The Anti-Proliferative Effects of Enterolactone in Prostate Cancer Cells: Evidence for the Role of DNA Licencing Genes, mi-R106b Cluster Expression, and PTEN Dosage

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Cell Culture and Enterolactone Preparation
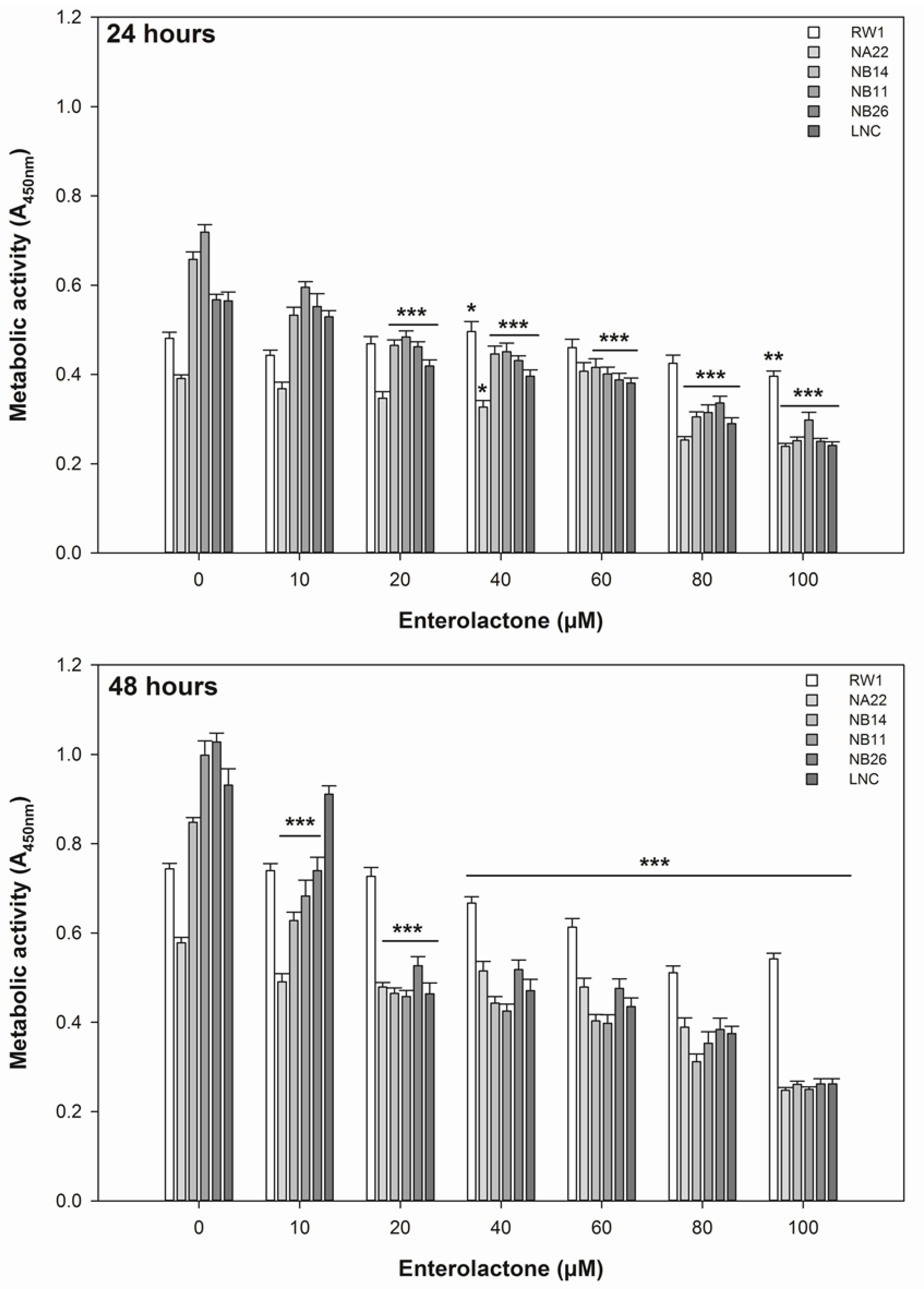
2.2. Cell Viability—Mitochondrial Activity Assay
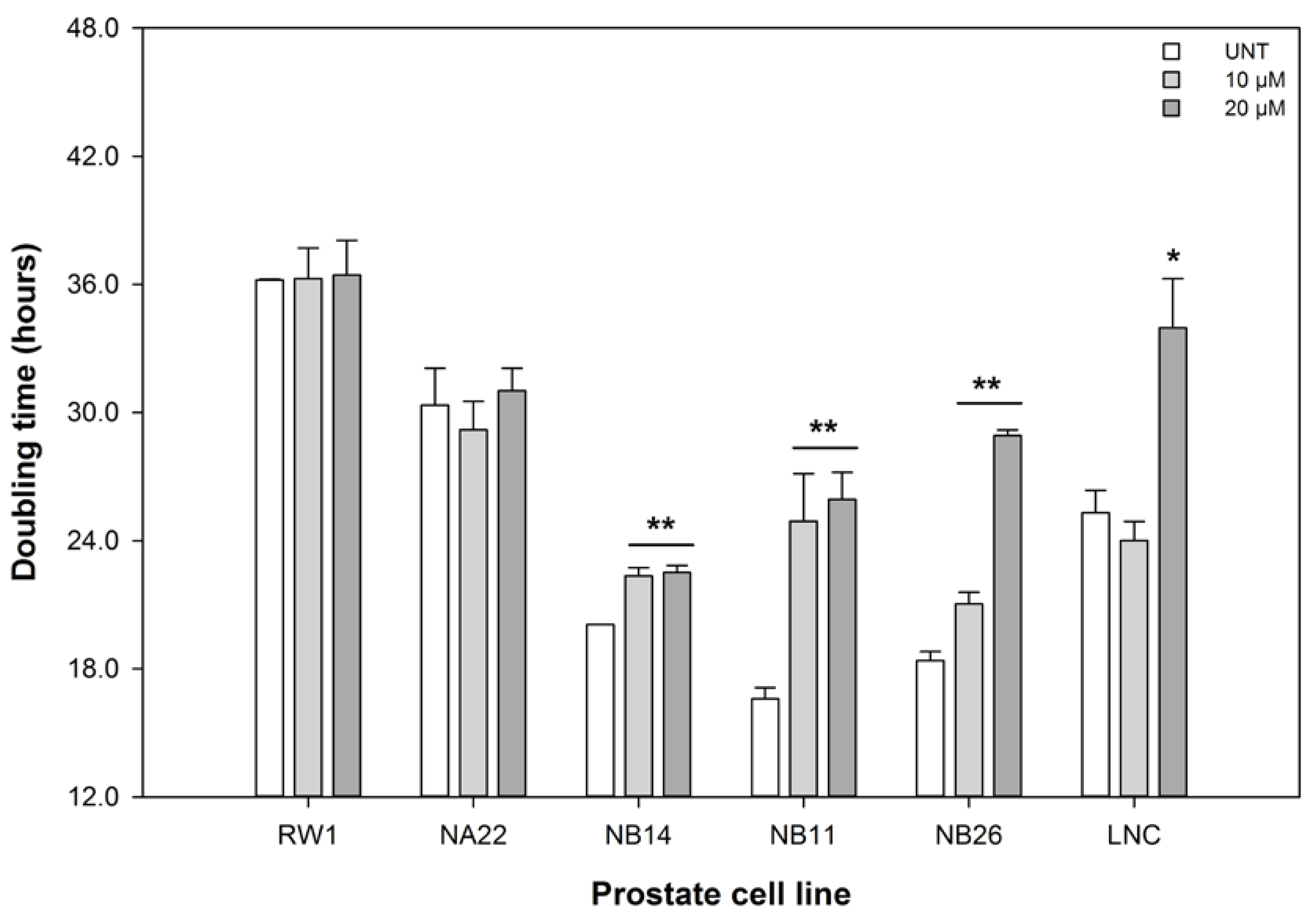
2.3. Cell Viability—Growth Kinetics Assay
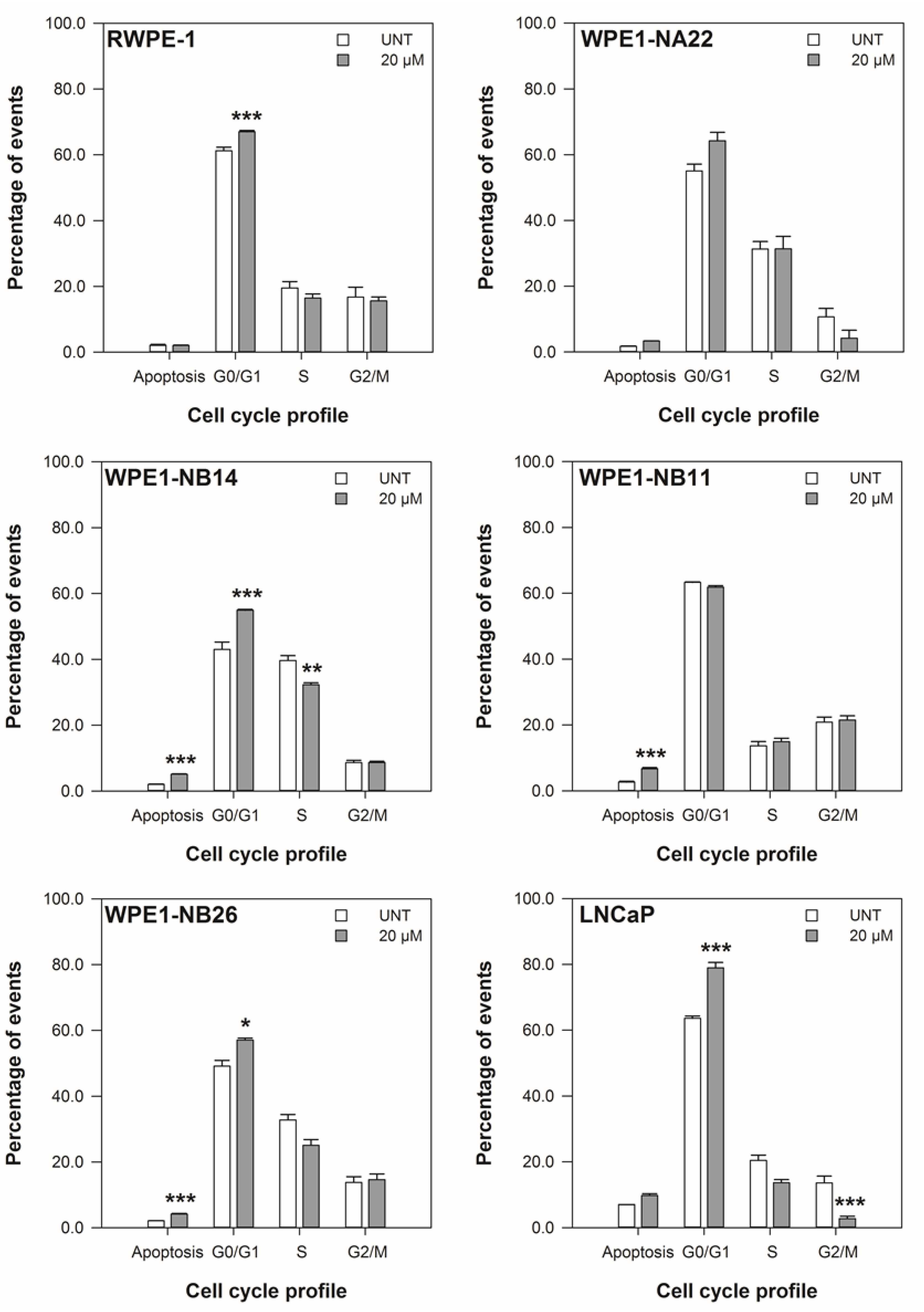
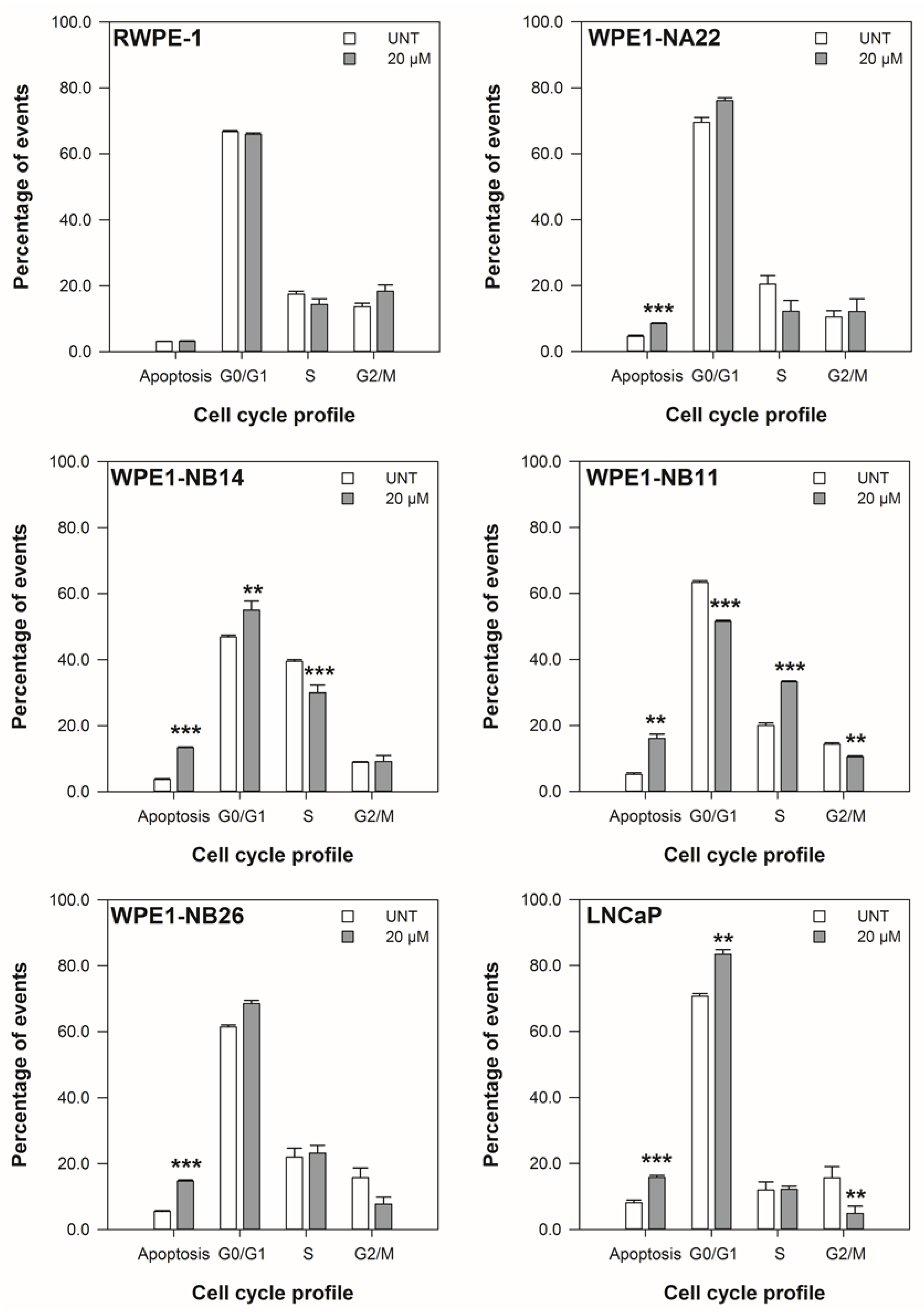
2.4. Cell Viability—Cell Cycle Profile Assay
2.5. Quantification of Gene Expression—mRNA and miRNA Genes
2.6. Statistical Analyses
3. Results
3.1. ENL Reduces the Viability of Mid to Later Stage Prostate Disease Cell Lines
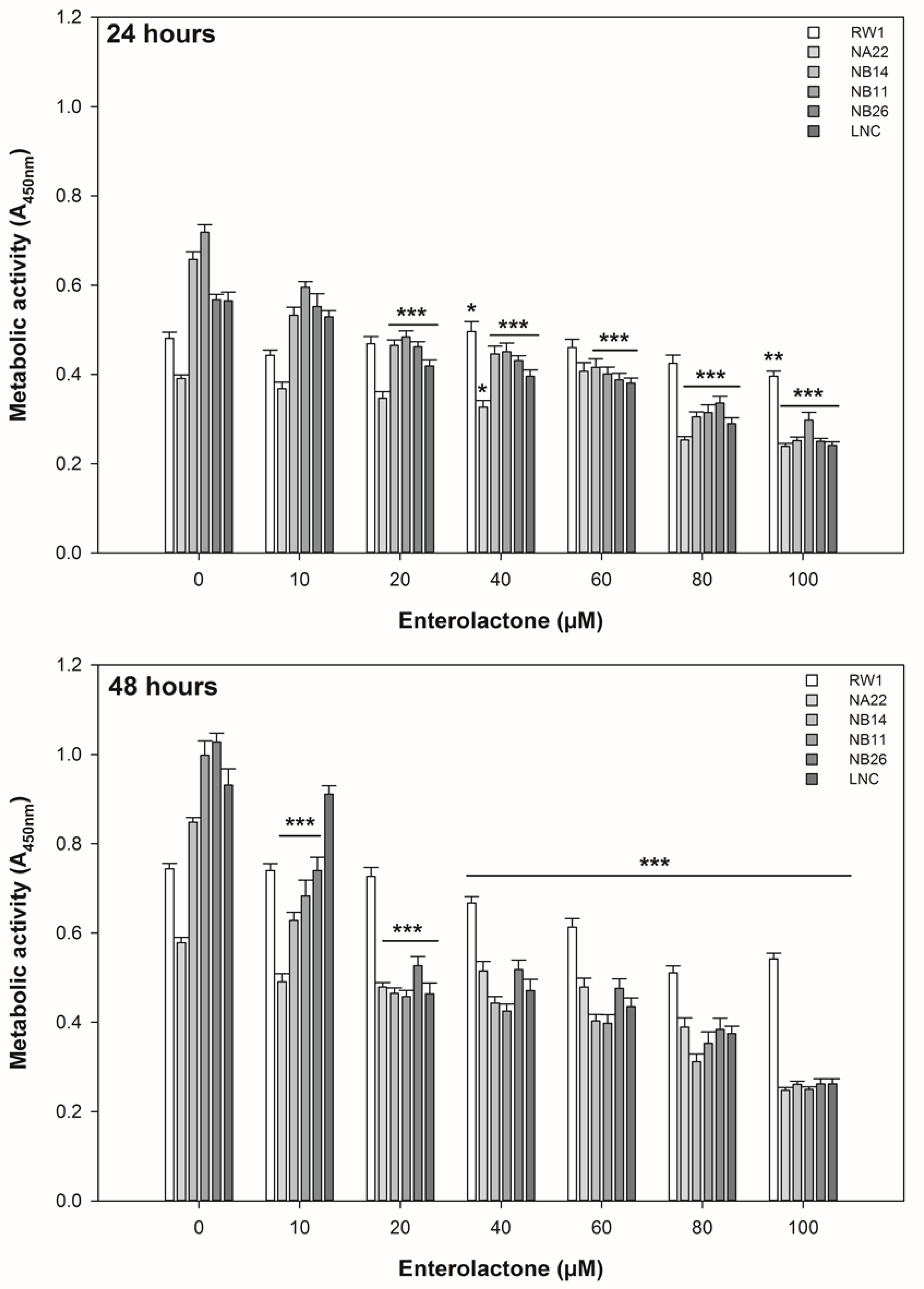
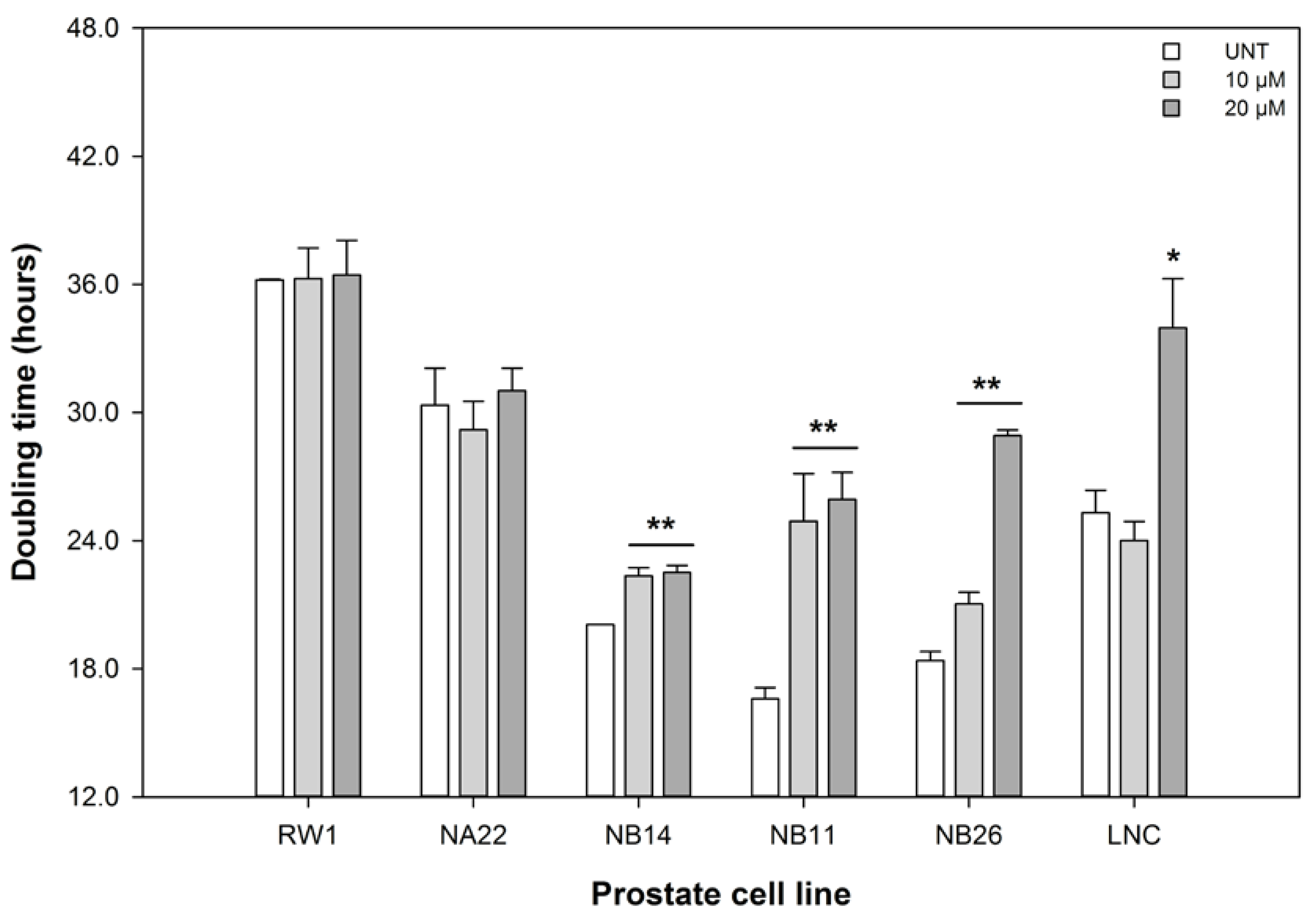
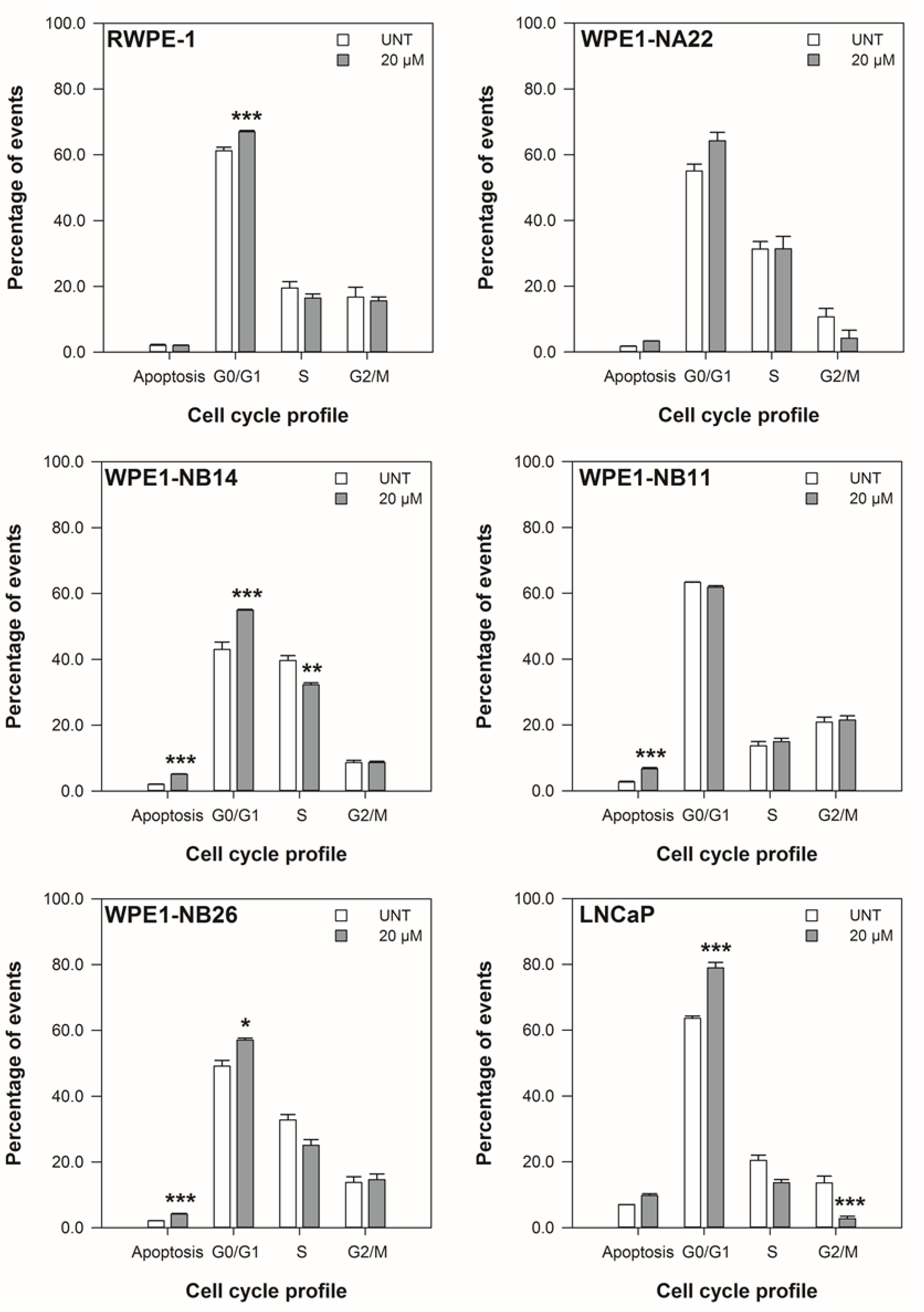
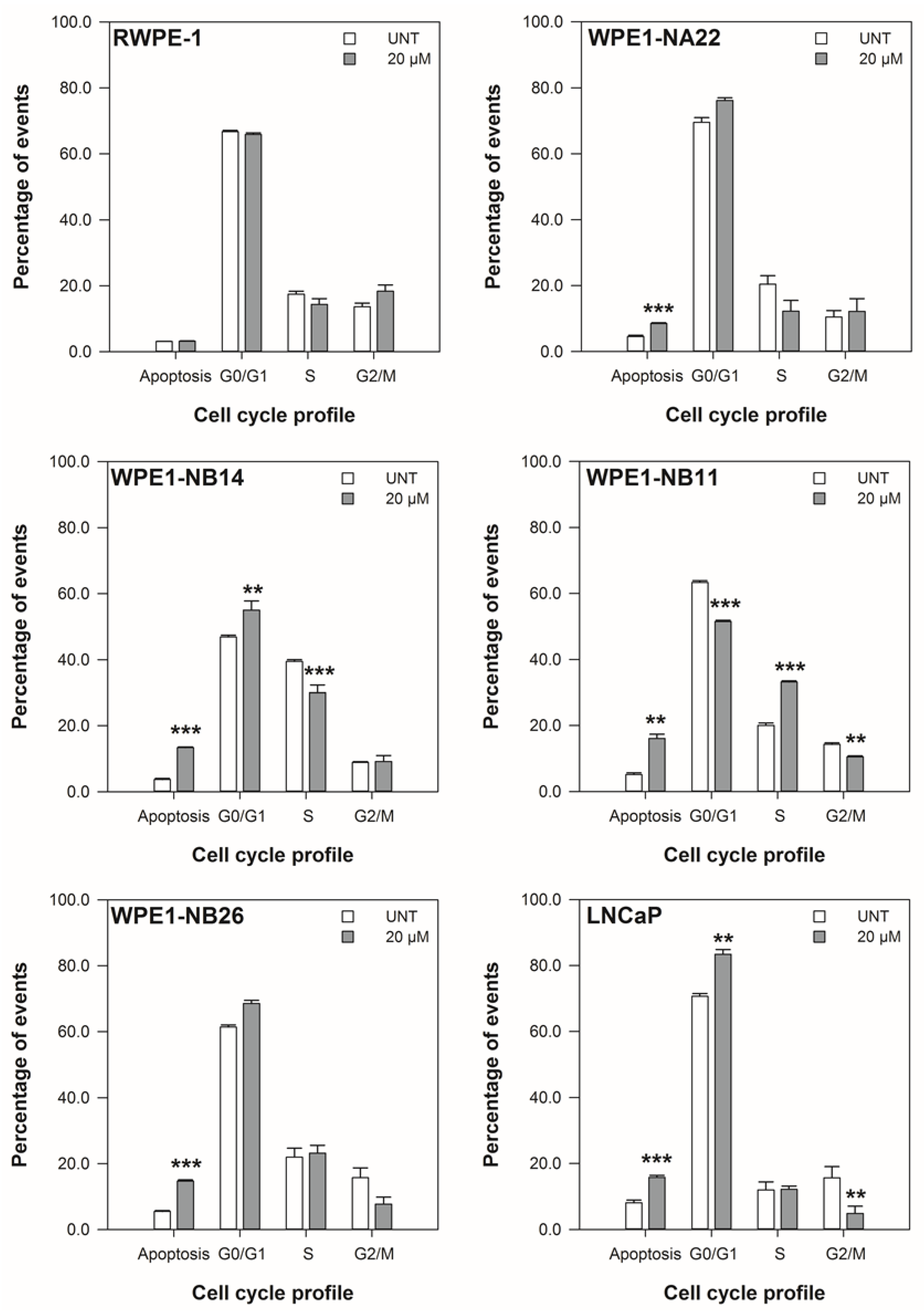
3.2. ENL Restricts the Cell Cycle of and Induces Apoptosis in Mid to Later Stage Prostate Disease Cell Lines
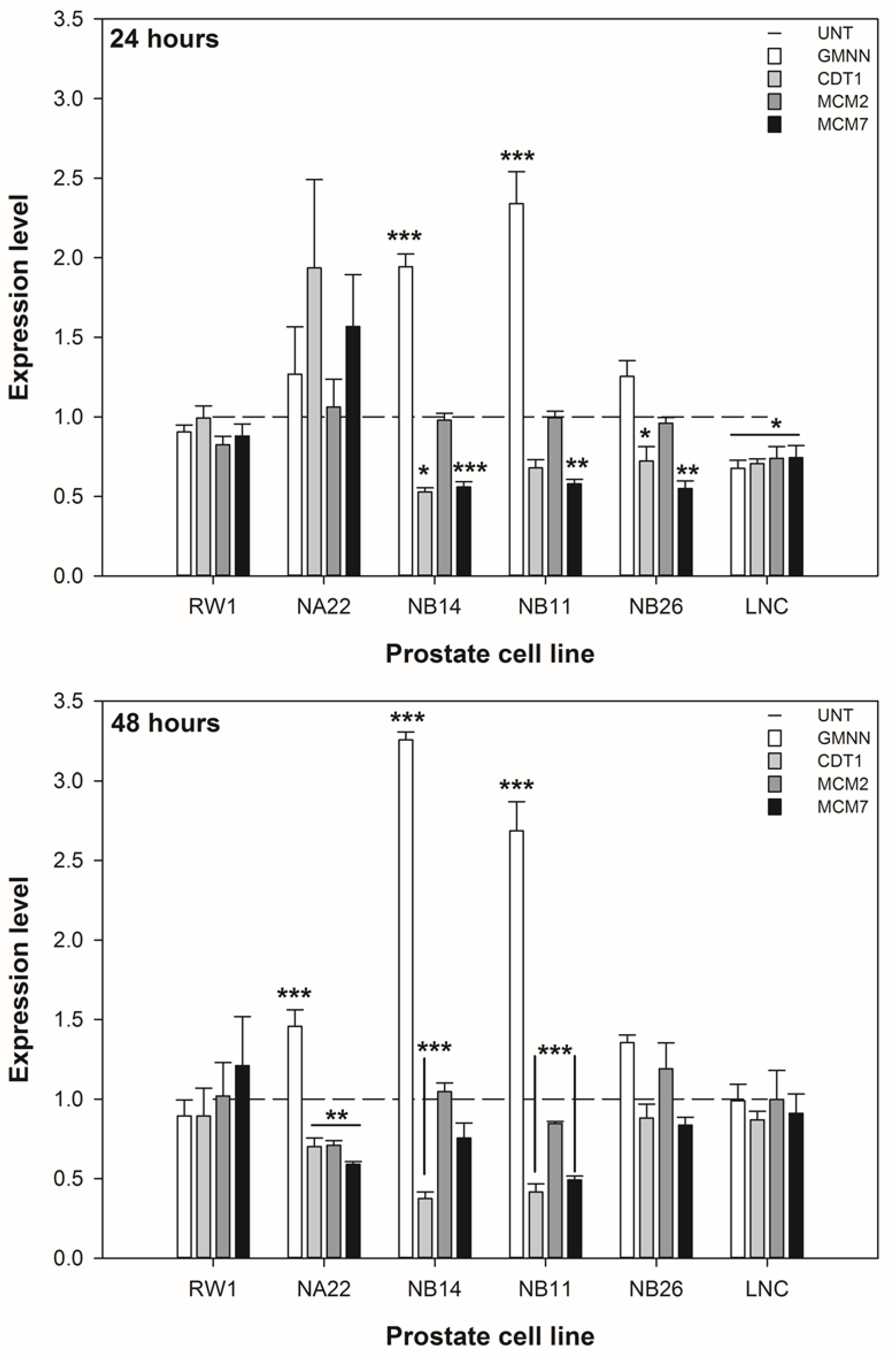
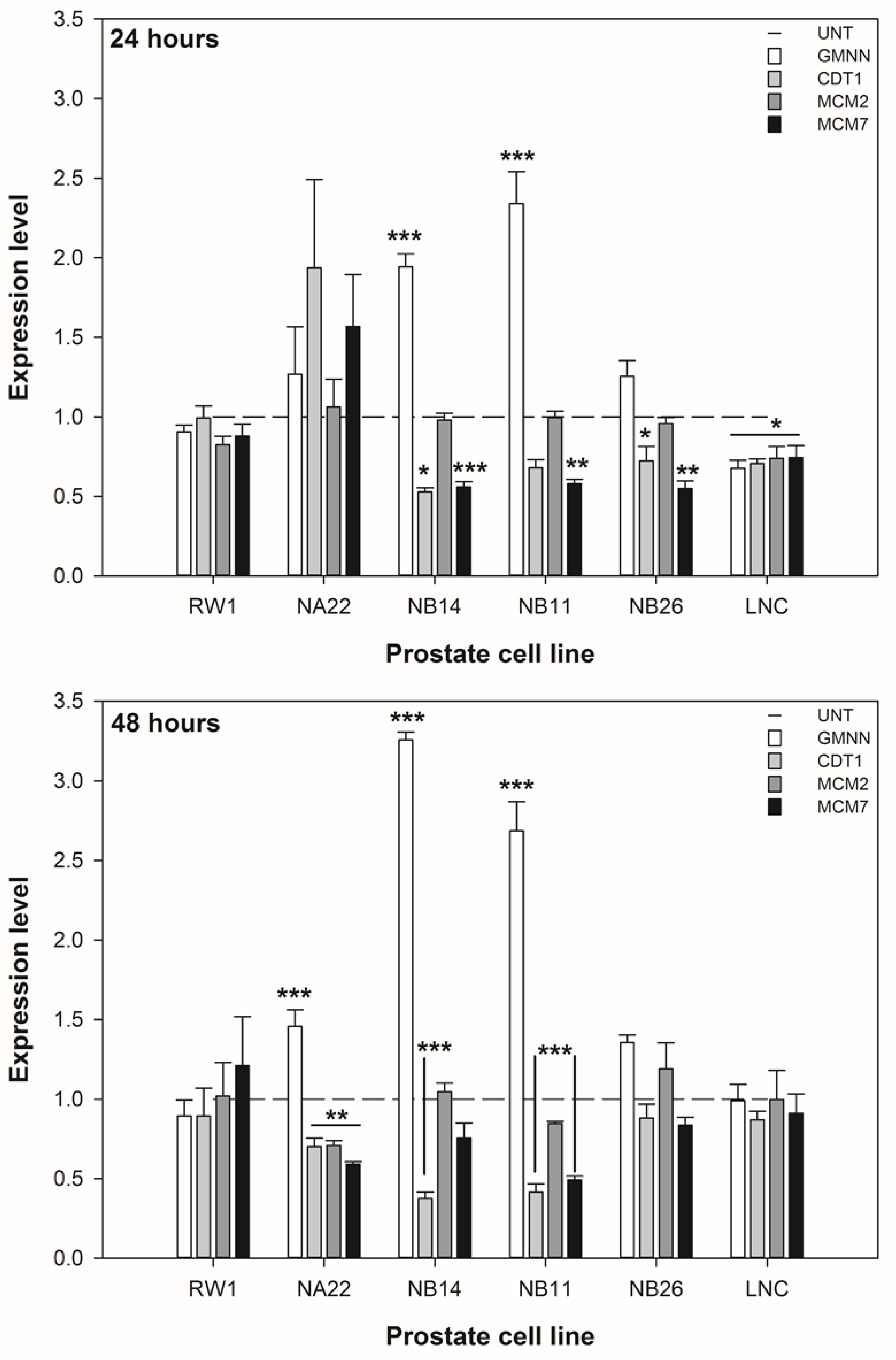
3.3. ENL Alters the Expression of DNA Licencing Genes in Mid to Later Stage Prostate Disease Cell Lines
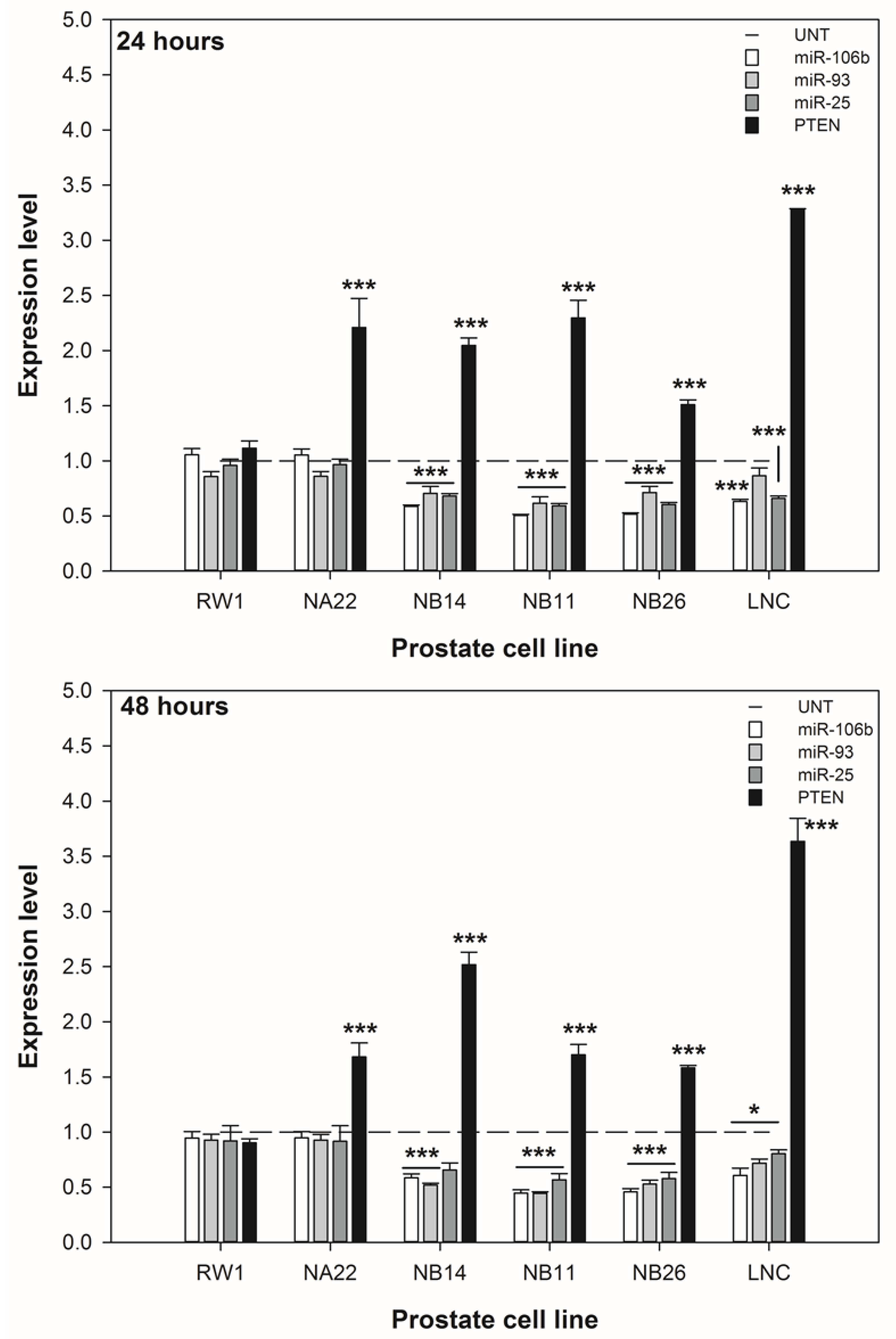
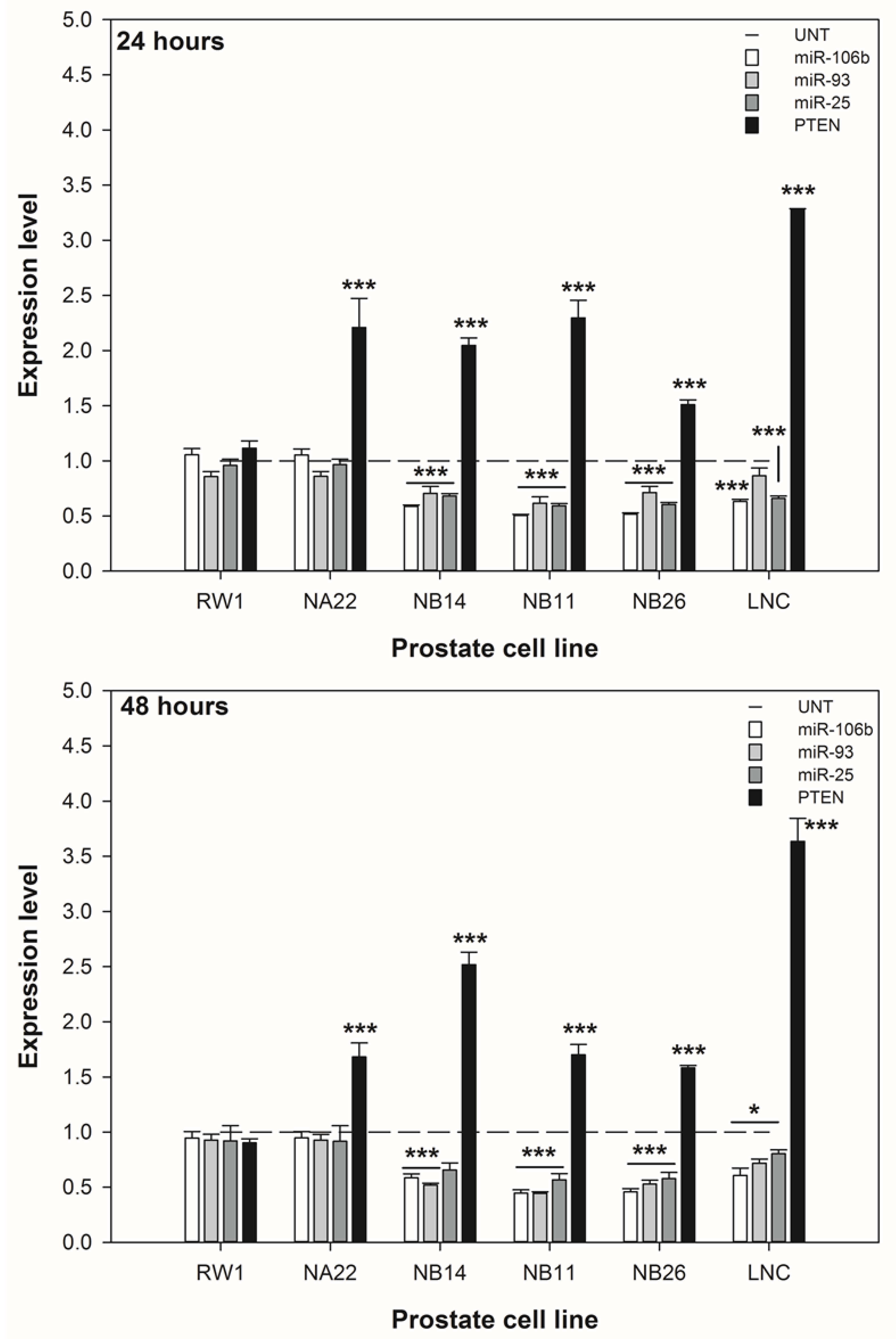
3.4. ENL Alters the Expression of the miR-106b Cluster Leading to Increased PTEN Expression
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Center, M.M.; Jemal, A.; Lortet-Tieulent, J.; Ward, E.; Ferlay, J.; Brawley, O.; Bray, F. International variation in prostate cancer incidence and mortality rates. Eur. Urol. 2012, 61, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Baade, P.D.; Youlden, D.R.; Krnjacki, L.J. International epidemiology of prostate cancer: Geographical distribution and secular trends. Mol. Nutr. Food Res. 2009, 53, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Cullen, J.; Elsamanoudi, S.; Brassell, S.A.; Chen, Y.; Colombo, M.; Srivastava, A.; McLeod, D.G. The burden of prostate cancer in Asian nations. J. Carcinog. 2012, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Venkateswaran, V.; Klotz, L.H. Diet and prostate cancer: Mechanisms of action and implications for chemoprevention. Nat. Rev. Urol. 2010, 7, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Afaq, F.; Mukhtar, H. Lifestyle as risk factor for cancer: Evidence from human studies. Cancer Lett. 2010, 293, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.C.; Severi, G.; Baglietto, L.; Krishnan, K.; English, D.R.; Hopper, J.L.; Giles, G.G. Dietary patterns and prostate cancer risk. Cancer Epidemiol. Biomark. Prev. 2009, 18, 3126–3129. [Google Scholar] [CrossRef]
- Saarinen, N.M.; Tuominen, J.; Pylkkanen, L.; Santti, R. Assessment of information to substantiate a health claim on the prevention of prostate cancer by lignans. Nutrients 2010, 2, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Adlercreutz, H. Lignans and human health. Crit. Rev. Clin. Lab. Sci. 2007, 44, 483–525. [Google Scholar] [CrossRef] [PubMed]
- McCann, M.J.; Gill, C.I.; McGlynn, H.; Rowland, I.R. Role of mammalian lignans in the prevention and treatment of prostate cancer. Nutr. Cancer 2005, 52, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kuhnle, G.G.; Dell’Aquila, C.; Aspinall, S.M.; Runswick, S.A.; Mulligan, A.A.; Bingham, S.A. Phytoestrogen content of foods of animal origin: Dairy products, eggs, meat, fish, and seafood. J. Agric. Food Chem. 2008, 56, 10099–10104. [Google Scholar] [CrossRef] [PubMed]
- Setchell, K.D.; Lawson, A.M.; Mitchell, F.L.; Adlercreutz, H.; Kirk, D.N.; Axelson, M. Lignans in man and in animal species. Nature 1980, 287, 740–742. [Google Scholar] [CrossRef] [PubMed]
- Woting, A.; Clavel, T.; Loh, G.; Blaut, M. Bacterial transformation of dietary lignans in gnotobiotic rats. FEMS Microbiol. Ecol. 2010, 72, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Clavel, T.; Borrmann, D.; Braune, A.; Dore, J.; Blaut, M. Occurrence and activity of human intestinal bacteria involved in the conversion of dietary lignans. Anaerobe 2006, 12, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Clavel, T.; Henderson, G.; Engst, W.; Dore, J.; Blaut, M. Phylogeny of human intestinal bacteria that activate the dietary lignan secoisolariciresinol diglucoside. FEMS Microbiol. Ecol. 2006, 55, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Saarinen, N.M.; Thompson, L.U. Prolonged administration of secoisolariciresinol diglycoside increases lignan excretion and alters lignan tissue distribution in adult male and female rats. Br. J. Nutr. 2010, 104, 833–841. [Google Scholar] [CrossRef] [PubMed]
- McCann, M.J.; Gill, C.I.; Linton, T.; Berrar, D.; McGlynn, H.; Rowland, I.R. Enterolactone restricts the proliferation of the LNCaP human prostate cancer cell line in vitro. Mol. Nutr. Food Res. 2008, 52, 567–580. [Google Scholar] [CrossRef] [PubMed]
- McCann, M.J.; Rowland, I.R.; Roy, N.C. Anti-proliferative effects of physiological concentrations of enterolactone in models of prostate tumourigenesis. Mol. Nutr. Food Res. 2013, 57, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Fang, J.; Li, H.; Demark-Wahnefried, W.; Lin, X. Enterolactone induces apoptosis in human prostate carcinoma LNCaP cells via a mitochondrial-mediated, caspase-dependent pathway. Mol. Cancer Ther. 2007, 6, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Switzer, B.R.; Demark-Wahnefried, W. Effect of mammalian lignans on the growth of prostate cancer cell lines. Anticancer Res. 2001, 21, 3995–3999. [Google Scholar] [PubMed]
- Morton, M.S.; Chan, P.S.; Cheng, C.; Blacklock, N.; Matos-Ferreira, A.; Abranches-Monteiro, L.; Correia, R.; Lloyd, S.; Griffiths, K. Lignans and isoflavonoids in plasma and prostatic fluid in men: Samples from Portugal, Hong Kong, and the United Kingdom. Prostate 1997, 32, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Adlercreutz, H.; Bannwart, C.; Wahala, K.; Makela, T.; Brunow, G.; Hase, T.; Arosemena, P.J.; Kellis, J.T., Jr.; Vickery, L.E. Inhibition of human aromatase by mammalian lignans and isoflavonoid phytoestrogens. J. Steroid Biochem. Mol. Biol. 1993, 44, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Masai, H.; Matsumoto, S.; You, Z.; Yoshizawa-Sugata, N.; Oda, M. Eukaryotic chromosome DNA replication: Where, when, and how? Annu. Rev. Biochem. 2010, 79, 89–130. [Google Scholar] [CrossRef]
- Luo, J.H. Oncogenic activity of MCM7 transforming cluster. World J. Clin. Oncol. 2011, 2, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Hook, S.S.; Lin, J.J.; Dutta, A. Mechanisms to control rereplication and implications for cancer. Curr. Opin. Cell Biol. 2007, 19, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.; Tsuji, T.; Guo, L.; Lu, S.H.; Jiang, W. The role of pre-replicative complex (pre-RC) components in oncogenesis. FASEB J. 2007, 21, 3786–3794. [Google Scholar] [CrossRef] [PubMed]
- Montanari, M.; Macaluso, M.; Cittadini, A.; Giordano, A. Role of geminin: from normal control of DNA replication to cancer formation and progression? Cell Death Differ. 2006, 13, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Yu, G.; Tseng, G.C.; Cieply, K.; Gavel, T.; Nelson, J.; Michalopoulos, G.; Yu, Y.P.; Luo, J.H. MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene 2006, 25, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Honeycutt, K.A.; Chen, Z.; Koster, M.I.; Miers, M.; Nuchtern, J.; Hicks, J.; Roop, D.R.; Shohet, J.M. Deregulated minichromosomal maintenance protein MCM7 contributes to oncogene driven tumorigenesis. Oncogene 2006, 25, 4027–4032. [Google Scholar] [CrossRef] [PubMed]
- Xouri, G.; Lygerou, Z.; Nishitani, H.; Pachnis, V.; Nurse, P.; Taraviras, S. Cdt1 and geminin are down-regulated upon cell cycle exit and are over-expressed in cancer-derived cell lines. Eur. J. Biochem. 2004, 271, 3368–3378. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Pourmand, G.; Ziaee, A.A.; Abedi, A.R.; Mehrsai, A.; Alavi, H.A.; Ahmadi, A.; Saadati, H.R. Role of PTEN gene in progression of prostate cancer. Urol. J. 2007, 4, 95–100. [Google Scholar] [PubMed]
- Vlietstra, R.J.; van Alewijk, D.C.; Hermans, K.G.; van Steenbrugge, G.J.; Trapman, J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998, 58, 2720–2723. [Google Scholar] [PubMed]
- Poliseno, L.; Salmena, L.; Riccardi, L.; Fornari, A.; Song, M.S.; Hobbs, R.M.; Sportoletti, P.; Varmeh, S.; Egia, A.; Fedele, G.; et al. Identification of the miR-106b~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci. Signal. 2010, 3, ra29. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.M.; Quader, S.T.; Kleinman, H.K.; Bello-DeOcampo, D.; Storto, P.D.; Bice, G.; DeMendonca-Calaca, W.; Williams, D.E. Human cell lines as an in vitro/in vivo model for prostate carcinogenesis and progression. Prostate 2001, 47, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Horoszewicz, J.S.; Leong, S.S.; Chu, T.M.; Wajsman, Z.L.; Friedman, M.; Papsidero, L.; Kim, U.; Chai, L.S.; Kakati, S.; Arya, S.K.; et al. The LNCaP cell line—A new model for studies on human prostatic carcinoma. Prog. Clin. Biol. Res. 1980, 37, 115–132. [Google Scholar] [PubMed]
- Van Engeland, M.; Nieland, L.J.; Ramaekers, F.C.; Schutte, B.; Reutelingsperger, C.P. Annexin V-affinity assay: A review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry 1998, 31, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Arentson, E.; Faloon, P.; Seo, J.; Moon, E.; Studts, J.M.; Fremont, D.H.; Choi, K. Oncogenic potential of the DNA replication licensing protein CDT1. Oncogene 2002, 21, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Gillespie, P.J. Replication licensing and cancer—A fatal entanglement? Nat. Rev. Cancer 2008, 8, 799–806. [Google Scholar] [CrossRef]
- Lei, M. The MCM complex: Its role in DNA replication and implications for cancer therapy. Curr. Cancer Drug Targets 2005, 5, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Sterner, J.M.; Dew-Knight, S.; Musahl, C.; Kornbluth, S.; Horowitz, J.M. Negative regulation of DNA replication by the retinoblastoma protein is mediated by its association with MCM7. Mol. Cell. Biol. 1998, 18, 2748–2757. [Google Scholar] [PubMed]
- Mukherjee, P.; Winter, S.L.; Alexandrow, M.G. Cell cycle arrest by transforming growth factor beta1 near G1/S is mediated by acute abrogation of prereplication complex activation involving an Rb-MCM interaction. Mol. Cell. Biol. 2010, 30, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.K.; Yu, Y.P.; Zhu, Z.H.; Han, Y.C.; Ren, B.; Nelson, J.B.; Luo, J.H. MCM7 interacts with androgen receptor. Am. J. Pathol. 2008, 173, 1758–1767. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.P.; Lara, P.N., Jr. Prostate cancer: Predicting response to androgen receptor signalling inhibition. Nat. Rev. Urol. 2014, 11, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Balk, S.P. Androgen receptor functions in prostate cancer development and progression. Asian J. Androl. 2014, 16, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [PubMed]
- Sarker, D.; Reid, A.H.; Yap, T.A.; de Bono, J.S. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin. Cancer Res. 2009, 15, 4799–4805. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.H.; Knudson, A.G.; Pandolfi, P.P. A continuum model for tumour suppression. Nature 2011, 476, 163–169. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCann, M.J.; Rowland, I.R.; Roy, N.C. The Anti-Proliferative Effects of Enterolactone in Prostate Cancer Cells: Evidence for the Role of DNA Licencing Genes, mi-R106b Cluster Expression, and PTEN Dosage. Nutrients 2014, 6, 4839-4855. https://doi.org/10.3390/nu6114839
McCann MJ, Rowland IR, Roy NC. The Anti-Proliferative Effects of Enterolactone in Prostate Cancer Cells: Evidence for the Role of DNA Licencing Genes, mi-R106b Cluster Expression, and PTEN Dosage. Nutrients. 2014; 6(11):4839-4855. https://doi.org/10.3390/nu6114839
Chicago/Turabian StyleMcCann, Mark J., Ian R. Rowland, and Nicole C. Roy. 2014. "The Anti-Proliferative Effects of Enterolactone in Prostate Cancer Cells: Evidence for the Role of DNA Licencing Genes, mi-R106b Cluster Expression, and PTEN Dosage" Nutrients 6, no. 11: 4839-4855. https://doi.org/10.3390/nu6114839
APA StyleMcCann, M. J., Rowland, I. R., & Roy, N. C. (2014). The Anti-Proliferative Effects of Enterolactone in Prostate Cancer Cells: Evidence for the Role of DNA Licencing Genes, mi-R106b Cluster Expression, and PTEN Dosage. Nutrients, 6(11), 4839-4855. https://doi.org/10.3390/nu6114839