Vitamin B12 Metabolism during Pregnancy and in Embryonic Mouse Models

Abstract
:1. Introduction
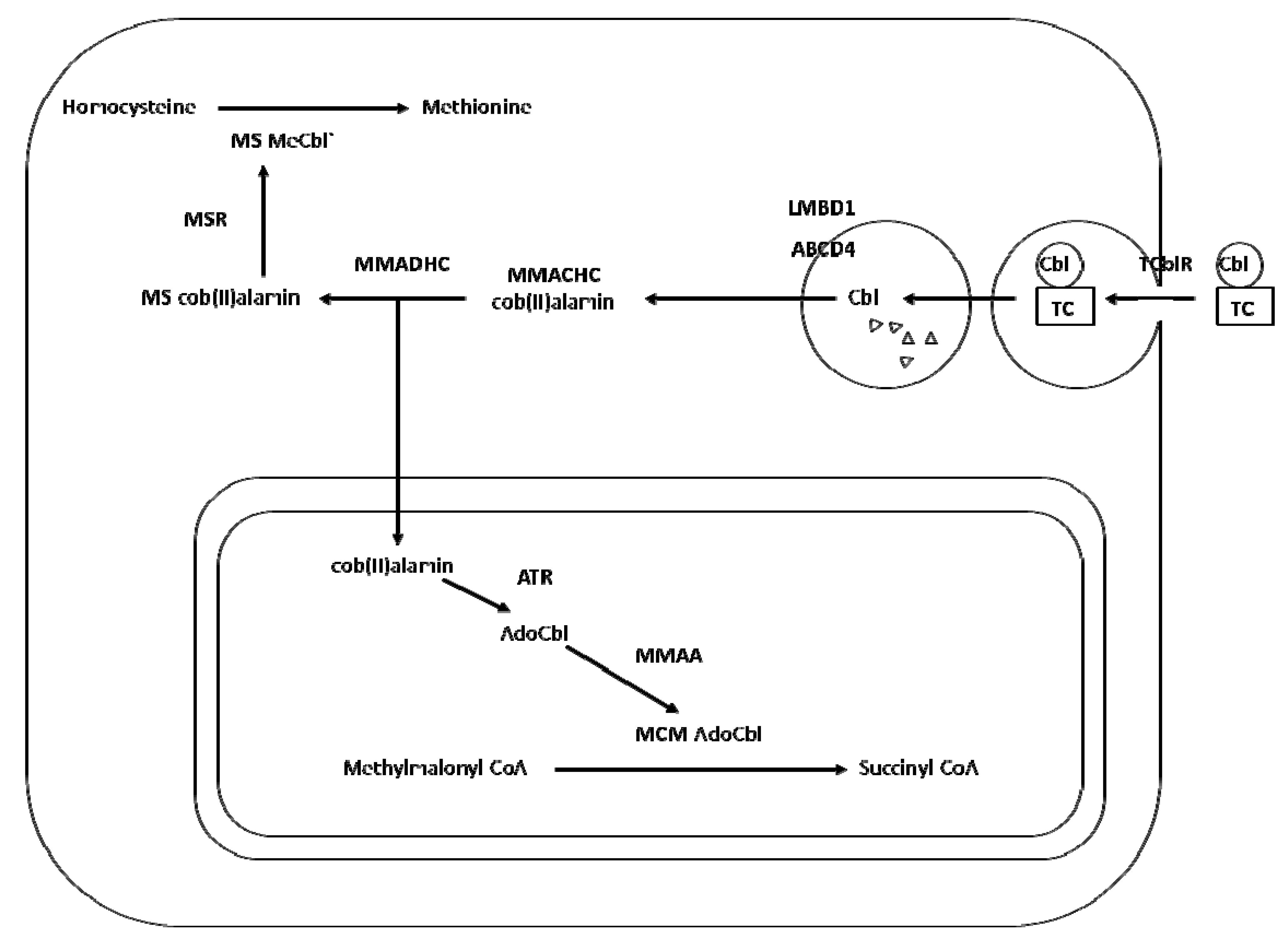
2. Transport and Metabolism of Cobalamin
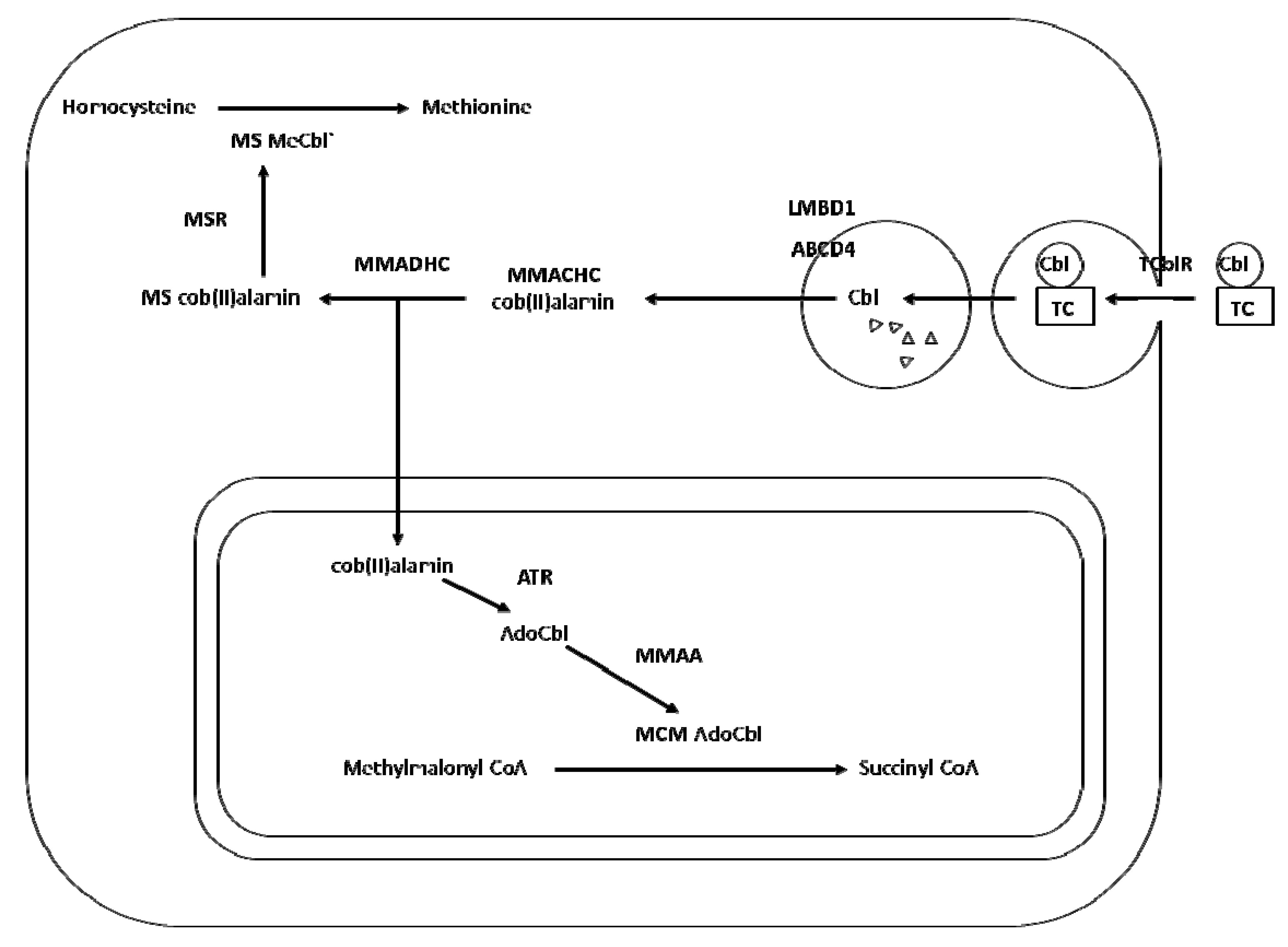
3. Cobalamin Deficiency, Polymorphism and Risk of Neural Tube Defects
4. Expression Pattern of Cbl Genes during Mouse Organogenesis

{kind=link}
| Genes | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Organs | Abcd4 1 | Mmachc 2 | Mmadhc 3 * | Mtrr 4 * | Mtr 5 | Mmaa 6 * | Mmab 7 * | Mut 8 * | |
| Branchial arches | + | + | + | + | |||||
| Mouth | + | + | + | ||||||
| Nose | + | + | + | ||||||
| Palate | + | + | + | ||||||
| nasal cavity | + | + | + | + | + | ||||
| Tongue | + | + | + | + | + | ||||
| Teeth | + | ||||||||
| Head | |||||||||
| Head mesenchyme | + | + | + | + | |||||
| Endothelial vessel of head | + | + | + | ||||||
| Neural crest cells | |||||||||
| Drg | + | + | + | + | + | + | + | ||
| Neural tube | + | + | + | + | + | + | |||
| Brain | + | + | + | ||||||
| spinal cord | + | + | + | + | |||||
| Forebrain | + | + | + | ||||||
| Midbrain | + | + | |||||||
| Hindbrain | + | ||||||||
| Rathkete’s Pouch | + | + | |||||||
| Pituitary | + | + | + | ||||||
| Eye and ear | |||||||||
| Eye | + | ||||||||
| Retina | + | ||||||||
| Somite | + | ||||||||
| Intersomitic blood vessels | + | + | |||||||
| Condensing somites | + | ||||||||
| Notochord | + | + | + | ||||||
| Heart | + | + | + | + | + | + | |||
| Atria | + | ||||||||
| Bulbus cordi | + | ||||||||
| Endothelium | + | + | |||||||
| Gut | + | + | |||||||
| Liver | + | + | + | + | + | + | |||
| Esphagus | + | + | + | ||||||
| Stomach | + | + | + | ||||||
| Pancreas | + | ||||||||
| Intestine | + | + | |||||||
| Rectum | + | ||||||||
| Anus | + | ||||||||
| Limbs | |||||||||
| Forelimb | + | ||||||||
| Hindlimb | + | ||||||||
| Urogenital sinus | + | ||||||||
| Kidneys | + | + | + | + | |||||
| Ureters | + | ||||||||
| Bladder | + | + | + | ||||||
| Urethra | |||||||||
| Genital sinus | + | ||||||||
| Integumental | + | ||||||||
| Skin | + | ||||||||
| Respiratory system | |||||||||
| Lungs | + | + | + | + | + | + | |||
5. Mouse Models
5.1. Cobalamin Absorption
5.2. Cobalamin and Folic Acid Pathways
5.3. Cobalamin and Methylmalonyl-CoA Mutase (MCM)
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- MRC Vitamin Study Research Group. Prevention of neural tube defects: Results of the medical research council vitamin study. Lancet 1991, 338, 131–137. [CrossRef]
- Czeizel, A.E.; Dudas, I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N. Engl. J. Med. 1992, 327, 1832–1835. [Google Scholar] [CrossRef]
- Heseker, H.B.; Mason, J.B.; Selhub, J.; Rosenberg, I.H.; Jacques, P.F. Not all cases of neural-tube defect can be prevented by increasing the intake of folic acid. Br. J. Nutr. 2009, 102, 173–180. [Google Scholar] [CrossRef]
- Black, M.M. Effects of vitamin B12 and folate deficiency on brain development in children. Food Nutr. Bull. 2008, 29, S126–S131. [Google Scholar]
- Gadhok, A.K.; Sinha, M.; Khunteta, R.; Vardey, S.K.; Upadhyaya, C.; Sharma, T.K.; Jha, M. Serum homocysteine level and its association with folic acid and vitamin B12 in the third trimester of pregnancies complicated with intrauterine growth restriction. Clin. Lab. 2011, 57, 933–938. [Google Scholar]
- Hozyasz, K.K.; Oltarzewski, M.; Lugowska, I.; Szymanski, M.; Surowiec, Z. Whole blood propionylcarnitine in newborns with orofacial cleft. Matern. Child Nutr. 2011, 7, 100–103. [Google Scholar] [CrossRef]
- Muthayya, S.; Kurpad, A.V.; Duggan, C.P.; Bosch, R.J.; Dwarkanath, P.; Mhaskar, A.; Mhaskar, R.; Thomas, A.; Vaz, M.; Bhat, S.; et al. Low maternal vitamin B12 status is associated with intrauterine growth retardation in urban South Indians. Eur. J. Clin. Nutr. 2006, 60, 791–801. [Google Scholar] [CrossRef]
- Froese, D.S.; Healy, S.; McDonald, M.; Kochan, G.; Oppermann, U.; Niesen, F.H.; Gravel, R.A. Thermolability of mutant MMACHC protein in the vitamin B12-responsive cblC disorder. Mol. Genet. Metab. 2010, 100, 29–36. [Google Scholar] [CrossRef]
- Fowler, B. Genetic defects of folate and cobalamin metabolism. Eur. J. Pediatr. 1998, 157, S60–S66. [Google Scholar] [CrossRef]
- Fyfe, J.C.; Madsen, M.; Hojrup, P.; Christensen, E.I.; Tanner, S.M.; de la Chapelle, A.; He, Q.; Moestrup, S.K. The functional cobalamin (vitamin B12)-intrinsic factor receptor is a novel complex of cubilin and amnionless. Blood 2004, 103, 1573–1579. [Google Scholar] [CrossRef]
- Moestrup, S.K.; Kozyraki, R.; Kristiansen, M.; Kaysen, J.H.; Rasmussen, H.H.; Brault, D.; Pontillon, F.; Goda, F.O.; Christensen, E.I.; Hammond, T.G.; et al. The intrinsic factor-vitamin B12 receptor and target of teratogenic antibodies is a megalin-binding peripheral membrane protein with homology to developmental proteins. J. Biol. Chem. 1998, 273, 5235–5242. [Google Scholar] [CrossRef]
- Quadros, E.V.; Sequeira, J.M. Cellular uptake of cobalamin: Transcobalamin and the TCblR/CD320 receptor. Biochimie 2013, 95, 1008–1018. [Google Scholar] [CrossRef]
- Youngdahl-Turner, P.; Rosenberg, L.E.; Allen, R.H. Binding and uptake of transcobalamin II by human fibroblasts. J. Clin. Investig. 1978, 61, 133–141. [Google Scholar] [CrossRef]
- Youngdahl-Turner, P.; Mellman, I.S.; Allen, R.H.; Rosenberg, L.E. Protein mediated vitamin uptake. Exp. Cell. Res. 1979, 118, 127–134. [Google Scholar] [CrossRef]
- Laframboise, R.; Cooper, B.A.; Rosenblatt, D.S. Malabsorption of vitamin B12 from the intestine in a child with cblF disease: Evidence for lysosomal-mediated absorption. Blood 1992, 80, 291–292. [Google Scholar]
- Miousse, I.R.; Watkins, D.; Rosenblatt, D.S. Novel splice site mutations and a large deletion in three patients with the cblF inborn error of vitamin B12 metabolism. Mol. Genet. Metab. 2011, 102, 505–507. [Google Scholar] [CrossRef]
- Oladipo, O.; Rosenblatt, D.S.; Watkins, D.; Miousse, I.R.; Sprietsma, L.; Dietzen, D.J.; Shinawi, M. Cobalamin F disease detected by newborn screening and follow-up on a 14-year-old patient. Pediatrics 2011, 128, 2010–3518. [Google Scholar]
- Rutsch, F.; Gailus, S.; Miousse, I.R.; Suormala, T.; Sagne, C.; Toliat, M.R.; Nurnberg, G.; Wittkampf, T.; Buers, I.; Sharifi, A.; et al. Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism. Nat. Genet. 2009, 41, 234–239. [Google Scholar] [CrossRef]
- Shih, V.E.; Axel, S.M.; Tewksbury, J.C.; Watkins, D.; Cooper, B.A.; Rosenblatt, D.S. Defective lysosomal release of vitamin B12 (cb1F): A hereditary cobalamin metabolic disorder associated with sudden death. Am. J. Med. Genet. 1989, 33, 555–563. [Google Scholar] [CrossRef]
- Vassiliadis, A.; Rosenblatt, D.S.; Cooper, B.A.; Bergeron, J.J. Lysosomal cobalamin accumulation in fibroblasts from a patient with an inborn error of cobalamin metabolism (cblF complementation group): Visualization by electron microscope radioautography. Exp. Cell. Res. 1991, 195, 295–302. [Google Scholar] [CrossRef]
- Waggoner, D.J.; Ueda, K.; Mantia, C.; Dowton, S.B. Methylmalonic aciduria (cblF): Case report and response to therapy. Am. J. Med. Genet. 1998, 79, 373–375. [Google Scholar] [CrossRef]
- Watkins, D.; Rosenblatt, D.S. Failure of lysosomal release of vitamin B12: A new complementation group causing methylmalonic aciduria (cblF). Am. J. Hum. Genet. 1986, 39, 404–408. [Google Scholar]
- Coelho, D.; Kim, J.C.; Miousse, I.R.; Fung, S.; du Moulin, M.; Buers, I.; Suormala, T.; Burda, P.; Frapolli, M.; Stucki, M.; et al. Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat. Genet. 2012, 44, 1152–1155. [Google Scholar] [CrossRef]
- Lerner-Ellis, J.P.; Tirone, J.C.; Pawelek, P.D.; Dore, C.; Atkinson, J.L.; Watkins, D.; Morel, C.F.; Fujiwara, T.M.; Moras, E.; Hosack, A.R.; et al. Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat. Genet. 2006, 38, 93–100. [Google Scholar] [CrossRef]
- Kim, J.; Gherasim, C.; Banerjee, R. Decyanation of vitamin B12 by a trafficking chaperone. Proc. Natl. Acad. Sci. USA 2008, 105, 14551–14554. [Google Scholar] [CrossRef]
- Hannibal, L.; Kim, J.; Brasch, N.E.; Wang, S.; Rosenblatt, D.S.; Banerjee, R.; Jacobsen, D.W. Processing of alkylcobalamins in mammalian cells: A role for the MMACHC (cblC) gene product. Mol. Genet. Metab. 2009, 97, 260–266. [Google Scholar] [CrossRef]
- Plesa, M.; Kim, J.; Paquette, S.G.; Gagnon, H.; Ng-Thow-Hing, C.; Gibbs, B.F.; Hancock, M.A.; Rosenblatt, D.S.; Coulton, J.W. Interaction between MMACHC and MMADHC, two human proteins participating in intracellular vitamin B12 metabolism. Mol. Genet. Metab. 2011, 102, 139–148. [Google Scholar] [CrossRef]
- Takahashi-Iniguez, T.; Garcia-Arellano, H.; Trujillo-Roldan, M.A.; Flores, M.E. Protection and reactivation of human methylmalonyl-CoA mutase by MMAA protein. Biochem. Biophys. Res. Commun. 2011, 404, 443–447. [Google Scholar] [CrossRef]
- Takahashi-Iniguez, T.; Garcia-Hernandez, E.; Arreguin-Espinosa, R.; Flores, M.E. Role of vitamin B12 on methylmalonyl-CoA mutase activity. J. Zhejiang Univ. Sci. B 2012, 13, 423–437. [Google Scholar] [CrossRef]
- Dobson, C.M.; Wai, T.; Leclerc, D.; Kadir, H.; Narang, M.; Lerner-Ellis, J.P.; Hudson, T.J.; Rosenblatt, D.S.; Gravel, R.A. Identification of the gene responsible for the cblB complementation group of vitamin B12-dependent methylmalonic aciduria. Hum. Mol. Genet. 2002, 11, 3361–3369. [Google Scholar] [CrossRef]
- Leal, N.A.; Park, S.D.; Kima, P.E.; Bobik, T.A. Identification of the human and bovine ATP:Cob(I)alamin adenosyltransferase cDNAs based on complementation of a bacterial mutant. J. Biol. Chem. 2003, 278, 9227–9234. [Google Scholar]
- Yamanishi, M.; Vlasie, M.; Banerjee, R. Adenosyltransferase: An enzyme and an escort for coenzyme B12? Trends Biochem. Sci. 2005, 30, 304–308. [Google Scholar] [CrossRef]
- Gaber, K.R.; Farag, M.K.; Soliman, S.E.; El-Bassyouni, H.T.; El-Kamah, G. Maternal vitamin B12 and the risk of fetal neural tube defects in Egyptian patients. Clin. Lab. 2007, 53, 69–75. [Google Scholar]
- Molloy, A.M.; Kirke, P.N.; Troendle, J.F.; Burke, H.; Sutton, M.; Brody, L.C.; Scott, J.M.; Mills, J.L. Maternal vitamin B12 status and risk of neural tube defects in a population with high neural tube defect prevalence and no folic Acid fortification. Pediatrics 2009, 123, 917–923. [Google Scholar] [CrossRef]
- Ray, J.G.; Wyatt, P.R.; Thompson, M.D.; Vermeulen, M.J.; Meier, C.; Wong, P.Y.; Farrell, S.A.; Cole, D.E. Vitamin B12 and the risk of neural tube defects in a folic-acid-fortified population. Epidemiology 2007, 18, 362–366. [Google Scholar] [CrossRef]
- Thompson, M.D.; Cole, D.E.; Ray, J.G. Vitamin B-12 and neural tube defects: The Canadian experience. Am. J. Clin. Nutr. 2009, 89, 697S–701S. [Google Scholar] [CrossRef]
- Zhang, T.; Xin, R.; Gu, X.; Wang, F.; Pei, L.; Lin, L.; Chen, G.; Wu, J.; Zheng, X. Maternal serum vitamin B12, folate and homocysteine and the risk of neural tube defects in the offspring in a high-risk area of China. Public Health Nutr. 2009, 12, 680–686. [Google Scholar] [CrossRef]
- Ceyhan, S.T.; Beyan, C.; Atay, V.; Yaman, H.; Alanbay, I.; Kaptan, K.; Baser, I. Serum vitamin B12 and homocysteine levels in pregnant women with neural tube defect. Gynecol. Endocrinol. 2010, 26, 578–581. [Google Scholar] [CrossRef]
- Economides, D.L.; Ferguson, J.; Mackenzie, I.Z.; Darley, J.; Ware, II; Holmes-Siedle, M. Folate and vitamin B12 concentrations in maternal and fetal blood, and amniotic fluid in second trimester pregnancies complicated by neural tube defects. Br. J. Obstet. Gynaecol. 1992, 99, 23–25. [Google Scholar]
- Mills, J.L.; Tuomilehto, J.; Yu, K.F.; Colman, N.; Blaner, W.S.; Koskela, P.; Rundle, W.E.; Forman, M.; Toivanen, L.; Rhoads, G.G. Maternal vitamin levels during pregnancies producing infants with neural tube defects. J. Pediatr. 1992, 120, 863–871. [Google Scholar] [CrossRef]
- Molloy, A.M.; Kirke, P.; Hillary, I.; Weir, D.G.; Scott, J.M. Maternal serum folate and vitamin B12 concentrations in pregnancies associated with neural tube defects. Arch. Dis. Child. 1985, 60, 660–665. [Google Scholar] [CrossRef]
- Schorah, C.J.; Smithells, R.W.; Scott, J. Vitamin B12 and anencephaly. Lancet 1980, 315, 880. [Google Scholar] [CrossRef]
- Stoll, C.; Dott, B.; Alembik, Y.; Koehl, C. Maternal trace elements, vitamin B12, vitamin A, folic acid, and fetal malformations. Reprod. Toxicol. 1999, 13, 53–57. [Google Scholar] [CrossRef]
- Suarez, L.; Hendricks, K.; Felkner, M.; Gunter, E. Maternal serum B12 levels and risk for neural tube defects in a Texas-Mexico border population. Ann. Epidemiol. 2003, 13, 81–88. [Google Scholar] [CrossRef]
- Thorand, B.; Pietrzik, K.; Prinz-Langenohl, R.; Hages, M.; Holzgreve, W. Maternal and fetal serum and red blood cell folate and vitamin B12 concentrations in pregnancies affected by neural tube defects. Z. Geburtshilfe Neonatol. 1996, 200, 176–180. [Google Scholar]
- Van der Put, N.M.; Thomas, C.M.; Eskes, T.K.; Trijbels, F.J.; Steegers-Theunissen, R.P.; Mariman, E.C.; de Graaf-Hess, A.; Smeitink, J.A.; Blom, H.J. Altered folate and vitamin B12 metabolism in families with spina bifida offspring. QJM 1997, 90, 505–510. [Google Scholar] [CrossRef]
- Wild, J.; Schorah, C.J.; Sheldon, T.A.; Smithells, R.W. Investigation of factors influencing folate status in women who have had a neural tube defect-affected infant. Br. J. Obstet. Gynaecol. 1993, 100, 546–549. [Google Scholar] [CrossRef]
- Ray, J.G.; Blom, H.J. Vitamin B12 insufficiency and the risk of fetal neural tube defects. QJM 2003, 96, 289–295. [Google Scholar] [CrossRef]
- Brouns, R.; Ursem, N.; Lindemans, J.; Hop, W.; Pluijm, S.; Steegers, E.; Steegers-Theunissen, R. Polymorphisms in genes related to folate and cobalamin metabolism and the associations with complex birth defects. Prenat. Diagn. 2008, 28, 485–493. [Google Scholar] [CrossRef]
- Dawson, E.B.; Evans, D.R.; Harris, W.A.; van Hook, J.W. Amniotic fluid B12, calcium, and lead levels associated with neural tube defects. Am. J. Perinatol. 1999, 16, 373–378. [Google Scholar] [CrossRef]
- Dawson, E.B.; Evans, D.R.; van Hook, J.W. Amniotic fluid B12 and folate levels associated with neural tube defects. Am. J. Perinatol. 1998, 15, 511–514. [Google Scholar] [CrossRef]
- Gardiki-Kouidou, P.; Seller, M.J. Amniotic fluid folate, vitamin B12 and transcobalamins in neural tube defects. Clin. Genet. 1988, 33, 441–448. [Google Scholar] [CrossRef]
- Steegers-Theunissen, R.P.; Boers, G.H.; Blom, H.J.; Nijhuis, J.G.; Thomas, C.M.; Borm, G.F.; Eskes, T.K. Neural tube defects and elevated homocysteine levels in amniotic fluid. Am. J. Obstet. Gynecol. 1995, 172, 1436–1441. [Google Scholar] [CrossRef]
- Weekes, E.W.; Tamura, T.; Davis, R.O.; Birch, R.; Vaughn, W.H.; Franklin, J.C.; Barganier, C.; Cosper, P.; Finley, S.C.; Finley, W.H. Nutrient levels in amniotic fluid from women with normal and neural tube defect pregnancies. Biol. Neonate 1992, 61, 226–231. [Google Scholar] [CrossRef]
- Magnus, P.; Magnus, E.M.; Berg, K. Increased levels of apo-transcobalamins I and II in amniotic fluid from pregnant women with previous neural tube defect offspring. Clin. Genet. 1986, 30, 167–172. [Google Scholar]
- Magnus, P.; Magnus, E.M.; Berg, K. Transcobalamins in the etiology of neural tube defects. Clin. Genet. 1991, 39, 309–310. [Google Scholar] [CrossRef]
- Lindgren, A.; Kilander, A.; Bagge, E.; Nexo, E. Holotranscobalamin—A sensitive marker of cobalamin malabsorption. Eur. J. Clin. Investig. 1999, 29, 321–329. [Google Scholar]
- Tisman, G.; Vu, T.; Amin, J.; Luszko, G.; Brenner, M.; Ramos, M.; Flener, V.; Cordts, V.; Bateman, R.; Malkin, S.; et al. Measurement of red blood cell-vitamin B12: A study of the correlation between intracellular B12 content and concentrations of plasma holotranscobalamin II. Am. J. Hematol. 1993, 43, 226–229. [Google Scholar] [CrossRef]
- Al Aisari, F.; Al-Hashmi, H.; Mula-Abed, W.A. Comparison between serum holotranscobalamin and total vitamin B12 as indicators of vitamin B12 status. Oman Med. J. 2010, 25, 9–12. [Google Scholar]
- Van Rooij, I.A.; Swinkels, D.W.; Blom, H.J.; Merkus, H.M.; Steegers-Theunissen, R.P. Vitamin and homocysteine status of mothers and infants and the risk of nonsyndromic orofacial clefts. Am. J. Obstet. Gynecol. 2003, 189, 1155–1160. [Google Scholar] [CrossRef]
- Bille, C.; Olsen, J.; Vach, W.; Knudsen, V.K.; Olsen, S.F.; Rasmussen, K.; Murray, J.C.; Andersen, A.M.; Christensen, K. Oral clefts and life style factors—A case-cohort study based on prospective Danish data. Eur. J. Epidemiol. 2007, 22, 173–181. [Google Scholar] [CrossRef]
- Krapels, I.P.; van Rooij, I.A.; Ocke, M.C.; van Cleef, B.A.; Kuijpers-Jagtman, A.M.; Steegers-Theunissen, R.P. Maternal dietary B vitamin intake, other than folate, and the association with orofacial cleft in the offspring. Eur. J. Nutr. 2004, 43, 7–14. [Google Scholar] [CrossRef]
- Franke, B.; Vermeulen, S.H.; Steegers-Theunissen, R.P.; Coenen, M.J.; Schijvenaars, M.M.; Scheffer, H.; den Heijer, M.; Blom, H.J. An association study of 45 folate-related genes in spina bifida: Involvement of cubilin (CUBN) and tRNA aspartic acid methyltransferase 1 (TRDMT1). Birth Defects Res. 2009, 85, 216–226. [Google Scholar] [CrossRef]
- Gos, M.; Sliwerska, E.; Szpecht-Potocka, A. Mutation incidence in folate metabolism genes and regulatory genes in Polish families with neural tube defects. J. Appl. Genet. 2004, 45, 363–368. [Google Scholar]
- Ouyang, S.; Li, Y.; Liu, Z.; Chang, H.; Wu, J. Association between MTR A2756G and MTRR A66G polymorphisms and maternal risk for neural tube defects: A meta-analysis. Gene 2013, 515, 308–312. [Google Scholar] [CrossRef]
- Pietrzyk, J.J.; Bik-Multanowski, M.; Sanak, M.; Twardowska, M. Polymorphisms of the 5,10-methylenetetrahydrofolate and the methionine synthase reductase genes as independent risk factors for spina bifida. J. Appl. Genet. 2003, 44, 111–113. [Google Scholar]
- Van der Linden, I.J.; den Heijer, M.; Afman, L.A.; Gellekink, H.; Vermeulen, S.H.; Kluijtmans, L.A.; Blom, H.J. The methionine synthase reductase 66A>G polymorphism is a maternal risk factor for spina bifida. J. Mol. Med. 2006, 84, 1047–1054. [Google Scholar] [CrossRef]
- Wilson, A.; Platt, R.; Wu, Q.; Leclerc, D.; Christensen, B.; Yang, H.; Gravel, R.A.; Rozen, R. A common variant in methionine synthase reductase combined with low cobalamin (vitamin B12) increases risk for spina bifida. Mol. Genet. Metab. 1999, 67, 317–323. [Google Scholar] [CrossRef]
- Zhu, H.; Wicker, N.J.; Shaw, G.M.; Lammer, E.J.; Hendricks, K.; Suarez, L.; Canfield, M.; Finnell, R.H. Homocysteine remethylation enzyme polymorphisms and increased risks for neural tube defects. Mol. Genet. Metab. 2003, 78, 216–221. [Google Scholar] [CrossRef]
- Doolin, M.T.; Barbaux, S.; McDonnell, M.; Hoess, K.; Whitehead, A.S.; Mitchell, L.E. Maternal genetic effects, exerted by genes involved in homocysteine remethylation, influence the risk of spina bifida. Am. J. Hum. Genet. 2002, 71, 1222–1226. [Google Scholar] [CrossRef]
- Relton, C.L.; Wilding, C.S.; Pearce, M.S.; Laffling, A.J.; Jonas, P.A.; Lynch, S.A.; Tawn, E.J.; Burn, J. Gene-gene interaction in folate-related genes and risk of neural tube defects in a UK population. J. Med. Genet. 2004, 41, 256–260. [Google Scholar] [CrossRef]
- Brandalize, A.P.; Bandinelli, E.; Borba, J.B.; Felix, T.M.; Roisenberg, I.; Schuler-Faccini, L. Polymorphisms in genes MTHFR, MTR and MTRR are not risk factors for cleft lip/palate in South Brazil. Braz. J. Med. Biol. Res. 2007, 40, 787–791. [Google Scholar] [CrossRef]
- Candito, M.; Rivet, R.; Herbeth, B.; Boisson, C.; Rudigoz, R.C.; Luton, D.; Journel, H.; Oury, J.F.; Roux, F.; Saura, R.; et al. Nutritional and genetic determinants of vitamin B and homocysteine metabolisms in neural tube defects: A multicenter case-control study. Am. J. Med. Genet. 2008, 1, 1128–1133. [Google Scholar]
- Ouyang, S.; Liu, Z.; Li, Y.; Wu, J. Meta-analyses on the association of MTR A2756G and MTRR A66G polymorphisms with neural tube defect risks in Caucasian children. J. Matern. Fetal Neonatal Med. 2013, 26, 1166–1170. [Google Scholar] [CrossRef]
- Relton, C.L.; Wilding, C.S.; Laffling, A.J.; Jonas, P.A.; Burgess, T.; Binks, K.; Tawn, E.J.; Burn, J. Low erythrocyte folate status and polymorphic variation in folate-related genes are associated with risk of neural tube defect pregnancy. Mol. Genet. Metab. 2004, 81, 273–281. [Google Scholar] [CrossRef]
- Van Beynum, I.M.; Kouwenberg, M.; Kapusta, L.; den Heijer, M.; van der Linden, I.J.; Daniels, O.; Blom, H.J. MTRR 66A>G polymorphism in relation to congenital heart defects. Clin. Chem. Lab. Med. 2006, 44, 1317–1323. [Google Scholar]
- Mostowska, A.; Hozyasz, K.K.; Jagodzinski, P.P. Maternal MTR genotype contributes to the risk of non-syndromic cleft lip and palate in the Polish population. Clin. Genet. 2006, 69, 512–517. [Google Scholar]
- Al Farra, H.Y. Methionine synthase polymorphisms (MTR 2756A>G and MTR 2758C>G) frequencies and distribution in the Jordanian population and their correlation with neural tube defects in the population of the northern part of Jordan. Indian J. Hum. Genet. 2010, 16, 138–143. [Google Scholar] [CrossRef]
- Gueant-Rodriguez, R.M.; Rendeli, C.; Namour, B.; Venuti, L.; Romano, A.; Anello, G.; Bosco, P.; Debard, R.; Gerard, P.; Viola, M.; et al. Transcobalamin and methionine synthase reductase mutated polymorphisms aggravate the risk of neural tube defects in humans. Neurosci. Lett. 2003, 344, 189–192. [Google Scholar] [CrossRef]
- Pangilinan, F.; Mitchell, A.; VanderMeer, J.; Molloy, A.M.; Troendle, J.; Conley, M.; Kirke, P.N.; Sutton, M.; Sequeira, J.M.; Quadros, E.V.; et al. Transcobalamin II receptor polymorphisms are associated with increased risk for neural tube defects. J. Med. Genet. 2010, 47, 677–685. [Google Scholar] [CrossRef]
- Martinelli, M.; Scapoli, L.; Palmieri, A.; Pezzetti, F.; Baciliero, U.; Padula, E.; Carinci, P.; Morselli, P.G.; Carinci, F. Study of four genes belonging to the folate pathway: Transcobalamin 2 is involved in the onset of non-syndromic cleft lip with or without cleft palate. Hum. Mutat. 2006, 27, 294. [Google Scholar]
- Cherukad, J.; Wainwright, V.; Watson, E.D. Spatial and temporal expression of folate-related transporters and metabolic enzymes during mouse placental development. Placenta 2012, 33, 440–448. [Google Scholar]
- Diez-Roux, G.; Banfi, S.; Sultan, M.; Geffers, L.; Anand, S.; Rozado, D.; Magen, A.; Canidio, E.; Pagani, M.; Peluso, I.; et al. A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. 2011, 9, e1000582. [Google Scholar]
- Elmore, C.L.; Wu, X.; Leclerc, D.; Watson, E.D.; Bottiglieri, T.; Krupenko, N.I.; Krupenko, S.A.; Cross, J.C.; Rozen, R.; Gravel, R.A.; et al. Metabolic derangement of methionine and folate metabolism in mice deficient in methionine synthase reductase. Mol. Genet. Metab. 2007, 91, 85–97. [Google Scholar]
- Moreno-Garcia, M.A.; Rosenblatt, D.S.; Jerome-Majewska, L.A. The methylmalonic aciduria related genes, Mmaa, Mmab, and Mut, are broadly expressed in placental and embryonic tissues during mouse organogenesis. Mol. Genet. Metab. 2012, 107, 368–374. [Google Scholar]
- Pupavac, M.; Garcia, M.A.; Rosenblatt, D.S.; Jerome-Majewska, L.A. Expression of Mmachc and Mmadhc during mouse organogenesis. Mol. Genet. Metab. 2011, 103, 401–405. [Google Scholar] [CrossRef]
- Sansom, S.N.; Griffiths, D.S.; Faedo, A.; Kleinjan, D.J.; Ruan, Y.; Smith, J.; van Heyningen, V.; Rubenstein, J.L.; Livesey, F.J. The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet. 2009, 5, e1000511. [Google Scholar] [CrossRef]
- Visel, A.; Thaller, C.; Eichele, G. GenePaint.org: An atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 2004, 32, D552–D556. [Google Scholar] [CrossRef]
- Li, F.; Watkins, D.; Rosenblatt, D.S. Vitamin B(12) and birth defects. Mol. Genet. Metab. 2009, 98, 166–172. [Google Scholar] [CrossRef]
- Seetharam, B.; Christensen, E.I.; Moestrup, S.K.; Hammond, T.G.; Verroust, P.J. Identification of rat yolk sac target protein of teratogenic antibodies, gp280, as intrinsic factor-cobalamin receptor. J. Clin. Investig. 1997, 99, 2317–2322. [Google Scholar] [CrossRef]
- Tanner, S.M.; Aminoff, M.; Wright, F.A.; Liyanarachchi, S.; Kuronen, M.; Saarinen, A.; Massika, O.; Mandel, H.; Broch, H.; de la Chapelle, A. Amnionless, essential for mouse gastrulation, is mutated in recessive hereditary megaloblastic anemia. Nat. Genet. 2003, 33, 426–429. [Google Scholar] [CrossRef]
- Wang, X.; Bornslaeger, E.A.; Haub, O.; Tomihara-Newberger, C.; Lonberg, N.; Dinulos, M.B.; Disteche, C.M.; Copeland, N.; Gilbert, D.J.; Jenkins, N.A.; et al. A candidate gene for the amnionless gastrulation stage mouse mutation encodes a TRAF-related protein. Dev. Biol. 1996, 177, 274–290. [Google Scholar] [CrossRef]
- Tomihara-Newberger, C.; Haub, O.; Lee, H.G.; Soares, V.; Manova, K.; Lacy, E. The amn gene product is required in extraembryonic tissues for the generation of middle primitive streak derivatives. Dev. Biol. 1998, 204, 34–54. [Google Scholar] [CrossRef]
- Densupsoontorn, N.; Sanpakit, K.; Vijarnsorn, C.; Pattaragarn, A.; Kangwanpornsiri, C.; Jatutipsompol, C.; Tirapongporn, H.; Jirapinyo, P.; Shah, N.P.; Sturm, A.C.; et al. Imerslund-grasbeck syndrome: New mutation in amnionless. Pediatr. Int. 2012, 54, e19–e21. [Google Scholar] [CrossRef]
- Namour, F.; Dobrovoljski, G.; Chery, C.; Audonnet, S.; Feillet, F.; Sperl, W.; Gueant, J.L. Luminal expression of cubilin is impaired in Imerslund-Grasbeck syndrome with compound AMN mutations in intron 3 and exon 7. Haematologica 2011, 96, 1715–1719. [Google Scholar] [CrossRef]
- Smith, B.T.; Mussell, J.C.; Fleming, P.A.; Barth, J.L.; Spyropoulos, D.D.; Cooley, M.A.; Drake, C.J.; Argraves, W.S. Targeted disruption of cubilin reveals essential developmental roles in the structure and function of endoderm and in somite formation. BMC Dev. Biol. 2006, 6, 30. [Google Scholar] [CrossRef]
- Kozyraki, R.; Kristiansen, M.; Silahtaroglu, A.; Hansen, C.; Jacobsen, C.; Tommerup, N.; Verroust, P.J.; Moestrup, S.K. The human intrinsic factor-vitamin B12 receptor, cubilin: Molecular characterization and chromosomal mapping of the gene to 10 p within the autosomal recessive megaloblastic anemia (MGA1) region. Blood 1998, 91, 3593–3600. [Google Scholar]
- Kristiansen, M.; Aminoff, M.; Jacobsen, C.; de La Chapelle, A.; Krahe, R.; Verroust, P.J.; Moestrup, S.K. Cubilin P1297L mutation associated with hereditary megaloblastic anemia 1 causes impaired recognition of intrinsic factor-vitamin B(12) by cubilin. Blood 2000, 96, 405–409. [Google Scholar]
- Storm, T.; Emma, F.; Verroust, P.J.; Hertz, J.M.; Nielsen, R.; Christensen, E.I. A patient with cubilin deficiency. N. Engl. J. Med. 2011, 364, 89–91. [Google Scholar] [CrossRef]
- Hammad, S.M.; Stefansson, S.; Twal, W.O.; Drake, C.J.; Fleming, P.; Remaley, A.; Brewer, H.B., Jr.; Argraves, W.S. Cubilin, the endocytic receptor for intrinsic factor-vitamin B(12) complex, mediates high-density lipoprotein holoparticle endocytosis. Proc. Natl. Acad. Sci. USA 1999, 96, 10158–10163. [Google Scholar] [CrossRef]
- Honein, M.A.; Paulozzi, L.J.; Mathews, T.J.; Erickson, J.D.; Wong, L.Y. Impact of folic acid fortification of the US food supply on the occurrence of neural tube defects. JAMA 2001, 285, 2981–2986. [Google Scholar] [CrossRef]
- Garcia, M.M.; Gueant-Rodriguez, R.M.; Pooya, S.; Brachet, P.; Alberto, J.M.; Jeannesson, E.; Maskali, F.; Gueguen, N.; Marie, P.Y.; Lacolley, P.; et al. Methyl donor deficiency induces cardiomyopathy through altered methylation/acetylation of PGC-1alpha by PRMT1 and SIRT1. J. Pathol. 2011, 225, 324–335. [Google Scholar] [CrossRef]
- Beaudin, A.E.; Perry, C.A.; Stabler, S.P.; Allen, R.H.; Stover, P.J. Maternal Mthfd1 disruption impairs fetal growth but does not cause neural tube defects in mice. Am. J. Clin. Nutr. 2012, 95, 882–891. [Google Scholar] [CrossRef]
- Swanson, D.A.; Liu, M.L.; Baker, P.J.; Garrett, L.; Stitzel, M.; Wu, J.; Harris, M.; Banerjee, R.; Shane, B.; Brody, L.C. Targeted disruption of the methionine synthase gene in mice. Mol. Cell. Biol. 2001, 21, 1058–1065. [Google Scholar] [CrossRef]
- Deng, L.; Elmore, C.L.; Lawrance, A.K.; Matthews, R.G.; Rozen, R. Methionine synthase reductase deficiency results in adverse reproductive outcomes and congenital heart defects in mice. Mol. Genet. Metab. 2008, 94, 336–342. [Google Scholar] [CrossRef]
- Chen, Z.; Karaplis, A.C.; Ackerman, S.L.; Pogribny, I.P.; Melnyk, S.; Lussier-Cacan, S.; Chen, M.F.; Pai, A.; John, S.W.; Smith, R.S.; et al. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum. Mol. Genet. 2001, 10, 433–443. [Google Scholar] [CrossRef]
- Chen, Z.; Schwahn, B.C.; Wu, Q.; He, X.; Rozen, R. Postnatal cerebellar defects in mice deficient in methylenetetrahydrofolate reductase. Int. J. Dev. Neurosci. 2005, 23, 465–474. [Google Scholar] [CrossRef]
- Jadavji, N.M.; Deng, L.; Leclerc, D.; Malysheva, O.; Bedell, B.J.; Caudill, M.A.; Rozen, R. Severe methylenetetrahydrofolate reductase deficiency in mice results in behavioral anomalies with morphological and biochemical changes in hippocampus. Mol. Genet. Metab. 2012, 106, 149–159. [Google Scholar] [CrossRef]
- Li, D.; Pickell, L.; Liu, Y.; Wu, Q.; Cohn, J.S.; Rozen, R. Maternal methylenetetrahydrofolate reductase deficiency and low dietary folate lead to adverse reproductive outcomes and congenital heart defects in mice. Am. J. Clin. Nutr. 2005, 82, 188–195. [Google Scholar]
- Peters, H.; Nefedov, M.; Sarsero, J.; Pitt, J.; Fowler, K.J.; Gazeas, S.; Kahler, S.G.; Ioannou, P.A. A knock-out mouse model for methylmalonic aciduria resulting in neonatal lethality. J. Biol. Chem. 2003, 278, 52909–52913. [Google Scholar] [CrossRef]
- Chandler, R.J.; Zerfas, P.M.; Shanske, S.; Sloan, J.; Hoffmann, V.; DiMauro, S.; Venditti, C.P. Mitochondrial dysfunction in mut methylmalonic acidemia. FASEB J. 2009, 23, 1252–1261. [Google Scholar] [CrossRef]
- Chandler, R.J.; Venditti, C.P. Adenovirus-mediated gene delivery rescues a neonatal lethal murine model of Mut(0) methylmalonic acidemia. Hum. Gene Ther. 2008, 19, 53–60. [Google Scholar] [CrossRef]
- Chandler, R.J.; Venditti, C.P. Pre-clinical efficacy and dosing of an AAV8 vector expressing human methylmalonyl-CoA mutase in a murine model of methylmalonic acidemia (MMA). Mol. Genet. Metab. 2012, 107, 617–619. [Google Scholar] [CrossRef]
- Senac, J.S.; Chandler, R.J.; Sysol, J.R.; Li, L.; Venditti, C.P. Gene therapy in a murine model of methylmalonic acidemia using rAAV9-mediated gene delivery. Gene Ther. 2012, 19, 385–391. [Google Scholar] [CrossRef]
- Carrillo-Carrasco, N.; Chandler, R.J.; Chandrasekaran, S.; Venditti, C.P. Liver-directed recombinant adeno-associated viral gene delivery rescues a lethal mouse model of methylmalonic acidemia and provides long-term phenotypic correction. Hum. Gene Ther. 2010, 21, 1147–1154. [Google Scholar] [CrossRef]
- Peters, H.L.; Pitt, J.J.; Wood, L.R.; Hamilton, N.J.; Sarsero, J.P.; Buck, N.E. Mouse models for methylmalonic aciduria. PLoS One 2012, 7, e40609. [Google Scholar]
- Buck, N.E.; Dashnow, H.; Pitt, J.J.; Wood, L.R.; Peters, H.L. Development of transgenic mice containing an introduced stop codon on the human methylmalonyl-CoA mutase locus. PLoS One 2012, 7, e44974. [Google Scholar]
- Xu, Y.; Li, L.; Zhang, Z.; Li, Y. Effects of folinic acid and Vitamin B12 on ethanol-induced developmental toxicity in mouse. Toxicol. Lett. 2006, 167, 167–172. [Google Scholar] [CrossRef]
- Lu, S.J.; He, W.; Shi, B.; Meng, T.; Li, X.Y.; Liu, Y.R. A preliminary study on the teratogenesis of dexamethasone and the preventive effect of vitamin B12 on murine embryonic palatal shelf fusion in vitro. J. Zhejiang Univ. Sci. B 2008, 9, 306–312. [Google Scholar] [CrossRef]
- He, W.; Meng, T.; Wu, M.; Shi, B.; Lu, S.J.; Li, C.H. Perturbation of Fgf10 signal pathway in mouse embryonic palate by dexamethasone and vitamin B12 in vivo. J. Pediatr. Surg. 2010, 45, 2030–2035. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moreno-Garcia, M.A.; Rosenblatt, D.S.; Jerome-Majewska, L.A. Vitamin B12 Metabolism during Pregnancy and in Embryonic Mouse Models. Nutrients 2013, 5, 3531-3550. https://doi.org/10.3390/nu5093531
Moreno-Garcia MA, Rosenblatt DS, Jerome-Majewska LA. Vitamin B12 Metabolism during Pregnancy and in Embryonic Mouse Models. Nutrients. 2013; 5(9):3531-3550. https://doi.org/10.3390/nu5093531
Chicago/Turabian StyleMoreno-Garcia, Maira A., David S. Rosenblatt, and Loydie A. Jerome-Majewska. 2013. "Vitamin B12 Metabolism during Pregnancy and in Embryonic Mouse Models" Nutrients 5, no. 9: 3531-3550. https://doi.org/10.3390/nu5093531
APA StyleMoreno-Garcia, M. A., Rosenblatt, D. S., & Jerome-Majewska, L. A. (2013). Vitamin B12 Metabolism during Pregnancy and in Embryonic Mouse Models. Nutrients, 5(9), 3531-3550. https://doi.org/10.3390/nu5093531