Insights from Space: Potential Role of Diet in the Spatial Organization of Chromosomes



Abstract
:1. Introduction

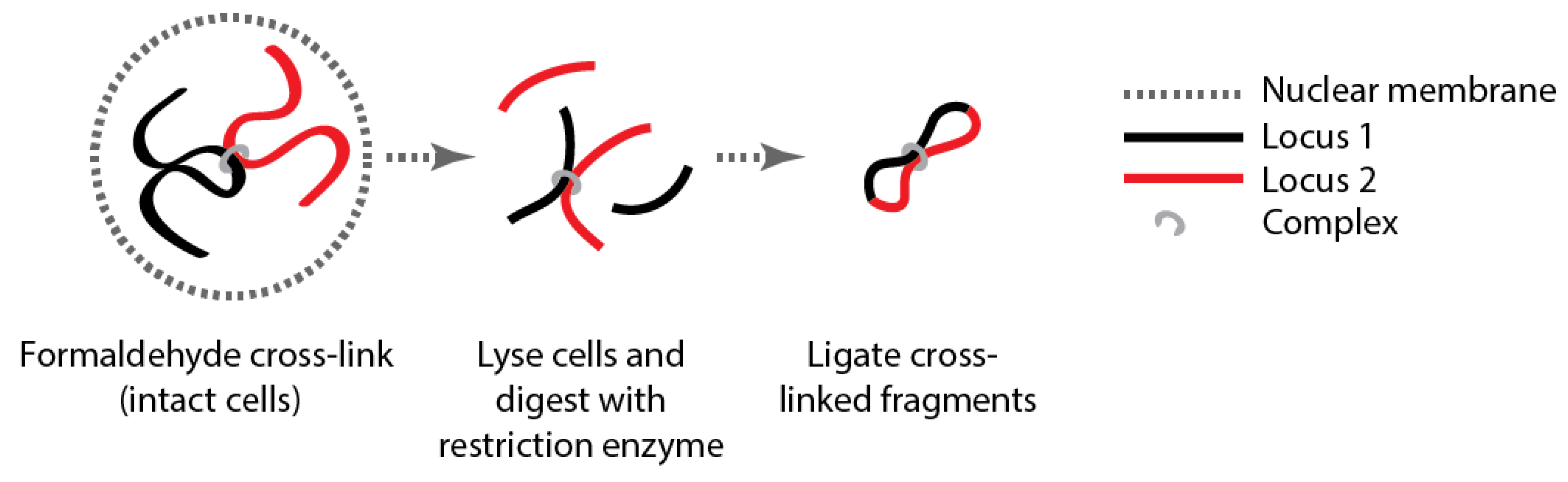{kind=link}
{kind=link}
| Gene Name | DNA Methylation | Histone Deacetylation | Histone Methylation | miRNAs Interference | Dietary Factor | Reference | ||
|---|---|---|---|---|---|---|---|---|
| Agouti | √ | folic acid, vitamin B12, betaine, choline & genistein, bisphenol A | [7,8] | |||||
| Axin-Fused | √ | folic acid, vitamin B12, betaine, choline | [9] | |||||
| LPLRAP1 | √ | rice diet | [10] | |||||
| GLUT4 | √ | calorie restriction & protein restriction during gestation | [11,12] | |||||
| P21 | √ | sulforaphane | [13] | |||||
| BAX | √ | sulforaphane | [13] | |||||
| IGF2 | √ | √ | √ | prenatal exposure to famine & protein restriction during gestation | [14,15] | |||
| INSIGF | √ | prenatal exposure to famine | [16] | |||||
| GNASAS | √ | prenatal exposure to famine | [16] | |||||
| MEG3 | √ | prenatal exposure to famine | [16] | |||||
| IL10 | √ | prenatal exposure to famine | [16] | |||||
| ABCA1 | √ | prenatal exposure to famine | [16] | |||||
| LEP | √ | prenatal exposure to famine & low calorie diet | [16,17] | |||||
| POMC | √ | √ | maternal undernutrition, twinning | [18] | ||||
| FASN | √ | high fat diet | [19] | |||||
| TNFα | √ | n-6 PUFA uptake | [20] | |||||
| Metastable epialleles (BOLA3, LOC654433, EXD3, ZFYVE28, RBM46ZNF678 | √ | seasonal periconceptual dietary intakes of methionine, choline, betaine, cofactors (folate, vitamins B2, B6, B12, active B12) | [4] | |||||
2. Genome Organization
3. How Are Genomes Organized?
Box 1 Genome organization.
| Method | Key Adaptation | Detects | Reference |
|---|---|---|---|
| 3C | A priori choice of both interacting sequences | Intereactions between two loci | [23] |
| 4C | A priori choice of one interacting sequence | Interactions | [38,39] |
| 5C | Design of primers for indepth interogation of structure of one region | One region | [40] |
| GCC3C-seq | Sequence all products in ligated library | All interactions in genome | [41,42] |
| HiC | Enrich for biotin labelled restriction sites after ligation | All interactions in genome | [43,44] |
| 6C ChIA-PET | Enrich for interactions involving protein of interest. Enrich for interactions involving transcription factors | All interactions mediated by protein of interest. All interactions with a given protein | [45,46] |
| Chip-Loop | Detects the role of specific transcription factors between a known promotor and enhancer | Two regions associated with a given protein | [47] |
| Enhanced 4C (e4C) | Analysis of transcriptional interactions genomewide | Interactions between transcribing genes | [48] |
| Associated Chromosome Trap (ACT) | Investigate mechnanisms of transcriptional regulation in trans | Detects distant regions interacting or in proximity with specific target | [49] |
4. Is Genome Organization Necessary for Cellular and Subsequent Levels of Function?
5. Can Chromatin Structure Respond to Nutritional Factors?
6. Estrogen Exposure and Chromatin Organization
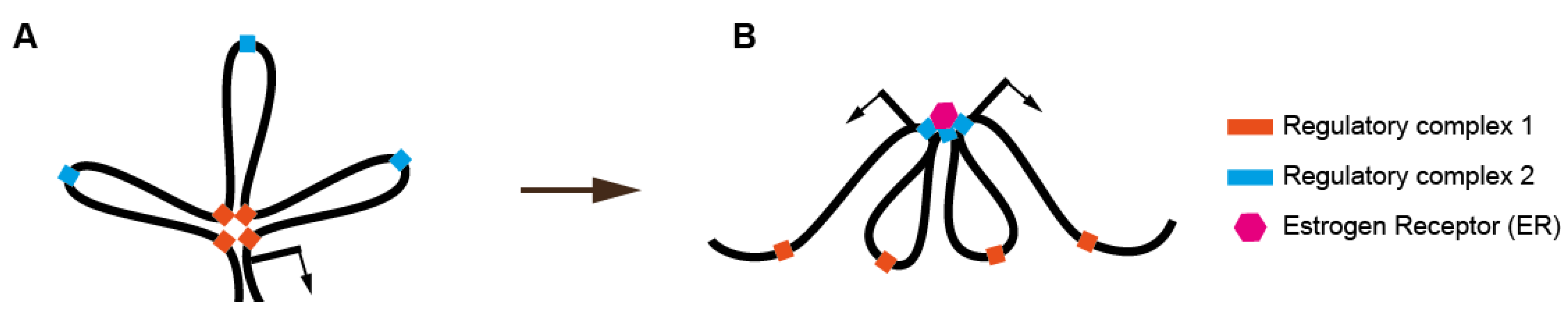
7. Organizing the Whole Genome or Part of the Whole?
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Choi, S.-W.; Friso, S. Epigenetics: A new bridge between nutrition and health. Adv. Nutr. 2010, 1, 8–16. [Google Scholar]
- Dauncey, M.J. Nutrition, the brain and cognitive decline: Insights from epigenetics. Eur. J. Clin. Nutr. 2014, 68, 1179–1185. [Google Scholar]
- Dauncey, M.J.; White, P.; Burton, K.A.; Katsumata, M. Nutrition—Hormone receptor—Gene interactions: Implications for development and disease. Proc. Nutr. Soc. 2007, 60, 63–72. [Google Scholar]
- Dominguez-Salas, P.; ominguez-Salas, P.; Moore, S.E.; Baker, M.S.; Bergen, A.W.; Cox, S.E.; Dyer, R.A.; Fulford, A.J.; Guan, Y.; Laritsky, E.; et al. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Ptashne, M. Epigenetics: Core misconcept. Proc. Natl. Acad. Sci. USA 2013, 110, 7101–7103. [Google Scholar]
- Drummond, E.M.; Gibney, E.R. Epigenetic regulation in obesity. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 392–397. [Google Scholar]
- Wolff, G.L.; Kodell, R.L.; Moore, S.R.; Cooney, C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998, 12, 949–957. [Google Scholar]
- Dolinoy, D.C. The agouti mouse model: An epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. Nutr. Rev. 2008, 66, 7–11. [Google Scholar]
- Waterland, R.A.; Dolinoy, D.C.; Lin, J.; Smith, C.A.; Shi, X.; Tahiliani, K.G. Maternal methyl supplements increase offspring DNA methylation at Axin Fused. Genesis 2006, 44, 401–406. [Google Scholar]
- Zhang, L.; Hou, D.; Chen, X.; Li, D.; Zhu, L.; Zhang, Y.; Li, J.; Bian, Z.; Liang, X.; Cai, X.; et al. Exogenous plant MIR168a specifically targets mammalian LDLRAP1: Evidence of cross-kingdom regulation by microRNA. Cell Res. 2012, 22, 107–126. [Google Scholar]
- Wheatley, K.E.; Nogueira, L.M.; Perkins, S.N.; Hursting, S.D. Differential effects of calorie restriction and exercise on the adipose transcriptome in diet-induced obese mice. J. Obes. 2011, 2011. [Google Scholar] [CrossRef]
- Zheng, S.; Rollet, M.; Pan, Y.-X. Protein restriction during gestation alters histone modifications at the glucose transporter 4 (GLUT4) promoter region and induces GLUT4 expression in skeletal muscle of female rat offspring. J. Nutr. Biochem. 2012, 23, 1064–1071. [Google Scholar]
- Myzak, M.C.; Hardin, K.; Wang, R.; Dashwood, R.H.; Ho, E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis 2006, 27, 811–819. [Google Scholar]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar]
- Zhou, D.; Pan, Y.-X. Gestational low protein diet selectively induces the amino acid response pathway target genes in the liver of offspring rats through transcription factor binding and histone modifications. Biochim. Biophys. Acta 2011, 1809, 549–556. [Google Scholar]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar]
- Cordero, P.; Campion, J.; Milagro, F.I.; Goyenechea, E.; Steemburgo, T.; Javierre, B.M.; Martinez, J.A. Leptin and TNF-alpha promoter methylation levels measured by MSP could predict the response to a low-calorie diet. J. Physiol. Biochem. 2011, 67, 463–470. [Google Scholar]
- Begum, G.; Stevens, A.; Smith, E.B.; Connor, K.; Challis, J.R.G.; Bloomfield, F.; White, A. Epigenetic changes in fetal hypothalamic energy regulating pathways are associated with maternal undernutrition and twinning. FASEB J. 2012, 26, 1694–1703. [Google Scholar]
- Lomba, A.; Martínez, J.A.; García-Díaz, D.F.; Paternain, L.; Marti, A.; ampión, J.; Milagro, F.I. Weight gain induced by an isocaloric pair-fed high fat diet: A nutriepigenetic study on FASN and NDUFB6 gene promoters. Mol. Genet. Metab. 2010, 101, 273–278. [Google Scholar]
- Hermsdorff, H.H.; Mansego, M.L.; Campión, J.; Milagro, F.I.; Zulet, M.A.; Martínez, J.A. TNF-alpha promoter methylation in peripheral white blood cells: Relationship with circulating TNFα, truncal fat and n-6 PUFA intake in young women. Cytokine 2013, 64, 265–271. [Google Scholar]
- Watson, J.D.; Crick, F.H.C. Molecular structure of nucleic acids. Nature 1953, 171, 737–738. [Google Scholar]
- O’Sullivan, J.M.; Sontam, D.M.; Grierson, R.; Jones, B. Repeated elements coordinate the spatial organization of the yeast genome. Yeast 2009, 26, 125–138. [Google Scholar]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar]
- Rosa, A. Topological constraints and chromosome organization in eukaryotes: A physical point of view. Biochem. Soc. Trans. 2013, 41, 612–615. [Google Scholar]
- Fudenberg, G.; Mirny, L.A. Higher-order chromatin structure: Bridging physics and biology. Curr. Opin. Genet. Dev. 2012, 22, 115–124. [Google Scholar]
- Matsuda, A.; Shao, L.; Boulanger, J.; Kervrann, C.; Carlton, P.M.; Kner, P.; Agard, D.; Sedat, J.W. Condensed mitotic chromosome structure at nanometer resolution using PALM and EGFP- histones. PLoS One 2010, 5, 1–12. [Google Scholar]
- Wang, W.; Li, G.-W.; Chen, C.; Xie, X.S.; Zhuang, X. Chromosome organization by a nucleoid-associated protein in live bacteria. Science 2011, 333, 1445–1449. [Google Scholar]
- Steglich, B.; Filion, G.J.; van Steensel, B.; Ekwall, K. The inner nuclear membrane proteins Man1 and Ima1 link to two different types of chromatin at the nuclear periphery in S. pombe. Nucleus 2012, 3, 77–87. [Google Scholar]
- Greil, F.; Moorman, C.; van Steensel, B. DamID: Mapping of in vivo protein-genome interactions using tethered DNA adenine methyltransferase. Methods Enzymol. 2006, 410, 342–359. [Google Scholar]
- Cullen, K.E.; Kladde, M.P.; Seyfred, M.A. Interaction between transcription regulatory regions of prolactin chromatin. Science 1993, 261, 203–206. [Google Scholar]
- Mukherjee, S.; Erickson, H.; Bastia, D. Detection of DNA looping due to simultaneous Interaction of a DNA-binding protein with 2 spatially separated binding-sites on DNA. Proc. Natl. Acad. Sci. USA 1988, 85, 6287–6291. [Google Scholar]
- Grand, R.S.; Gehlen, L.R.; O’Sullivan, J.M. Methods for the investigation of chromosome organization. In Advances in Genetics Research; Urbano, K.V., Ed.; Nova Science Publishers: New York, NY, USA, 2011; Volume 5, pp. 111–130. [Google Scholar]
- De Wit, E.; de Laat, W. A decade of 3C technologies : Insights into nuclear organization. Genes Dev. 2012, 26, 11–24. [Google Scholar]
- Dekker, J.; Marti-Renom, M.A.; Mirny, L.A. Exploring the three-dimensional organization of genomes: Interpreting chromatin interaction data. Nat. Rev. Genet. 2013, 14, 390–403. [Google Scholar]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64. [Google Scholar]
- O’Sullivan, J.; Hendy, M.; Pichugina, T.; Wake, G.; Langowski, J. The statistical-mechanics of chromosome conformation capture. Nucleus 2013, 4, 1–9. [Google Scholar]
- Baker, M. Technology feature: Genomes in three dimensions. Nature 2011, 470, 289–294. [Google Scholar]
- Simonis, M.; Klous, P.; Splinter, E.; Moshkin, Y.; Willemsen, R.; de Wit, E.; van Steensel, B.; de Laat, W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet 2006, 38, 1348–1354. [Google Scholar]
- Zhao, Z.; Tavoosidana, G.; Sjölinder, M.; Göndör, A.; Mariano, P.; Wang, S.; Kanduri, C.; Lezcano, M.; Sandhu, K.S.; Singh, U.; et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat. Genet. 2006, 38, 1341–1347. [Google Scholar]
- Dostie, J.; Richmond, T.A.; Arnaout, R.A.; Selzer, R.R.; Lee, W.L.; Honan, T.A.; Rubio, E.D.; Krumm, A.; Lamb, J.; Nusbaum, C.; et al. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006, 16, 1299–1309. [Google Scholar]
- Rodley, C.D.; Bertels, F.; Jones, B.; O’Sullivan, J.M. Global identification of yeast chromosome interactions using Genome conformation capture. Fungal Genet. Biol. 2009, 46, 879–886. [Google Scholar]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012, 148, 458–472. [Google Scholar]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar]
- Duan, Z.; Andronescu, M.; Schutz, K.; McIlwain, S.; Kim, Y.J.; Lee, C.; Shendure, J.; Fields, S.; Blau, C.A.; Noble, W.S. A three-dimensional model of the yeast genome. Nature 2010, 465, 363–367. [Google Scholar]
- Tiwari, V.K.; Cope, L.; McGarvey, K.M.; Ohm, J.E.; Baylin, S.B. A novel 6C assay uncovers Polycomb-mediated higher order chromatin conformations. Genome Res. 2008, 18, 1171–1179. [Google Scholar]
- Fullwood, M.J.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Mohamed, Y.B.; Orlov, Y.L.; Velkov, S.; Ho, A.; Mei, PH.; et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar]
- Horike, S.; Cai, S.; Miyano, M.; Cheng, J.-F.F.; Kohwi-Shigematsu, T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 2005, 37, 31–40. [Google Scholar]
- Schoenfelder, S.; Sexton, T.; Chakalova, L.; Cope, N.F.; Horton, A.; Andrews, S.; Kurukuti, S.; Mitchell, J.; Umlauf, D.; Dimitrova, D.S.; et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet. 2010, 42, 53–61. [Google Scholar]
- Ling, J.Q.; Li, T.; Hu, J.F.; Vu, T.H.; Chen, H.L.; Qiu, X.W.; Cherry, A.M.; Hoffman, A.R. CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science 2006, 312, 269–272. [Google Scholar]
- Pandita, T.K.; Hunt, C.R.; Sharma, G.G.; Yang, Q. Regulation of telomere movement by telomere chromatin structure. Cell. Mol. Life Sci. 2007, 64, 131–138. [Google Scholar]
- Kind, J.; Pagie, L.; Ortabozkoyun, H.; Boyle, S.; de Vries, S.S.; Janssen, H.; Amendola, M.; Nolen, L.D.; Bickmore, W.A.; van Steensel, B. Single-Cell dynamics of genome-nuclear lamina interactions. Cell 2013, 153, 178–192. [Google Scholar]
- Pickersgill, H.; Kalverda, B.; de Wit, E.; Talhout, W.; Fornerod, M.; van Steensel, B. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat. Genet. 2006, 38, 1005–1014. [Google Scholar]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar]
- De Ribeiro Almeida, C.; Stadhouders, R.; Thongjuea, S.; Soler, E.; Hendriks, R.W. DNA-binding factor CTCF and long-range gene interactions in V(D)J recombination and oncogene activation. Blood 2012, 119, 6209–6218. [Google Scholar]
- Cremer, T.; Cremer, M. Chromosome territories. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Rosa, A.; Everaers, R. Structure and dynamics of interphase chromosomes. PLoS Comput. Biol. 2008, 4. [Google Scholar] [CrossRef]
- Branco, M.R.; Pombo, A. Intermingling of chromosome territories in interphase suggests role in translocations and transcription-dependent associations. PLoS Biol. 2006, 4, 780–788. [Google Scholar]
- Davidson, S.; Macpherson, N.; Mitchell, J.A. Nuclear organization of RNA polymerase II transcription. Biochem. Cell. Biol. 2013, 91, 22–30. [Google Scholar]
- Cavalli, G.; Misteli, T. Functional implications of genome topology. Nat. Struct. Mol. Biol. 2013, 20, 290–299. [Google Scholar]
- Dekker, J. Gene regulation in the third dimension. Science 2008, 319, 1793–1794. [Google Scholar]
- Kurukuti, S.; Tiwari, V.K.; Tavoosidana, G.; Pugacheva, E.; Murrell, A.; Zhao, Z.; Lobanenkov, V.; Reik, W.; Ohlsson, R. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc. Natl. Acad. Sci. USA 2006, 103, 10684–10689. [Google Scholar]
- Noordermeer, D.; de Laat, W. Joining the loops : β-Globin gene regulation. IUBMB Life 2008, 60, 824–833. [Google Scholar]
- Tolhuis, B.; Palstra, R.J.; Splinter, E.; Grosveld, F.; de Laat, W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol. Cell 2002, 10, 1453–1465. [Google Scholar]
- Brown, J.M.; Leach, J.; Reittie, J.E.; Atzberger, A.; Lee-Prudhoe, J.; Wood, W.G.; Higgs, D.R.; Iborra, F.J.; Buckle, V.J. Coregulated human globin genes are frequently in spatial proximity when active. J. Cell Biol. 2006, 172, 177–187. [Google Scholar]
- Deng, W.; Lee, J.; Wang, H.; Miller, J.; Reik, A.; Gregory, P.D.; Blobel, G.A. Controlling long range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 2012, 149, 1233–1244. [Google Scholar]
- Deng, W.; Rupon, J.W.; Krivega, I.; Breda, L.; Motta, I.; Jahn, K.S.; Reik, A.; Gregory, P.D.; Rivella, S.; Dean, A.; et al. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell 2014, 158, 849–860. [Google Scholar]
- Spilianakis, C.G.; Flavell, R.A. Long-range intrachromosomal interactions in the T helper type 2 cytokine locus. Nat. Immunol. 2004, 5, 1017–1027. [Google Scholar]
- Spilianakis, C.G.; Lalioti, M.D.; Town, T.; Lee, G.R.; Flavell, R.A. Interchromosomal associations between alternatively expressed loci. Nature 2005, 435, 637–645. [Google Scholar]
- Lomvardas, S.; Barnea, G.; Pisapia, D.J.; Mendelsohn, M.; Kirkland, J.; Axel, R. Interchromosomal interactions and olfactory receptor choice. Cell 2006, 126, 403–413. [Google Scholar]
- Noordermeer, D.; Duboule, D. Chromatin looping and organization at developmentally regulated gene loci. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 615–630. [Google Scholar]
- Jin, F.; Li, Y.; Dixon, J.R.; Selvaraj, S.; Ye, Z.; Lee, A.Y.; Yen, C.-A.; Schmitt, A.D.; Espinoza, C.A.; Ren, B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 2013, 503, 290–294. [Google Scholar]
- De Wit, E.; Bouwman, B.A.; Zhu, Y.; Klous, P.; Splinter, E.; Verstegen, M.J.; Krijger, P.H.L.; Festuccia, N.; Nora, E.P.; Welling, M.; et al. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature 2013, 501, 227–231. [Google Scholar]
- Cruz, G.; Foster, W.; Paredes, A.; Yi, K.D.; Uzumcu, M. Long-term effects of early-life exposure to environmental oestrogens on ovarian function: Role of epigenetics. J. Neuroendocrinol. 2014, 26, 613–624. [Google Scholar]
- Chao, H.-H.; Zhang, X.-F.; Chen, B.; Pan, B.; Zhang, L.-J.; Li, L.; Sun, X.-F.; Shi, Q.-H.; Shen, W. Bisphenol A exposure modifies methylation of imprinted genes in mouse oocytes via the estrogen receptor signaling pathway. Histochem. Cell Biol. 2012, 137, 249–259. [Google Scholar]
- Susiarjo, M.; Sasson, I.; Mesaros, C.; Bartolomei, M.S. Bisphenol A exposure disrupts genomic imprinting in the mouse. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef]
- Veiga-Lopez, A.; Luense, L.J.; Christenson, L.K.; Padmanabhan, V. Developmental programming: Gestational bisphenol-A treatment alters trajectory of fetal ovarian gene expression. Endocrinology 2013, 154, 1873–1884. [Google Scholar]
- Zama, A.M.; Uzumcu, M. Fetal and neonatal exposure to the endocrine disruptor methoxychlor causes epigenetic alterations in adult ovarian genes. Endocrinology 2009, 150, 4681–4691. [Google Scholar]
- Schmidt, D.; Schwalie, P.C.; Ross-innes, C.S.; Hurtado, A.; Brown, G.D.; Carroll, J.S.; Flicek, P.; Odom, D.T. A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res. 2010; 20, 578–588. [Google Scholar]
- Merkenschlager, M.; Odom, D.T. CTCF and cohesin: Linking gene regulatory elements with their targets. Cell 2013, 152, 1285–1297. [Google Scholar]
- Seitan, V.C.; Faure, A.J.; Zhan, Y.; McCord, R.P.; Lajoie, B.R.; Ing-Simmons, E.; Lenhard, B.; Giorgetti, L.; Heard, E.; Fisher, A.G.; et al. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res. 2013, 23, 2066–2077. [Google Scholar]
- Antony, J.; Dasgupta, T.; Rhodes, J.M.; McEwan, M.V.; Print, C.; O’Sullivan, J.M.; Horsfield, J.A. Cohesin modulates transcription of estrogen-responsive genes. BBA Gene Regul. Mech. 2014, in press. [Google Scholar]
- Zhang, X.; Zhang, L.; Li, L.; Feng, Y.; Chen, B.; Ma, J.; Huynh, E.; Shi, Q.; de Felici, M.; Shen, W. Diethylhexyl phthalate exposure impairs follicular development and affects oocyte maturation in the mouse. Environ. Mol. Mutagen. 2013, 54, 354–361. [Google Scholar]
- Ghirlando, R.; Giles, K.; Gowher, H.; Xiao, T.; Xu, Z.; Yao, H.; Felsenfeld, G. Chromatin domains, insulators, and the regulation of gene expression. Biochim. Biophys. Acta 2012, 1819, 644–651. [Google Scholar]
- Vayssière, J.-L.; Cordeau-Lossouarn, L.; Larcher, J.; Basseville, M.; Gros, F.; Croizat, B. Participation of the mitochondrial genome in the differentiation of neuroblastoma cells. Vitr. Cell. Dev. Biol. Anim. 1992, 28, 763–772. [Google Scholar]
- Blank, H.M.; Li, C.; Mueller, J.E.; Bogomolnaya, L.M.; Bryk, M.; Polymenis, M. An increase in mitochondrial DNA promotes nuclear DNA replication in yeast. PLoS Genet. 2008, 4. [Google Scholar] [CrossRef]
- Clay Montier, L.L.; Deng, J.J.; Bai, Y. Number matters: Control of mammalian mitochondrial DNA copy number. J. Genet. Genomics 2009, 36, 125–131. [Google Scholar]
- Sharpley, M.S.; Marciniak, C.; Eckel-Mahan, K.; McManus, M.; Crimi, M.; Waymire, K.; Lin, C.S.; Masubuchi, S.; Friend, N.; Koike, M.; et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 2012, 151, 333–343. [Google Scholar]
- Samuels, D.C.; Li, C.; Li, B.; Song, Z.; Torstenson, E.; Boyd Clay, H.; Rokas, A.; Thornton-Wells, T.A.; Moore, J.H.; Hughes, T.M.; et al. Recurrent tissue-specific mtDNA mutations are common in humans. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef]
- Ricchetti, M.; Fairhead, C.; Dujon, B. Mitochondrial DNA repairs double-strand breaks in yeast chromosomes. Nature 1999, 402, 96–100. [Google Scholar]
- Ricchetti, M.; Tekaia, F.; Dujon, B. Continued colonization of the human genome by mitochondrial DNA. PLoS Biol. 2004, 2. [Google Scholar] [CrossRef]
- Cheng, X.; Ivessa, A.S. The migration of mitochondrial DNA fragments to the nucleus affects the chronological aging process of Saccharomyces cerevisiae. Aging Cell 2010, 9, 919–923. [Google Scholar]
- Rodley, C.D.; Grand, R.S.; Gehlen, L.R.; Greyling, G.; Jones, M.B.; O’Sullivan, J.M. Mitochondrial-nuclear DNA interactions contribute to the regulation of nuclear transcript levels as part of the inter-organelle communication system. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Grand, R.S.; Martienssen, R.; O’Sullivan, J.M. Functional roles for mitochondrial-nuclear DNA interactions in the Schizosaccharomyces pombe cell cycle. Mitochondrion 2014, 17, 141–149. [Google Scholar]
- Parikh, V.S.; Morgan, M.M.; Scott, R.; Clements, L.S.; Butow, R.A. The mitochondrial genotype can influence nuclear gene expression in yeast. Science 1987, 235, 576–580. [Google Scholar]
- Parikh, V.S.; Conrad-Webb, H.; Docherty, R.; Butow, R.A. Interaction between the yeast mitochondrial and nuclear genomes influences the abundance of novel transcripts derived from the spacer region of the nuclear ribosomal DNA repeat. Mol. Cell. Biol. 1989, 9, 1897–1907. [Google Scholar]
- Myers, R.M.; Stamatoyannopoulos, J.; Snyder, M.; Dunham, I.; Hardison, R.C.; Bernstein, B.E.; Gingeras, T.R.; Kent, W.J.; Birney, E.; Wold, B.; et al. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011, 9. [Google Scholar] [CrossRef]
- Schierding, W.; Cutfield, W.S.; O’Sullivan, J.M. The missing story behind Genome Wide Association Studies: Single nucleotide polymorphisms in gene deserts have a story to tell. Front. Genet. 2014, 5. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O'Sullivan, J.M.; Doynova, M.D.; Antony, J.; Pichlmuller, F.; Horsfield, J.A. Insights from Space: Potential Role of Diet in the Spatial Organization of Chromosomes. Nutrients 2014, 6, 5724-5739. https://doi.org/10.3390/nu6125724
O'Sullivan JM, Doynova MD, Antony J, Pichlmuller F, Horsfield JA. Insights from Space: Potential Role of Diet in the Spatial Organization of Chromosomes. Nutrients. 2014; 6(12):5724-5739. https://doi.org/10.3390/nu6125724
Chicago/Turabian StyleO'Sullivan, Justin M., Malina D. Doynova, Jisha Antony, Florian Pichlmuller, and Julia A. Horsfield. 2014. "Insights from Space: Potential Role of Diet in the Spatial Organization of Chromosomes" Nutrients 6, no. 12: 5724-5739. https://doi.org/10.3390/nu6125724
APA StyleO'Sullivan, J. M., Doynova, M. D., Antony, J., Pichlmuller, F., & Horsfield, J. A. (2014). Insights from Space: Potential Role of Diet in the Spatial Organization of Chromosomes. Nutrients, 6(12), 5724-5739. https://doi.org/10.3390/nu6125724