junction by stimulating expression of vascular endothelial growth factor and matrix metalloproteinase 9. J. Bone Miner. Res. 2002, 17, 1604–1612. [Google Scholar] [CrossRef] [PubMed]
- Riboldi, E.; Musso, T.; Moroni, E.; Urbinati, C.; Bernasconi, S.; Rusnati, M.; Adorini, L.; Presta, M.; Sozzani, S. Cutting edge: Proangiogenic properties of alternatively activated dendritic cells. J. Immunol. 2005, 175, 2788–2792. [Google Scholar] [CrossRef] [PubMed]
- Cardus, A.; Panizo, S.; Encinas, M.; Dolcet, X.; Gallego, C.; Aldea, M.; Fernandez, E.; Valdivielso, J.M. 1,25-dihydroxyvitamin D3 regulates VEGF production through a vitamin D response element in the VEGF promoter. Atherosclerosis 2009, 204, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Cardus, A.; Parisi, E.; Gallego, C.; Aldea, M.; Fernandez, E.; Valdivielso, J.M. 1,25-Dihydroxyvitamin D3 stimulates vascular smooth muscle cell proliferation through a VEGF-mediated pathway. Kidney Int. 2006, 69, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Quiroz, J.; Rivas-Suarez, M.; Garcia-Becerra, R.; Barrera, D.; Martinez-Reza, I.; Ordaz-Rosado, D.; Santos-Martinez, N.; Villanueva, O.; Santos-Cuevas, C.L.; Avila, E.; et al. Calcitriol reduces thrombospondin-1 and increases vascular endothelial growth factor in breast cancer cells: Implications for tumor angiogenesis. J. Steroid Biochem. Mol. Biol. 2014, 144, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Oades, G.M.; Dredge, K.; Kirby, R.S.; Colston, K.W. Vitamin D receptor-dependent antitumour effects of 1,25-dihydroxyvitamin D3 and two synthetic analogues in three in vivo models of prostate cancer. BJU Int. 2002, 90, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; Haidar, M.; Placzko, S.; Niendorf, R.; Darashchonak, N.; Hubel, C.A.; von Versen-Hoynck, F. Vitamin D improves the angiogenic properties of endothelial progenitor cells. Am. J. Physiol. Cell Physiol. 2012, 303, C954–C962. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Kozawa, O.; Tanabe, K.; Akamatsu, S.; Matsuno, H.; Dohi, S.; Hirose, H.; Uematsu, T. 1,25-dihydroxyvitamin D3 stimulates vascular endothelial growth factor release in aortic smooth muscle cells: Role of p38 mitogen-activated protein kinase. Arch. Biochem. Biophys. 2002, 398, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bassett, S.A.; Barnett, M.P. The role of dietary histone deacetylases (HDACs) inhibitors in health and disease. Nutrients 2014, 6, 4273–4301. [Google Scholar] [CrossRef] [PubMed]
- Burdelski, C.; Ruge, O.M.; Melling, N.; Koop, C.; Simon, R.; Steurer, S.; Sauter, G.; Kluth, M.; Hube-Magg, C.; Minner, S.; et al. HDAC1 overexpression independently predicts biochemical recurrence and is associated with rapid tumor cell proliferation and genomic instability in prostate cancer. Exp. Mol. Pathol. 2015, 98, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Chuang, M.J.; Wu, S.T.; Tang, S.H.; Lai, X.M.; Lai, H.C.; Hsu, K.H.; Sun, K.H.; Sun, G.H.; Chang, S.Y.; Yu, D.S.; et al. The HDAC inhibitor LBH589 induces ERK-dependent prometaphase arrest in prostate cancer via HDAC6 inactivation and down-regulation. PLoS ONE 2013, 8, e73401. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Kidane, A.I.; Yu, T.W.; Dashwood, W.M.; Bisson, W.H.; Lohr, C.V.; Ho, E.; Williams, D.E.; Dashwood, R.H. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics 2013, 8, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Min, S.K.; Park, H.R.; Kim, D.H.; Kwon, M.J.; Kim, L.S.; Ju, Y.S. Expression of histone deacetylases HDAC1, HDAC2, HDAC3, and HDAC6 in invasive ductal carcinomas of the breast. J. Breast Cancer 2014, 17, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Raha, P.; Thomas, S.; Thurn, K.T.; Park, J.; Munster, P.N. Combined histone deacetylase inhibition and tamoxifen induces apoptosis in tamoxifen resistant breast cancer models, by reversing Bcl-2 overexpression. Breast Cancer Res. BCR 2015, 17, 533. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhao, X.; Pei, F.; Ji, M.; Ma, W.; Wang, Y.; Jiang, G. Activation of the intrinsic apoptosis pathway contributes to the induction of apoptosis in hepatocellular carcinoma cells by valproic acid. Oncol. Lett. 2015, 9, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Gu, J.; Ma, J.; Xu, Z.; Tao, H. Histone deacetylase inhibitors repress chondrosarcoma cell proliferation. J. BUON 2015, 20, 269–274. [Google Scholar] [PubMed]
- Cao, K.; Wang, G.; Li, W.; Zhang, L.; Wang, R.; Huang, Y.; Du, L.; Jiang, J.; Wu, C.; He, X.; et al. Histone deacetylase inhibitors prevent activation-induced cell death and promote anti-tumor immunity. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Karlic, H.; Varga, F. Impact of vitamin D metabolism on clinical epigenetics. Clin. Epigenetics 2011, 2, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Janssens, W.; Decramer, M.; Mathieu, C.; Korf, H. Vitamin D and chronic obstructive pulmonary disease: Hype or reality? Lancet Respir. Med. 2013, 1, 804–812. [Google Scholar] [CrossRef]
- Zella, L.A.; Meyer, M.B.; Nerenz, R.D.; Lee, S.M.; Martowicz, M.L.; Pike, J.W. Multifunctional enhancers regulate mouse and human vitamin D receptor gene transcription. Mol. Endocrinol. 2010, 24, 128–147. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.F.; Moore, J.S.; Walker, E.; Driver, P.M.; Engel, J.; Edwards, C.E.; Brown, G.; Uskokovic, M.R.; Campbell, M.J. Synergistic growth inhibition of prostate cancer cells by 1α,25 Dihydroxyvitamin D3 and its 19-nor-hexafluoride analogs in combination with either sodium butyrate or trichostatin A. Oncogene 2001, 20, 1860–1872. [Google Scholar] [CrossRef] [PubMed]
- Thorne, J.L.; Maguire, O.; Doig, C.L.; Battaglia, S.; Fehr, L.; Sucheston, L.E.; Heinaniemi, M.; O’Neill, L.P.; McCabe, C.J.; Turner, B.M.; Carlberg, C.; Campbell, M.J. Epigenetic control of a VDR-governed feed-forward loop that regulates p21(waf1/cip1) expression and function in non-malignant prostate cells. Nucleic Acids Res. 2011, 39, 2045–2056. [Google Scholar] [CrossRef] [PubMed]
- Saramaki, A.; Diermeier, S.; Kellner, R.; Laitinen, H.; Vaisanen, S.; Carlberg, C. Cyclical chromatin looping and transcription factor association on the regulatory regions of the p21 (CDKN1A) gene in response to 1α,25-dihydroxyvitamin D3. J. Biol. Chem. 2009, 284, 8073–8082. [Google Scholar] [CrossRef] [PubMed]
- Abedin, S.A.; Banwell, C.M.; Colston, K.W.; Carlberg, C.; Campbell, M.J. Epigenetic corruption of VDR signalling in malignancy. Anticancer Res. 2006, 26, 2557–2566. [Google Scholar] [PubMed]
- An, B.S.; Tavera-Mendoza, L.E.; Dimitrov, V.; Wang, X.; Calderon, M.R.; Wang, H.J.; White, J.H. Stimulation of Sirt1-regulated FoxO protein function by the ligand-bound vitamin D receptor. Mol. Cell. Biol. 2010, 30, 4890–4900. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Zhu, P.; Yang, X.; Han, Z.; Jiang, J.; Zong, C.; Zhang, X.; Liu, W.; Zhao, Q.; Fan, T.; et al. Overexpression of SIRT1 promotes metastasis through epithelial-mesenchymal transition in hepatocellular carcinoma. BMC Cancer 2014, 14, 978. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.W.; Li, Y.C.; Wan, G.X.; Du, X.M.; Li, F. Clinicopathological and prognostic role of SIRT1 in breast cancer patients: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 616–624. [Google Scholar] [PubMed]
- Singh, S.; Kumar, P.U.; Thakur, S.; Kiran, S.; Sen, B.; Sharma, S.; Rao, V.V.; Poongothai, A.R.; Ramakrishna, G. Expression/localization patterns of sirtuins (SIRT1, SIRT2, and SIRT7) during progression of cervical cancer and effects of sirtuin inhibitors on growth of cervical cancer cells. Tumour Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Daniel, M.; Tollefsbol, T.O. Epigenetic regulation of caloric restriction in aging. BMC Med. 2011, 9, 98. [Google Scholar] [CrossRef] [PubMed]
- Khorchide, M.; Lechner, D.; Cross, H.S. Epigenetic regulation of vitamin D hydroxylase expression and activity in normal and malignant human prostate cells. J. Steroid Biochem. Mol. Biol. 2005, 93, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Whitlatch, L.W.; Young, M.V.; Schwartz, G.G.; Flanagan, J.N.; Burnstein, K.L.; Lokeshwar, B.L.; Rich, E.S.; Holick, M.F.; Chen, T.C. 25-Hydroxyvitamin D-1α-hydroxylase activity is diminished in human prostate cancer cells and is enhanced by gene transfer. J. Steroid Biochem. Mol. Biol. 2002, 81, 135–140. [Google Scholar] [CrossRef]
- Cross, H.S.; Bareis, P.; Hofer, H.; Bischof, M.G.; Bajna, E.; Kriwanek, S.; Bonner, E.; Peterlik, M. 25-Hydroxyvitamin D3-1α-hydroxylase and vitamin D receptor gene expression in human colonic mucosa is elevated during early cancerogenesis. Steroids 2001, 66, 287–292. [Google Scholar] [CrossRef]
- Ly, L.H.; Zhao, X.Y.; Holloway, L.; Feldman, D. Liarozole acts synergistically with 1α,25-dihydroxyvitamin D3 to inhibit growth of DU 145 human prostate cancer cells by blocking 24-hydroxylase activity. Endocrinology 1999, 140, 2071–2076. [Google Scholar] [PubMed]
- Hobaus, J.; Fetahu, I.; Khorchide, M.; Manhardt, T.; Kallay, E. Epigenetic regulation of the 1,25-dihydroxyvitamin D3 24-hydroxylase (CYP24A1) in colon cancer cells. J. Steroid Biochem. Mol. Biol. 2013, 136, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.G.; Nakane, M.; Ruan, X.A.; Kroeger, P.E.; Wu-Wong, J.R. Expression of VDR and CYP24A1 mRNA in human tumors. Cancer Chemother. Pharm. 2006, 57, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Karpf, A.R.; Deeb, K.K.; Muindi, J.R.; Morrison, C.D.; Johnson, C.S.; Trump, D.L. Epigenetic Regulation of Vitamin D 24-Hydroxylase/CYP24A1 in Human Prostate Cancer. Cancer Res. 2010, 70, 5953–5962. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Schottenfeld, D.; Beebe-Dimmer, J. Chronic inflammation: A common and important factor in the pathogenesis of neoplasia. CA Cancer J. Clin. 2006, 56, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Mantovani, A. Cancer-related inflammation: Common themes and therapeutic opportunities. Semin. Cancer Biol. 2011, 22, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114. [Google Scholar] [CrossRef] [PubMed]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal inflammation and cancer. Gastroenterology 2011, 140, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Sung, B.; Aggarwal, B.B. TNF: A master switch for inflammation to cancer. Front. Biosci. 2008, 13, 5094–5107. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Deng, J.; Rychahou, P.G.; Qiu, S.; Evers, B.M.; Zhou, B.P. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 2009, 15, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Levy, D.E.; Inghirami, G. STAT3: A multifaceted oncogene. Proc. Natl. Acad. Sci. USA 2006, 103, 10151–10152. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.; Bowman, T.L.; Niu, G.; Yu, H.; Minton, S.; Muro-Cacho, C.A.; Cox, C.E.; Falcone, R.; Fairclough, R.; Parsons, S.; et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 2001, 20, 2499–2513. [Google Scholar] [CrossRef] [PubMed]
- So, J.Y.; Smolarek, A.K.; Salerno, D.M.; Maehr, H.; Uskokovic, M.; Liu, F.; Suh, N. Targeting CD44-STAT3 signaling by Gemini vitamin D analog leads to inhibition of invasion in basal-like breast cancer. PLoS ONE 2013, 8, e54020. [Google Scholar] [CrossRef] [PubMed]
- Park, M.R.; Lee, J.H.; Park, M.S.; Hwang, J.E.; Shim, H.J.; Cho, S.H.; Chung, I.J.; Bae, W.K. Suppressive effect of 19-nor-1α-25-dihydroxyvitamin D2 on gastric cancer cells and peritoneal metastasis model. J. Korean Med. Sci. 2012, 27, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Stio, M.; Martinesi, M.; Bruni, S.; Treves, C.; Mathieu, C.; Verstuyf, A.; d’Albasio, G.; Bagnoli, S.; Bonanomi, A.G. The Vitamin D analogue TX 527 blocks NF-kappaB activation in peripheral blood mononuclear cells of patients with Crohn’s disease. J. Steroid Biochem. Mol. Biol. 2007, 103, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.P.; Bellido, T.; Manolagas, S.C. Down-regulation of NF-kappa B protein levels in activated human lymphocytes by 1,25-dihydroxyvitamin D3. Proc. Natl. Acad. Sci. USA 1995, 92, 10990–10994. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.V.; Feldman, D. Molecular pathways mediating the anti-inflammatory effects of calcitriol: Implications for prostate cancer chemoprevention and treatment. Endocr. Relat. Cancer 2010, 17, R19–R38. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Kong, J.; Duan, Y.; Szeto, F.L.; Liao, A.; Madara, J.L.; Li, Y.C. Increased NF-kappaB activity in fibroblasts lacking the vitamin D receptor. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E315–E322. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Ge, X.; Du, J.; Deb, D.K.; Li, Y.C. Vitamin D receptor inhibits nuclear factor kappaB activation by interacting with IkappaB kinase beta protein. J. Biol. Chem. 2013, 288, 19450–19458. [Google Scholar] [CrossRef] [PubMed]
- Riis, J.L.; Johansen, C.; Gesser, B.; Moller, K.; Larsen, C.G.; Kragballe, K.; Iversen, L. 1α,25(OH)2D3 regulates NF-kappaB DNA binding activity in cultured normal human keratinocytes through an increase in IkappaBα expression. Arch. Dermatol. Res. 2004, 296, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-kappaB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-kappaB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. NF-kappaB in cancer therapy. Arch. Toxicol. 2015, 89, 711–731. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.; Krishnan, A.V.; Swami, S.; Nonn, L.; Peehl, D.M.; Feldman, D. Regulation of prostaglandin metabolism by calcitriol attenuates growth stimulation in prostate cancer cells. Cancer Res. 2005, 65, 7917–7925. [Google Scholar] [PubMed]
- Diaz, L.; Noyola-Martinez, N.; Barrera, D.; Hernandez, G.; Avila, E.; Halhali, A.; Larrea, F. Calcitriol inhibits TNF-α-induced inflammatory cytokines in human trophoblasts. J. Reprod. Immunol. 2009, 81, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Noyola-Martinez, N.; Diaz, L.; Avila, E.; Halhali, A.; Larrea, F.; Barrera, D. Calcitriol downregulates TNF-α and IL-6 expression in cultured placental cells from preeclamptic women. Cytokine 2013, 61, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Leung, D.Y.; Richers, B.N.; Liu, Y.; Remigio, L.K.; Riches, D.W.; Goleva, E. Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J. Immunol. 2012, 188, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Beilfuss, J.; Berg, V.; Sneve, M.; Jorde, R.; Kamycheva, E. Effects of a 1-year supplementation with cholecalciferol on interleukin-6, tumor necrosis factor-α and insulin resistance in overweight and obese subjects. Cytokine 2012, 60, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Bessler, H.; Djaldetti, M. 1α,25-Dihydroxyvitamin D3 modulates the interaction between immune and colon cancer cells. Biomed. Pharmacother. 2012, 66, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Kulbe, H.; Thompson, R.; Wilson, J.L.; Robinson, S.; Hagemann, T.; Fatah, R.; Gould, D.; Ayhan, A.; Balkwill, F. The inflammatory cytokine tumor necrosis factor-α generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 2007, 67, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Defacque, H.; Piquemal, D.; Basset, A.; Marti, J.; Commes, T. Transforming growth factor-beta1 is an autocrine mediator of U937 cell growth arrest and differentiation induced by vitamin D3 and retinoids. J. Cell. Physiol. 1999, 178, 109–119. [Google Scholar] [CrossRef]
- Lambert, J.R.; Kelly, J.A.; Shim, M.; Huffer, W.E.; Nordeen, S.K.; Baek, S.J.; Eling, T.E.; Lucia, M.S. Prostate derived factor in human prostate cancer cells: Gene induction by vitamin D via a p53-dependent mechanism and inhibition of prostate cancer cell growth. J. Cell. Physiol. 2006, 208, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Olmos-Ortiz, A.; Noyola-Martinez, N.; Barrera, D.; Zaga-Clavellina, V.; Avila, E.; Halhali, A.; Biruete, B.; Larrea, F.; Diaz, L. IL-10 inhibits while calcitriol reestablishes placental antimicrobial peptides gene expression. J. Steroid Biochem. Mol. Biol. 2015, 148, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.T.; Nestel, F.P.; Bourdeau, V.; Nagai, Y.; Wang, Q.; Liao, J.; Tavera-Mendoza, L.; Lin, R.; Hanrahan, J.W.; Mader, S.; et al. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J. Immunol. 2004, 173, 2909–2912. [Google Scholar] [CrossRef] [PubMed]
- Paillard, F. Immunosuppression mediated by tumor cells: A challenge for immunotherapeutic approaches. Hum. Gene Ther. 2000, 11, 657–658. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Stolina, M.; Lin, Y.; Gardner, B.; Miller, P.W.; Kronenberg, M.; Dubinett, S.M. T cell-derived IL-10 promotes lung cancer growth by suppressing both T cell and APC function. J. Immunol. 1999, 163, 5020–5028. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz, L.; Díaz-Muñoz, M.; García-Gaytán, A.C.; Méndez, I. Mechanistic Effects of Calcitriol in Cancer Biology. Nutrients 2015, 7, 5020-5050. https://doi.org/10.3390/nu7065020
Díaz L, Díaz-Muñoz M, García-Gaytán AC, Méndez I. Mechanistic Effects of Calcitriol in Cancer Biology. Nutrients. 2015; 7(6):5020-5050. https://doi.org/10.3390/nu7065020
Chicago/Turabian StyleDíaz, Lorenza, Mauricio Díaz-Muñoz, Ana Cristina García-Gaytán, and Isabel Méndez. 2015. "Mechanistic Effects of Calcitriol in Cancer Biology" Nutrients 7, no. 6: 5020-5050. https://doi.org/10.3390/nu7065020
APA StyleDíaz, L., Díaz-Muñoz, M., García-Gaytán, A. C., & Méndez, I. (2015). Mechanistic Effects of Calcitriol in Cancer Biology. Nutrients, 7(6), 5020-5050. https://doi.org/10.3390/nu7065020