The Interaction between Epigenetics, Nutrition and the Development of Cancer



Abstract
:1. Introduction
2. Epigenetic Modifications
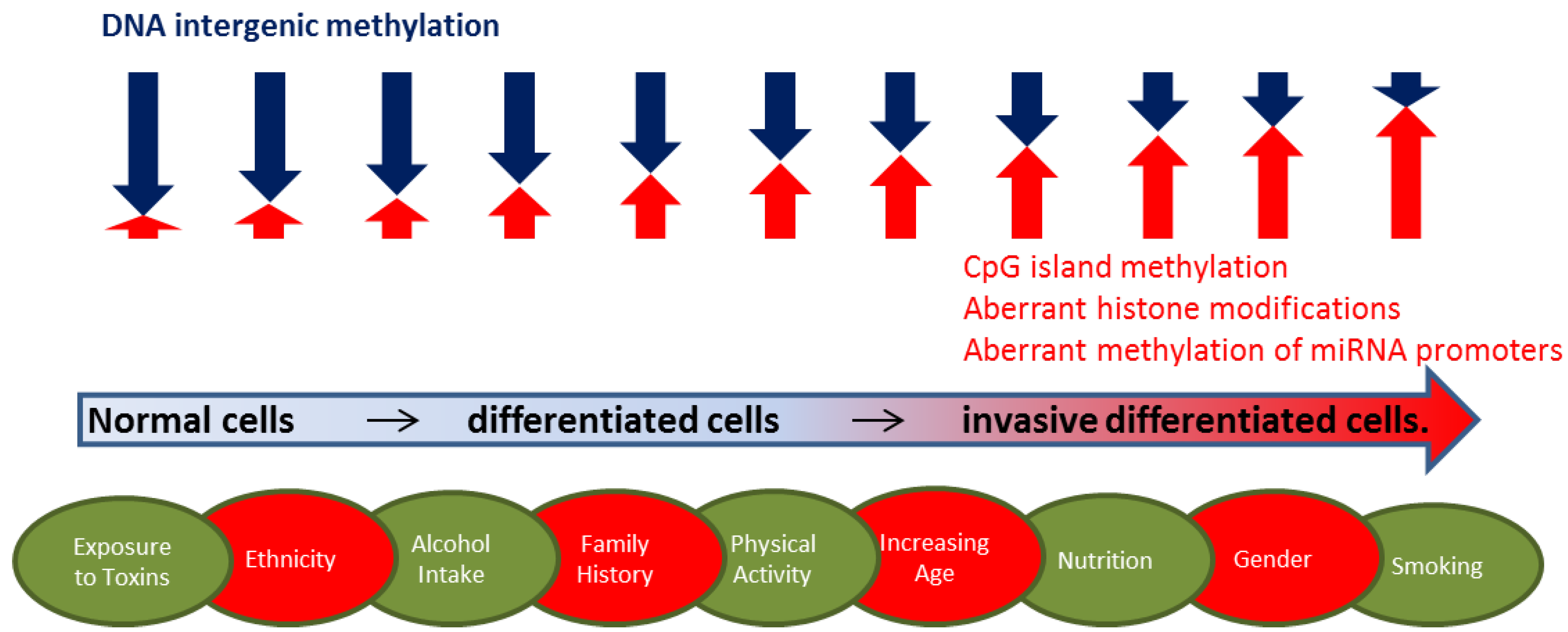
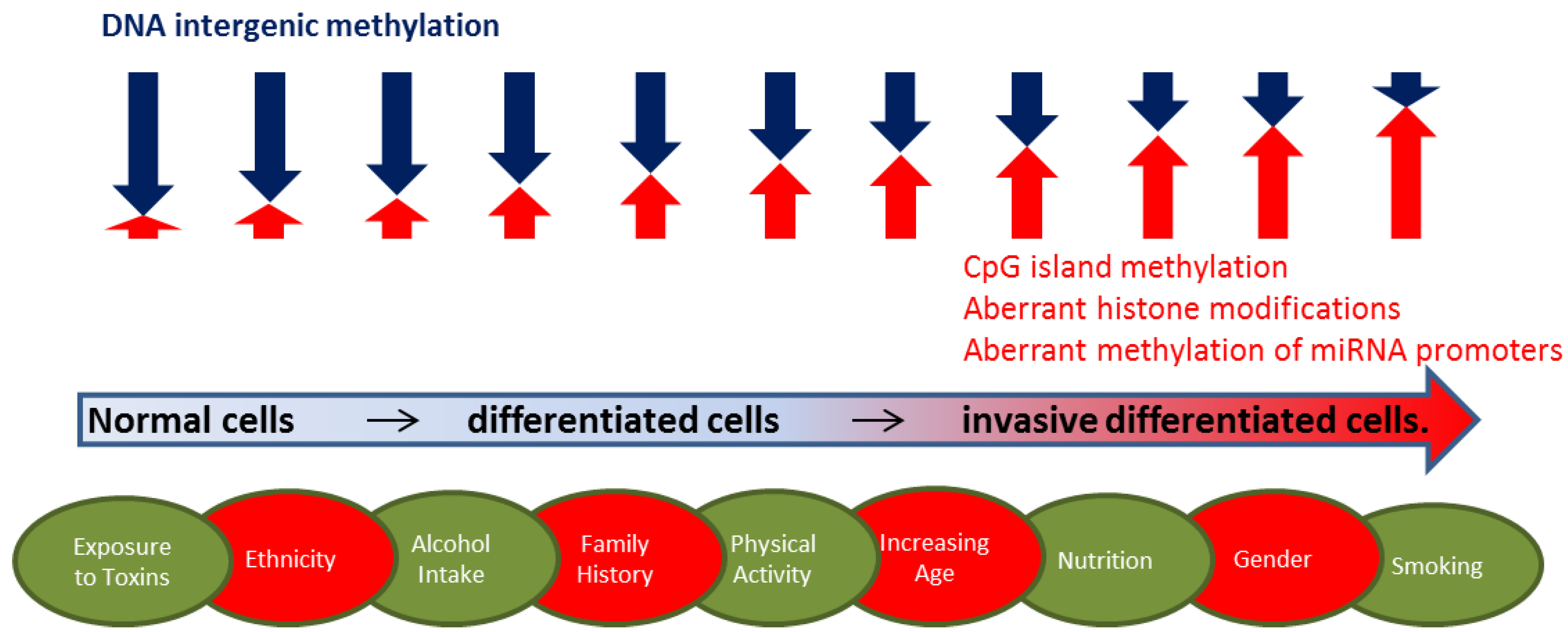
2.1. DNA Methylation
2.2. Histone Modifications
2.3. Small Non-Coding RNA
3. The Impact of Nutrition on Epigenetic Modifications and the Development and/or Progression of Cancers

{kind=link}
| Food Component | Source | Epigenetic or Cellular Effect | Cancer Effect | Reference |
|---|---|---|---|---|
| Polyphenols: Genistein | Soybeans | Suppress expression of the androgen receptor (ER-β); inhibition of DMNT; demethylation of RARβ, p16 and MGMT promoters; demethylation of promoters of miR-29a and miR-1256 | Inhibition of PCa cell proliferation and invasion; decreased risk of PCa and breast cancer | [2,66,71,72,73,74] |
| Polyphenols: Resveratrol | Grapes, peanuts | DNMT 3b inhibitor; decrease in RASSF-1α methylation with increasing circulating resveratrol; Suppress expression of the androgen receptor | Decreased risk of PCa and breast cancer | [36,70,75] |
| Polyphenols: Epigallocatechin-3-gallate | Green tea | Demethylation and/or suppressed methylation of TSG promoters (p15 and p16); inhibits HDAC activity. | Antioxidant activity; inhibition of angiogenesis; induction of apoptosis; inhibited invasive metastasis in a human pancreatic adenocarcinoma cell line. | [4,76,77,78,79] |
| Isothiocyanates | Cruciferous vegetables | Interaction with xenobiotic compounds, smoking and consumption of cruciferous vegetables | Anti-cancer effect: induced apoptosis and suppressed metastatic potential in lung cells. | [80,81,82] |
| Folate | Periconceptional folic acid supplementation; dark green leafy vegetables | Higher IGF2 methylation in offspring; higher hMLH1 promoter methylation | Lower birth weight; association with CRC risk. | [83,84] |
| Zinc | Seafood, beef, lamb | Zinc deficiency may induce protein kinase B and thus inhibit PTEN activity or inhibit alternative cancer associated inflammatory pathways. | Inhibition of cell proliferation in human prostatic carcinoma cell lines; evidence from cell line and mouse model studies (respectively): deficiency may contribute to prostate and oesophageal carcinomer risk and/or progression | [85,86,87] |
| α linoleic acid | Flaxseed | Decreased expression of COX 1 and COX 2 when fed to male Fischer rats; Decreased COX 2 expression when fed to hens; Changed expression of genes associated with brain | Tumour incidence, multiplicity and size decreased; reduction in ovarian cancer incidence and severity; influence on brain development. | [88,89,90,91] |
| development, memory and learning in mice—no correlation between gene expression and methylation status; In mice, maternal supplementation induced hypomethylation of the FADS2 promoter. | ||||
| Omega 3—EPA and DHA | Fish oils | Methylation of the COX 2 promoter in numerous cancer cell lines is linked to COX 2 silencing; Maternal intake of PUFA influences epigenetic regulation of FADS 2 in the offspring. | Fish oils increase apoptosis during tumour initiation and act through the COX 2 pathway; lower levels of COX 2 expression. | [92,93,94] |
| trans fatty acids | Industrially processed foods and low levels in meat. | DNA hypomethylation in the brains of offspring; histone modifications; hypomethylation at the SacII site in the ER gene in response to a diet high in omega 6 PUFA | during seven years of follow-up serum trans MUFA levels were associated with risk of invasive breast cancer. | [95] |
3.1. The Impact of Folate on Epigenetic Modifications Associated with Cancer
3.2. The Impact of Polyphenols on Epigenetic Modifications Associated with Cancer
3.2.1. Green Tea
3.2.2. Resveratrol
3.2.3. Caffeic Acid
3.2.4. Genistein/Daidzein
3.2.5. Selenium
3.2.6. Isothiocyanates
3.2.7. Vitamin D
3.2.8. Lycopene
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Burger, G.C.E.; Sandstead, H.R.; Drummond, J. Starvation in Western Holland: 1945. Lancet 1945, 246, 282–283. [Google Scholar] [CrossRef]
- Barrera, L.N.; Cassidy, A.; Johnson, I.T.; Bao, Y.; Belshaw, N.J. Epigenetic and antioxidant effects of dietary isothiocyanates and selenium: Potential implications for cancer chemoprevention. Proc. Nutr. Soc. 2012, 71, 237–245. [Google Scholar] [CrossRef]
- Bygren, L.O.; Tinghog, P.; Carstensen, J.; Edvinsson, S.; Kaati, G.; Pembrey, M.E.; Sjostrom, M. Change in paternal grandmothers’ early food supply influenced cardiovascular mortality of the female grandchildren. BMC Genet. 2014, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Berner, C.; Aumuller, E.; Gnauck, A.; Nestelberger, M.; Just, A.; Haslberger, A.G. Epigenetic control of estrogen receptor expression and tumor suppressor genes is modulated by bioactive food compounds. Ann. Nutr. Metab. 2010, 57, 183–189. [Google Scholar] [CrossRef]
- Youngson, N.A.; Whitelaw, E. Transgenerational epigenetic effects. Annu. Rev. Genomics Hum. Genet. 2008, 9, 233–257. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef]
- Supic, G.; Jagodic, M.; Magic, Z. Epigenetics: A New Link Between Nutrition and Cancer. Nutr. Cancer 2013, 65, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Faulk, C.; Dolinoy, D.C. Timing is everything: The when and how of environmentally induced changes in the epigenome of animals. Epigenetics Off. J. DNA Methylation Soc. 2011, 6, 791–797. [Google Scholar] [CrossRef]
- Jiménez-Chillarón, J.C.; Díaz, R.; Martínez, D.; Pentinat, T.; Ramón-Krauel, M.; Ribó, S.; Plösch, T. The role of nutrition on epigenetic modifications and their implications on health. Biochimie 2012, 94, 2242–2263. [Google Scholar]
- Hughes, L.A.E.; van den Brandt, P.A.; de Bruïne, A.P.; Wouters, K.A.D.; Hulsmans, S.; Spiertz, A.; Goldbohm, R.A.; de Goeij, A.F.P.M.; Herman, J.G.; Weijenberg, M.P.; et al. Early Life Exposure to Famine and Colorectal Cancer Risk: A Role for Epigenetic Mechanisms. PLoS One 2009, 4, e7951. [Google Scholar]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef]
- Schulz, L.C. The Dutch Hunger Winter and the developmental origins of health and disease. Proc. Natl. Acad. Sci. USA 2010, 107, 16757–16758. [Google Scholar] [CrossRef]
- Kyle, U.G.; Pichard, C. The Dutch Famine of 1944–1945: A pathophysiological model of long-term consequences of wasting disease. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 388–394. [Google Scholar] [CrossRef]
- Pembrey, M.; Bygren, L.; Golding, J. The Nature of Human Transgenerational Responses. In Environmental Epigenomics in Health and Disease; Springer Berlin Heidelberg: Berlin, Germany, 2013; pp. 257–271. [Google Scholar]
- Lillycrop, K.A.; Burdge, G.C. Epigenetic mechanisms linking early nutrition to long term health. Best Pract. Res. Clin. Endocr. Metab. 2012, 26, 667–676. [Google Scholar] [CrossRef]
- Rayman, M.P. Selenium in cancer prevention: A review of the evidence and mechanism of action. Proc. Nutr. Soc. 2005, 64, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, T.; Inoue, M.; Sasazuki, S.; Iwasaki, M.; Kurahashi, N.; Shimazu, T.; Tsugane, S.; Japan Public Health Center-based Prospective Study Group. Fruit and vegetable consumption and squamous cell carcinoma of the esophagus in Japan: The JPHC study. Int. J. Cancer 2008, 123, 1935–1940. [Google Scholar]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: A comprehensive review. Cell. Mol. Life Sci. 2007, 64, 1105–1127. [Google Scholar] [CrossRef]
- American Institute for Cancer Research. Preventing Colon Cancer: Six Steps to Reduce Your Risk. Available online: http://www.aicr.org/press/press-releases/preventing-colon-cancer-6-steps.html (accessed on 7 July 2014).
- Teegarden, D.; Romieu, I.; Lelièvre, S.A. Redefining the impact of nutrition on breast cancer incidence: Is epigenetics involved? Nutr. Res. Rev. 2012, 25, 68–95. [Google Scholar] [CrossRef]
- Ramadas, A.; Chan, C.K.; Oldenburg, B.; Hussien, Z.; Quek, K. A Web-Based Dietary Intervention for People with Type 2 Diabetes: Development, Implementation, and Evaluation. Int. J. Behav. Med. 2014, 1–9. [Google Scholar] [CrossRef]
- Kristal, A.R.; Beresford, S.A.; Lazovich, D. Assessing change in diet-intervention research. Am. J. Clin. Nutr. 1994, 59, 185S–189S. [Google Scholar]
- Kristal, A.R.; Peters, U.; Potter, J.D. Is it time to abandon the food frequency questionnaire? Cancer Epidemiol. Biomark. Prev. 2005, 14, 2826–2828. [Google Scholar] [CrossRef]
- Brunner, E.; Stallone, D.; Juneja, M.; Bingham, S.; Marmot, M. Dietary assessment in Whitehall II: Comparison of 7 d diet diary and food-frequency questionnaire and validity against biomarkers. Br. J. Nutr. 2001, 86, 405–414. [Google Scholar]
- Buzzard, I.M.; Faucett, C.L.; Jeffery, R.W.; McBane, L.; McGovern, P.; Baxter, J.S.; Shapiro, A.C.; Blackburn, G.L.; Chlebowski, R.T.; Elashoff, R.M.; et al. Monitoring dietary change in a low-fat diet intervention study: Advantages of using 24-h dietary recalls vs. food records. J. Am. Diet. Assoc. 1996, 96, 574–579. [Google Scholar] [CrossRef]
- Day, N.; McKeown, N.; Wong, M.; Welch, A.; Bingham, S. Epidemiological assessment of diet: A comparison of a 7-day diary with a food frequency questionnaire using urinary markers of nitrogen, potassium and sodium. Int. J. Epidemiol. 2001, 30, 309–317. [Google Scholar] [CrossRef]
- Shukla, S.; Meeran, S.M.; Katiyar, S.K. Epigenetic regulation by selected dietary phytochemicals in cancer chemoprevention. Cancer Lett. 2014, 355, 9–17. [Google Scholar]
- Cooper, D.N.; Youssoufian, H. The CpG dinucleotide and human genetic disease. Hum. Genet. 1988, 78, 151–155. [Google Scholar]
- Cui, H.; Horon, I.L.; Ohlsson, R.; Hamilton, S.R.; Feinberg, A.P. Loss of imprinting in normal tissue of colorectal cancer patients with microsatellite instability. Nat. Med. 1998, 4, 1276–1280. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Correa, M.; Cui, H.; Giardiello, F.M.; Powe, N.R.; Hylind, L.; Robinson, A.; Hutcheon, D.F.; Kafonek, D.R.; Brandenburg, S.; Wu, Y.; et al. Loss of imprinting of insulin growth factor II gene: A potential heritable biomarker for colon neoplasia predisposition. Gastroenterology 2004, 126, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Hernandez, R.; Zhang, X.Y.; Qu, G.Z.; Frady, A.; Varela, M.; Ehrlich, M. DNA demethylation and pericentromeric rearrangements of chromosome 1. Mutat. Res. 1997, 379, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Suter, C.; Martin, D.; Ward, R. Hypomethylation of L1 retrotransposons in colorectal cancer and adjacent normal tissue. Int. J. Colorectal Dis. 2004, 19, 95–101. [Google Scholar] [CrossRef]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar]
- Cheng, J.C.; Matsen, C.B.; Gonzales, F.A.; Ye, W.; Greer, S.; Marquez, V.E.; Jones, P.A.; Selker, E.U. Inhibition of DNA Methylation and Reactivation of Silenced Genes by Zebularine. J. Natl Cancer Inst. 2003, 95, 399–409. [Google Scholar] [CrossRef]
- Davis, C.D.; Uthus, E.O. Dietary Selenite and Azadeoxycytidine Treatments Affect Dimethylhydrazine-Induced Aberrant Crypt Formation in Rat Colon and DNA Methylation in HT-29 Cells. J. Nutr. 2002, 132, 292–297. [Google Scholar]
- Qin, W.; Zhang, K.; Clarke, K.; Weiland, T.; Sauter, E.R. Methylation and miRNA Effects of Resveratrol on Mammary Tumors vs. Normal Tissue. Nutr. Cancer 2014, 66, 270–277. [Google Scholar]
- Jin, W.; Lee, J.J.; Kim, M.S.; Son, B.H.; Cho, Y.K.; Kim, H.P. DNA methylation-dependent regulation of TrkA, TrkB, and TrkC genes in human hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2011, 406, 89–95. [Google Scholar] [CrossRef]
- Yang, J.; Ji, W.; Qu, Y.; He, L.; Zhao, X. Study of RASSF1A expression and promoter demethylation in Hep-2 cell line. J. Clin. Otorhinolaryngol. Head Neck Surg. 2011, 25, 64–66. [Google Scholar]
- Rodriguez, R.M.; Huidobro, C.; Urdinguio, R.G.; Mangas, C.; Soldevilla, B.; Domínguez, G.; Bonilla, F.; Fernandez, A.F.; Fraga, M.F. Aberrant epigenetic regulation of bromodomain Brd4 in human colon cancer. J. Mol. Med. 2012, 90, 587–595. [Google Scholar]
- Fandy, T.E.; Herman, J.G.; Kerns, P.; Jiemjit, A.; Sugar, E.A.; Choi, S.-H.; Yang, A.S.; Aucott, T.; Dauses, T.; Odchimar-Reissig, R.; et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood 2009, 114, 2764–2773. [Google Scholar]
- Kim, M.S.; Louwagie, J.; Carvalho, B.; Terhaar Sive Droste, J.S.; Park, H.L.; Chae, Y.K.; Yamashita, K.; Liu, J.; Ostrow, K.L.; Ling, S.; et al. Promoter DNA methylation of oncostatin m receptor-beta as a novel diagnostic and therapeutic marker in colon cancer. PLoS One 2009, 4, e6555. [Google Scholar] [CrossRef]
- Ahmed, D.; Danielsen, S.A.; Aagesen, T.H.; Bretthauer, M.; Thiis-Evensen, E.; Hoff, G.; Rognum, T.O.; Nesbakken, A.; Lothe, R.A.; Lind, G.E. A tissue-based comparative effectiveness analysis of biomarkers for early detection of colorectal tumors. Clin. Transl. Gastroenterol. 2012, 3, e27. [Google Scholar] [CrossRef]
- Deng, G.; Kakar, S.; Okudiara, K.; Choi, E.; Sleisenger, M.H.; Kim, Y.S. Unique Methylation Pattern of Oncostatin M Receptor Gene in Cancers of Colorectum and Other Digestive Organs. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 1519–1526. [Google Scholar] [CrossRef]
- Hibi, K.; Goto, T.; Sakuraba, K.; Shirahata, A.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; Sanada, Y. Methylation of OSMR Gene Is Frequently Observed in Non-invasive Colorectal Cancer. Anticancer Res. 2011, 31, 1293–1295. [Google Scholar]
- Duthie, S.J. Epigenetic modifications and human pathologies: Cancer and CVD. Proc. Nutr. Soc. 2011, 70, 47–56. [Google Scholar]
- Kim, M.S.; Lee, J.; Sidransky, D. DNA methylation markers in colorectal cancer. Cancer Metastasis Rev. 2010, 29, 181–206. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T.; Reinberg, D. Overview and Concepts. In Epigenetics; John Inglis: New York, NY, USA, 2007; pp. 23–61. [Google Scholar]
- Kramer, J.M. Epigenetic regulation of memory: Implications in human cognitive disorders. BMC 2013, 4, 1–12. [Google Scholar]
- Luger, K. Structure and dynamic behavior of nucleosomes. Curr. Opin. Genet. Dev. 2003, 13, 127–135. [Google Scholar] [CrossRef]
- Toraño, E.G.; Fernandez, A.F.; Urdinguio, R.G.; Fraga, M.F. Role of Epigenetics in Neural Differentiation: Implications for Health and Disease. In Molecular Mechanisms and Physiology of Disease; Springer: New York, NY, USA, 2014. [Google Scholar]
- Pan, Y.-X.; Zhang, Y.; Chen, D. Epigenetic Mechanisms of Colon Cancer Prevention: What Can Nutrition Do? In Molecular Mechanisms and Physiology of Disease; Springer: New York, NY, USA, 2014; pp. 277–353. [Google Scholar]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar]
- Mathews, L.A.; Crea, F.; Farrar, W.L. Epigenetic gene regulation in stem cells and correlation to cancer. Differ. Res. Biol. Divers. 2009, 78, 1–17. [Google Scholar] [CrossRef]
- Paolicchi, E.; Crea, F.; Farrar, W.L.; Green, J.E.; Danesi, R. Histone lysine demethylases in breast cancer. Crit. Rev. Oncol. Hematol. 2013, 86, 97–103. [Google Scholar] [CrossRef]
- Bianco-Miotto, T.; Chiam, K.; Buchanan, G.; Jindal, S.; Day, T.K.; Thomas, M.; Pickering, M.A.; O’Loughlin, M.A.; Ryan, N.K.; Raymond, W.A.; et al. Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2611–2622. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, L.; Wang, Q.; Li, W. Histone modifications and chromatin organization in prostate cancer. Epigenomics 2010, 2, 551–560. [Google Scholar]
- Rajendran, P.; Delage, B.; Dashwood, W.M.; Yu, T.W.; Wuth, B.; Williams, D.E.; Ho, E.; Dashwood, R.H. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: Competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol. Cancer 2011, 10, 68. [Google Scholar]
- Ashktorab, H.; Belgrave, K.; Hosseinkhah, F.; Brim, H.; Nouraie, M.; Takkikto, M.; Hewitt, S.; Lee, E.; Dashwood, R.H.; Smoot, D. Global Histone H4 Acetylation and HDAC2 Expression in Colon Adenoma and Carcinoma. Dig. Dis. Sci. 2009, 54, 2109–2117. [Google Scholar]
- Liloglou, T.; Bediaga, N.G.; Brown, B.R.; Field, J.K.; Davies, M.P. Epigenetic biomarkers in lung cancer. Cancer Lett. 2014, 342, 200–212. [Google Scholar]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar]
- Lopez-Serra, P.; Esteller, M. DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer. Oncogene 2012, 31, 1609–1622. [Google Scholar]
- Lujambio, A.; Ropero, S.; Ballestar, E.; Fraga, M.F.; Cerrato, C.; Setién, F.; Casado, S.; Suarez-Gauthier, A.; Sanchez-Cespedes, M.; Gitt, A.; et al. Genetic Unmasking of an Epigenetically Silenced microRNA in Human Cancer Cells. Cancer Res. 2007, 67, 1424–1429. [Google Scholar]
- Wilting, S.; van Boerdonk, R.; Henken, F.; Meijer, C.; Diosdado, B.; Meijer, G.; le Sage, C.; Agami, R.; Snijders, P.; Steenbergen, R. Methylation-mediated silencing and tumour suppressive function of hsa-miR-124 in cervical cancer. Mol. Cancer 2010, 9, 167. [Google Scholar]
- Ando, T.; Yoshida, T.; Enomoto, S.; Asada, K.; Tatematsu, M.; Ichinose, M.; Sugiyama, T.; Ushijima, T. DNA methylation of microRNA genes in gastric mucosae of gastric cancer patients: Its possible involvement in the formation of epigenetic field defect. Int. J. Cancer J. Int. Cancer 2009, 124, 2367–2374. [Google Scholar] [CrossRef]
- Van Wolfswinkel, J.C.; Ketting, R.F. The role of small non-coding RNAs in genome stability and chromatin organization. J. Cell Sci. 2010, 123, 1825–1839. [Google Scholar]
- Li, Y.; Kong, D.; Ahmad, A.; Bao, B.; Dyson, G.; Sarkar, F.H. Epigenetic deregulation of miR-29a and miR-1256 by isoflavone contributes to the inhibition of prostate cancer cell growth and invasion. Epigenetics Off. J. DNA Methylation Soc. 2012, 7, 940–949. [Google Scholar] [CrossRef]
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007, 449, 682–688. [Google Scholar]
- Suh, S.O.; Chen, Y.; Zaman, M.S.; Hirata, H.; Yamamura, S.; Shahryari, V.; Liu, J.; Tabatabai, Z.L.; Kakar, S.; Deng, G.; et al. MicroRNA-145 is regulated by DNA methylation and p53 gene mutation in prostate cancer. Carcinogenesis 2011, 32, 772–778. [Google Scholar]
- Lujambio, A.; Esteller, M. How epigenetics can explain human metastasis: A new role for microRNAs. Cell Cycle 2009, 8, 377–382. [Google Scholar]
- Verma, M. Cancer control and prevention by nutrition and epigenetic approaches. Antioxid. Redox Signal. 2012, 17, 355–364. [Google Scholar] [CrossRef]
- Fang, M.Z.; Chen, D.; Sun, Y.; Jin, Z.; Christman, J.K.; Yang, C.S. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin. Cancer Res. 2005, 11, 7033–7041. [Google Scholar] [CrossRef]
- Chen, M.; Rao, Y.; Zheng, Y.; Wei, S.; Li, Y.; Guo, T.; Yin, P. Association between soy isoflavone intake and breast cancer risk for pre- and post-menopausal women: A meta-analysis of epidemiological studies. PLoS One 2014, 9, e89288. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Weidman, J.R.; Waterland, R.A.; Jirtle, R.L. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ. Health Perspect. 2006, 114, 567–572. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Huang, D.; Jirtle, R.L. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. USA 2007, 104, 13056–13061. [Google Scholar] [CrossRef]
- Zhu, W.; Qin, W.; Zhang, K.; Rottinghaus, G. E.; Chen, Y.-C.; Kliethermes, B.; Sauter, E.R. Trans-Resveratrol Alters Mammary Promoter Hypermethylation in Women at Increased Risk for Breast Cancer. Nutr. Cancer 2012, 64, 393–400. [Google Scholar]
- Berletch, J.B.; Liu, C.; Love, W.K.; Andrews, L.G.; Katiyar, S.K.; Tollefsbol, T.O. Epigenetic and genetic mechanisms contribute to telomerase inhibition by EGCG. J. Cell. Biochem. 2008, 103, 509–519. [Google Scholar]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (−)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar]
- Kim, S.O.; Kim, M.R. (−)-Epigallocatechin 3-gallate inhibits invasion by inducing the expression of Raf kinase inhibitor protein in AsPC1 human pancreatic adenocarcinoma cells through the modulation of histone deacetylase activity. Int. J. Oncol. 2013, 42, 349–358. [Google Scholar]
- Holst, B.; Williamson, G. A critical review of the bioavailability of glucosinolates and related compounds. Nat. Prod. Rep. 2004, 21, 425–447. [Google Scholar] [CrossRef]
- Verkerk, R.; Schreiner, M.; Krumbein, A.; Ciska, E.; Holst, B.; Rowland, I.; de Schrijver, R.; Hansen, M.; Gerhauser, C.; Mithen, R.; Dekker, M. Glucosinolates in Brassica vegetables: The influence of the food supply chain on intake, bioavailability and human health. Mol. Nutr. Food Res. 2009, 53 (Suppl. 2), S219. [Google Scholar] [CrossRef]
- Yan, H.; Zhu, Y.; Liu, B.; Wu, H.; Li, Y.; Wu, X.; Zhou, Q.; Xu, K. Mitogen-activated protein kinase mediates the apoptosis of highly metastatic human non-small cell lung cancer cells induced by isothiocyanates. Br. J. Nutr. 2011, 106, 1779–1791. [Google Scholar]
- Steegers-Theunissen, R.P.; Obermann-Borst, S.A.; Kremer, D.; Lindemans, J.; Siebel, C.; Steegers, E.A.; Slagboom, P.E.; Heijmans, B.T. Periconceptional maternal folic acid use of 400 microg per day is related to increased methylation of the IGF2 gene in the very young child. PLoS One 2009, 4, e7845. [Google Scholar] [CrossRef]
- Coppedè, F. Epigenetic biomarkers of colorectal cancer: Focus on DNA methylation. Cancer Lett. 2014, 342, 238–247. [Google Scholar]
- Han, C.T.; Schoene, N.W.; Lei, K.Y. Influence of zinc deficiency on Akt-Mdm2-p53 and Akt-p21 signaling axes in normal and malignant human prostate cells. Am. J. Physiol. Cell Physiol. 2009, 297, C1188–C1199. [Google Scholar] [CrossRef]
- Wan, S.G.; Taccioli, C.; Jiang, Y.; Chen, H.; Smalley, K.J.; Huang, K.; Liu, X.P.; Farber, J.L.; Croce, C.M.; Fong, L.Y. Zinc deficiency activates S100A8 inflammation in the absence of COX-2 and promotes murine oral-esophageal tumor progression. Int. J. Cancer 2011, 129, 331–345. [Google Scholar]
- Liang, J.Y.; Liu, Y.Y.; Zou, J.; Franklin, R.B.; Costello, L.C.; Feng, P. Inhibitory effect of zinc on human prostatic carcinoma cell growth. Prostate 1999, 40, 200–207. [Google Scholar]
- Bommareddy, A.; Arasada, B.L.; Mathees, D.P.; Dwivedi, C. Chemopreventive effects of dietary flaxseed on colon tumor development. Nutr. Cancer 2006, 54, 216–222. [Google Scholar]
- Eilati, E.; Bahr, J.; Hales, D. Long term consumption of flaxseed enriched diet decreased ovarian cancer incidence and prostaglandin E2 in hens. Gynecol. Oncol. 2013, 130, 620–628. [Google Scholar] [CrossRef]
- He, F.; Lupu, D.S.; Niculescu, M.D. Perinatal α-linolenic acid availability alters the expression of genes related to memory and to epigenetic machinery, and the Mecp2 DNA methylation in the whole brain of mouse offspring. Int. J. Dev. Neurosci. 2014, 36, 38–44. [Google Scholar] [CrossRef]
- Makrides, M.; Collins, C.T.; Gibson, R.A. Impact of fatty acid status on growth and neurobehavioural development in humans. Matern. Child. Nutr. 2011, 7, 80–88. [Google Scholar]
- Hong, S.K.; Kim, J.H.; Lin, M.F.; Park, J.I. The Raf/MEK/extracellular signal-regulated kinase 1/2 pathway can mediate growth inhibitory and differentiation signaling via androgen receptor downregulation in prostate cancer cells. Exp. Cell Res. 2011, 317, 2671–2682. [Google Scholar] [CrossRef]
- Romagnolo, D.F.; Papoutsis, A.J.; Selmin, O. Nutritional Targeting of Cyclooxygenase-2 for Colon Cancer Prevention. Inflamm. Allergy Drug Targets 2010, 9, 181–191. [Google Scholar] [CrossRef]
- Hoile, S.P.; Irvine, N.A.; Kelsall, C.J.; Sibbons, C.; Feunteun, A.; Collister, A.; Torrens, C.; Calder, P.C.; Hanson, M.A.; Lillycrop, K.A.; et al. Maternal fat intake in rats alters 20:4n-6 and 22:6n-3 status and the epigenetic regulation of Fads2 in offspring liver. J. Nutr. Biochem. 2013, 24, 1213–1220. [Google Scholar]
- Chajes, V.; Thiebaut, A.C.; Rotival, M.; Gauthier, E.; Maillard, V.; Boutron-Ruault, M.C.; Joulin, V.; Lenoir, G.M.; Clavel-Chapelon, F. Association between serum trans-monounsaturated fatty acids and breast cancer risk in the E3N-EPIC Study. Am. J. Epidemiol. 2008, 167, 1312–1320. [Google Scholar]
- Soubry, A.; Murphy, S.K.; Wang, F.; Huang, Z.; Vidal, A.C.; Fuemmeler, B.F.; Kurtzberg, J.; Murtha, A.; Jirtle, R.L.; Schildkraut, J.M.; et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int. J. Obes. (Lond.) 2013. [Google Scholar] [CrossRef]
- Rampersaud, G.C.; Kauwell, G.P.; Hutson, A.D.; Cerda, J.J.; Bailey, L.B. Genomic DNA methylation decreases in response to moderate folate depletion in elderly women. Am. J. Clin. Nutr. 2000, 72, 998–1003. [Google Scholar]
- Scalbert, A.; Manach, C.; Morand, C.; Rémésy, C.; Jiménez, L. Dietary Polyphenols and the Prevention of Diseases. Crit. Rev. Food Sci. Nutr. 2005, 45, 287–306. [Google Scholar] [CrossRef]
- Li, Y.; Chang, S.C.; Goldstein, B.Y.; Scheider, W.L.; Cai, L.; You, N.C.; Tarleton, H.P.; Ding, B.; Zhao, J.; Wu, M.; et al. Green tea consumption, inflammation and the risk of primary hepatocellular carcinoma in a Chinese population. Cancer Epidemiol. 2011, 35, 362–368. [Google Scholar] [CrossRef]
- Khan, N.; Mukhtar, H. Cancer and metastasis: Prevention and treatment by green tea. Cancer Metastasis Rev. 2010, 29, 435–445. [Google Scholar] [CrossRef]
- Steegers-Theunissen, R.P.; Twigt, J.; Pestinger, V.; Sinclair, K.D. The periconceptional period, reproduction and long-term health of offspring: The importance of one-carbon metabolism. Hum. Reprod. Update 2013, 19, 640–655. [Google Scholar] [CrossRef]
- Zeisel, S.H. Epigenetic mechanisms for nutrition determinants of later health outcomes. Am. J. Clin. Nutr. 2009, 89, 1488S–1493S. [Google Scholar] [CrossRef]
- Chang, H.; Zhang, T.; Zhang, Z.; Bao, R.; Fu, C.; Wang, Z.; Bao, Y.; Li, Y.; Wu, L.; Zheng, X.; et al. Tissue-specific distribution of aberrant DNA methylation associated with maternal low-folate status in human neural tube defects. J. Nutr. Biochem. 2011, 22, 1172–1177. [Google Scholar] [CrossRef]
- Van den Donk, M.; Pellis, L.; Crott, J.W.; van Engeland, M.; Friederich, P.; Nagengast, F.M.; van Bergeijk, J.D.; de Boer, S.Y.; Mason, J.B.; Kok, F.J.; et al. Folic acid and vitamin B-12 supplementation does not favorably influence uracil incorporation and promoter methylation in rectal mucosa DNA of subjects with previous colorectal adenomas. J. Nutr. 2007, 137, 2114–2120. [Google Scholar]
- Choi, S.W.; Friso, S. Epigenetics: A New Bridge between Nutrition and Health. Adv. Nutr. 2010, 1, 8–16. [Google Scholar]
- Chen, J.; Gammon, M.D.; Chan, W.; Palomeque, C.; Wetmur, J.G.; Kabat, G.C.; Teitelbaum, S.L.; Britton, J.A.; Terry, M.B.; Neugut, A.I.; et al. One-carbon metabolism, MTHFR polymorphisms, and risk of breast cancer. Cancer Res. 2005, 65, 1606–1614. [Google Scholar]
- Kim, Y.I. Folate and colorectal cancer: An evidence-based critical review. Mol. Nutr. Food Res. 2007, 51, 267–292. [Google Scholar] [CrossRef]
- James, S.J.; Pogribny, I.P.; Pogribna, M.; Miller, B.J.; Jernigan, S.; Melnyk, S. Mechanisms of DNA damage, DNA hypomethylation, and tumor progression in the folate/methyl-deficient rat model of hepatocarcinogenesis. J. Nutr. 2003, 133, 3740S–3747S. [Google Scholar]
- Trasler, J.; Deng, L.; Melnyk, S.; Pogribny, I.; Hiou-Tim, F.; Sibani, S.; Oakes, C.; Li, E.; James, S.J.; Rozen, R. Impact of Dnmt1 deficiency, with and without low folate diets, on tumor numbers and DNA methylation in Min mice. Carcinogenesis 2003, 24, 39–45. [Google Scholar]
- Lelievre, S.A. Contributions of extracellular matrix signaling and tissue architecture to nuclear mechanisms and spatial organization of gene expression control. Biochim. Biophys. Acta 2009, 1790, 925–935. [Google Scholar] [CrossRef]
- Stempak, J.M.; Sohn, K.J.; Chiang, E.P.; Shane, B.; Kim, Y.I. Cell and stage of transformation-specific effects of folate deficiency on methionine cycle intermediates and DNA methylation in an in vitro model. Carcinogenesis 2005, 26, 981–990. [Google Scholar]
- Song, J.; Medline, A.; Mason, J.B.; Gallinger, S.; Kim, Y.I. Effects of dietary folate on intestinal tumorigenesis in the apcMin mouse. Cancer Res. 2000, 60, 5434–5440. [Google Scholar]
- Supic, G.; Jovic, N.; Kozomara, R.; Zeljic, K.; Magic, Z. Interaction between the MTHFR C677T polymorphism and alcohol--impact on oral cancer risk and multiple DNA methylation of tumor-related genes. J. Dent. Res. 2011, 90, 65–70. [Google Scholar]
- Frisco, S.; Choi, S.W.; Girelli, D.; Mason, J.B.; Dolnikowski, G.G.; Bagley, P.J.; Olivieri, O.; Jacques, P.F.; Rosenberg, I.H.; Corrocher, R.; et al. A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc. Natl. Acad. Sci. USA 2002, 99, 5606–5611. [Google Scholar] [CrossRef]
- Van Engeland, M.; Weijenberg, M.P.; Roemen, G.M.; Brink, M.; de Bruine, A.P.; Goldbohm, R.A.; van den Brandt, P.A.; Baylin, S.B.; de Goeij, A.F.; Herman, J.G. Effects of dietary folate and alcohol intake on promoter methylation in sporadic colorectal cancer: The Netherlands cohort study on diet and cancer. Cancer Res. 2003, 63, 3133–3137. [Google Scholar]
- Shrubsole, M.J.; Shu, X.O.; Li, H.-L.; Cai, H.; Yang, G.; Gao, Y.-T.; Gao, J.; Zheng, W. Dietary B Vitamin and Methionine Intakes and Breast Cancer Risk Among Chinese Women. Am. J. Epidemiol. 2011, 173, 1171–1182. [Google Scholar] [CrossRef]
- Duthie, S.J. Folate and cancer: How DNA damage, repair and methylation impact on colon carcinogenesis. J. Inherit. Metab. Dis. 2011, 34, 101–109. [Google Scholar] [CrossRef]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar]
- Mantena, S.K.; Meeran, S.M.; Elmets, C.A.; Katiyar, S.K. Orally Administered Green Tea Polyphenols Prevent Ultraviolet Radiation-Induced Skin Cancer in Mice through Activation of Cytotoxic T Cells and Inhibition of Angiogenesis in Tumors. J. Nutr. 2005, 135, 2871–2877. [Google Scholar]
- Yang, C.S.; Landau, J.M.; Huang, M.T.; Newmark, H.L. Inhibition of carcinogenesis by dietary polyphenolic compounds. Annu. Rev. Nutr. 2001, 21, 381–406. [Google Scholar] [CrossRef]
- Paluszczak, J.; Krajka-Kuzniak, V.; Baer-Dubowska, W. The effect of dietary polyphenols on the epigenetic regulation of gene expression in MCF7 breast cancer cells. Toxicol. Lett. 2010, 192, 119–125. [Google Scholar]
- Binda, O.; Nassif, C.; Branton, P.E. SIRT1 negatively regulates HDAC1-dependent transcriptional repression by the RBP1 family of proteins. Oncogene 2008, 27, 3384–3392. [Google Scholar]
- Ndiaye, M.; Kumar, R.; Ahmad, N. Resveratrol in cancer management: Where are we and where we go from here? Ann. N. Y. Acad. Sci. 2011, 1215, 144–149. [Google Scholar] [CrossRef]
- Fang, M.; Chen, D.; Yang, C.S. Dietary Polyphenols May Affect DNA Methylation. J. Nutr. 2007, 137, 223S–228S. [Google Scholar]
- Geybels, M.S.; Neuhouser, M.L.; Wright, J.L.; Stott-Miller, M.; Stanford, J.L. Coffee and tea consumption in relation to prostate cancer prognosis. Cancer Causes Control 2013, 24, 1947–1954. [Google Scholar] [CrossRef]
- De, P.; Baltas, M.; Bedos-Belval, F. Cinnamic Acid Derivatives as Anticancer Agents-A Review. Curr. Med. Chem. 2011, 18, 1672–1703. [Google Scholar] [CrossRef]
- Constantinou, A.I.; Lantvit, D.; Hawthorne, M.; Xu, X.; van Breemen, R.B.; Pezzuto, J.M. Chemopreventive effects of soy protein and purified soy isoflavones on DMBA-induced mammary tumors in female Sprague-Dawley rats. Nutr. Cancer 2001, 41, 75–81. [Google Scholar]
- Duffy, C.; Cyr, M. Phytoestrogens: Potential Benefits and Implications for Breast Cancer Survivors. J. Women’s Health 2003, 12, 617–631. [Google Scholar]
- Korde, L.A.; Wu, A.H.; Fears, T.; Nomura, A.M.; West, D.W.; Kolonel, L.N.; Pike, M.C.; Hoover, R.N.; Ziegler, R.G. Childhood soy intake and breast cancer risk in Asian American women. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1050–1059. [Google Scholar] [CrossRef]
- Thanos, J.; Cotterchio, M.; Boucher, B.A.; Kreiger, N.; Thompson, L.U. Adolescent dietary phytoestrogen intake and breast cancer risk (Canada). Cancer Causes Control 2006, 17, 1253–1261. [Google Scholar] [CrossRef]
- Qin, W.; Zhu, W.; Shi, H.; Hewett, J.E.; Ruhlen, R.L.; MacDonald, R.S.; Rottinghaus, G.E.; Chen, Y.-C.; Sauter, E.R. Soy Isoflavones Have an Antiestrogenic Effect and Alter Mammary Promoter Hypermethylation in Healthy Premenopausal Women. Nutr. Cancer 2009, 61, 238–244. [Google Scholar]
- Desjobert, C.; Renalier, M.H.; Bergalet, J.; Dejean, E.; Joseph, N.; Kruczynski, A.; Soulier, J.; Espinos, E.; Meggetto, F.; Cavaille, J.; et al. MiR-29a down-regulation in ALK-positive anaplastic large cell lymphomas contributes to apoptosis blockade through MCL-1 overexpression. Blood 2011, 117, 6627–6637. [Google Scholar]
- Lippman, S.M.; Klein, E.A.; Goodman, P.J.; Lucia, M.S.; Thompson, I.M.; Ford, L.G.; Parnes, H.L.; Minasian, L.M.; Gaziano, J.M.; Hartline, J.A.; et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009, 301, 39–51. [Google Scholar]
- Clark, L.C.; Combs, G.F., Jr.; Turnbull, B.W.; Slate, E.H.; Chalker, D.K.; Chow, J.; Davis, L.S.; Glover, R.A.; Graham, G.F.; Gross, E.G.; et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA 1996, 276, 1957–1963. [Google Scholar]
- Lee, S.O.; Yeon Chun, J.; Nadiminty, N.; Trump, D.L.; Ip, C.; Dong, Y.; Gao, A.C. Monomethylated selenium inhibits growth of LNCaP human prostate cancer xenograft accompanied by a decrease in the expression of androgen receptor and prostate-specific antigen (PSA). Prostate 2006, 66, 1070–1075. [Google Scholar]
- Unni, E.; Koul, D.; Yung, W.K.; Sinha, R. Se-methylselenocysteine inhibits phosphatidylinositol 3-kinase activity of mouse mammary epithelial tumor cells in vitro. Breast Cancer Res. Treat. 2005, 7, R699–R707. [Google Scholar]
- Tsavachidou, D.; McDonnell, T.J.; Wen, S.; Wang, X.; Vakar-Lopez, F.; Pisters, L.L.; Pettaway, C.A.; Wood, C.G.; Do, K.A.; Thall, P.F.; et al. Selenium and vitamin E: Cell type- and intervention-specific tissue effects in prostate cancer. J. Natl. Cancer Inst. 2009, 101, 306–320. [Google Scholar] [CrossRef]
- Lee, J.-I.; Nian, H.; Cooper, A.J.L.; Sinha, R.; Dai, J.; Bisson, W.H.; Dashwood, R.H.; Pinto, J.T. α-Keto Acid Metabolites of Naturally Occurring Organoselenium Compounds as Inhibitors of Histone Deacetylase in Human Prostate Cancer Cells. Cancer Prev. Res. 2009, 2, 683–693. [Google Scholar] [CrossRef]
- Arikawa, A.Y.; Gallaher, D.D. Cruciferous vegetables reduce morphological markers of colon cancer risk in dimethylhydrazine-treated rats. J. Nutr. 2008, 138, 526–532. [Google Scholar]
- Suzuki, R.; Iwasaki, M.; Hara, A.; Inoue, M.; Sasazuki, S.; Sawada, N.; Yamaji, T.; Shimazu, T.; Tsugane, S.; Japan Public Health Center-based Prospective Study Group. Fruit and vegetable intake and breast cancer risk defined by estrogen and progesterone receptor status: The JPHC study. Cancer Causes Control 2013, 24, 2117–2128. [Google Scholar]
- Traka, M.; Gasper, A.V.; Smith, J.A.; Hawkey, C.J.; Bao, Y.; Mithen, R.F. Transcriptome analysis of human colon Caco-2 cells exposed to sulforaphane. J. Nutr. 2005, 135, 1865–1872. [Google Scholar]
- Hsu, A.; Wong, C.P.; Yu, Z.; Williams, D.E.; Dashwood, R.H.; Ho, E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clin. Epigenetics 2011, 3, 3. [Google Scholar] [CrossRef]
- Meeran, S.M.; Patel, S.N.; Li, Y.; Shukla, S.; Tollefsbol, T.O. Bioactive dietary supplements reactivate ER expression in ER-negative breast cancer cells by active chromatin modifications. PLoS One 2012, 7, e37748. [Google Scholar] [CrossRef]
- Banwell, C.M.; MacCartney, D.P.; Guy, M.; Miles, A.E.; Uskokovic, M.R.; Mansi, J.; Stewart, P.M.; O’Neill, L.P.; Turner, B.M.; Colston, K.W.; et al. Altered nuclear receptor corepressor expression attenuates vitamin D receptor signaling in breast cancer cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 2004–2013. [Google Scholar] [CrossRef]
- Luo, W.; Karpf, A.R.; Deeb, K.K.; Muindi, J.R.; Morrison, C.D.; Johnson, C.S.; Trump, D.L. Epigenetic regulation of vitamin D 24-hydroxylase/CYP24A1 in human prostate cancer. Cancer Res. 2010, 70, 5953–5962. [Google Scholar]
- Trump, D.L.; Muindi, J.; Fakih, M.; Yu, W.-D.; Johnson, C.S. Vitamin D Compounds: Clinical Development as Cancer Therapy and Prevention Agents. Anticancer Res. 2006, 26, 2551–2556. [Google Scholar]
- Kim, M.S.; Fujiki, R.; Kitagawa, H.; Kato, S. 1alpha,25(OH)2D3-induced DNA methylation suppresses the human CYP27B1 gene. Mol. Cell Endocrinol. 2007, 265–266, 168–173. [Google Scholar]
- McGregor, L.M.; McCune, B.K.; Graff, J.R.; McDowell, P.R.; Romans, K.E.; Yancopoulos, G.D.; Ball, D.W.; Baylin, S.B.; Nelkin, B.D. Roles of trk family neurotrophin receptors in medullary thyroid carcinoma development and progression. Proc. Natl. Acad. Sci. USA 1999, 96, 4540–4545. [Google Scholar] [CrossRef]
- Au, C.W.H.; Siu, M.K.Y.; Liao, X.; Wong, E.S.Y.; Ngan, H.Y.S.; Tam, K.F.; Chan, D.C.W.; Chan, Q.K.Y.; Cheung, A.N.Y. Tyrosine kinase B receptor and BDNF expression in ovarian cancers—Effect on cell migration, angiogenesis and clinical outcome. Cancer Lett. 2009, 281, 151–161. [Google Scholar]
- Bistulfi, G.; Pozzi, S.; Ren, M.; Rossetti, S.; Sacchi, N. A repressive epigenetic domino effect confers susceptibility to breast epithelial cell transformation: Implications for predicting breast cancer risk. Cancer Res. 2006, 66, 10308–10314. [Google Scholar]
- Deeb, K.K.; Luo, W.; Karpf, A.R.; Omilian, A.R.; Bshara, W.; Tian, L.; Tangrea, M.A.; Morrison, C.D.; Johnson, C.S.; Trump, D.L. Differential vitamin D 24-hydroxylase/CYP24A1 gene promoter methylation in endothelium from benign and malignant human prostate. Epigenetics Off. J. DNA Methylation Soc. 2011, 6, 994–1000. [Google Scholar] [CrossRef]
- King-Batoon, A.; Leszczynska, J.M.; Klein, C.B. Modulation of gene methylation by genistein or lycopene in breast cancer cells. Environ. Mol. Mutagen. 2008, 49, 36–45. [Google Scholar] [CrossRef]
- Ferlay, J.; Shin, H.-R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer J. Int. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Burdge, G.C.; Slater-Jefferies, J.; Torrens, C.; Phillips, E.S.; Hanson, M.A.; Lillycrop, K.A. Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Br. J. Nutr. 2007, 97, 435–439. [Google Scholar]
- Rantakallio, P. The longitudinal study of the Northern Finland birth cohort of 1966. Paediatr. Perinat. Epidemiol. 1988, 2, 59–88. [Google Scholar] [CrossRef]
- Morton, S.M.B.; Atatoa Carr, P.E.; Grant, C.C.; Robinson, E.M.; Bandara, D.K.; Bird, A.; Ivory, V.C.; Kingi, T.K.R.; Liang, R.; Marks, E.J.; et al. Cohort Profile: Growing Up in New Zealand. Int. J. Epidemiol. 2013, 42, 65–75. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bishop, K.S.; Ferguson, L.R. The Interaction between Epigenetics, Nutrition and the Development of Cancer. Nutrients 2015, 7, 922-947. https://doi.org/10.3390/nu7020922
Bishop KS, Ferguson LR. The Interaction between Epigenetics, Nutrition and the Development of Cancer. Nutrients. 2015; 7(2):922-947. https://doi.org/10.3390/nu7020922
Chicago/Turabian StyleBishop, Karen S., and Lynnette R. Ferguson. 2015. "The Interaction between Epigenetics, Nutrition and the Development of Cancer" Nutrients 7, no. 2: 922-947. https://doi.org/10.3390/nu7020922
APA StyleBishop, K. S., & Ferguson, L. R. (2015). The Interaction between Epigenetics, Nutrition and the Development of Cancer. Nutrients, 7(2), 922-947. https://doi.org/10.3390/nu7020922