Calcium Regulation and Bone Mineral Metabolism in Elderly Patients with Chronic Kidney Disease

Abstract
:1. Introduction
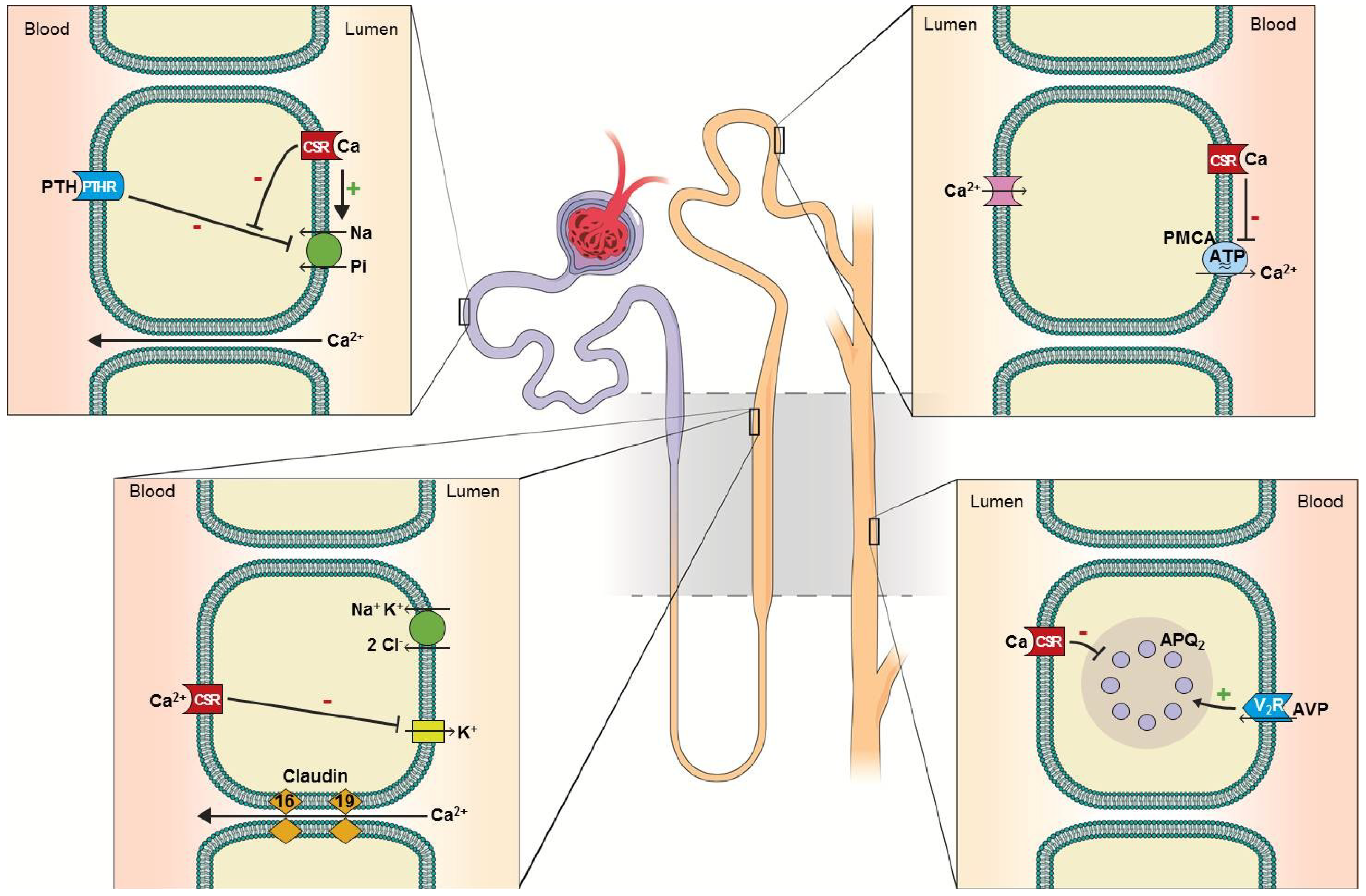
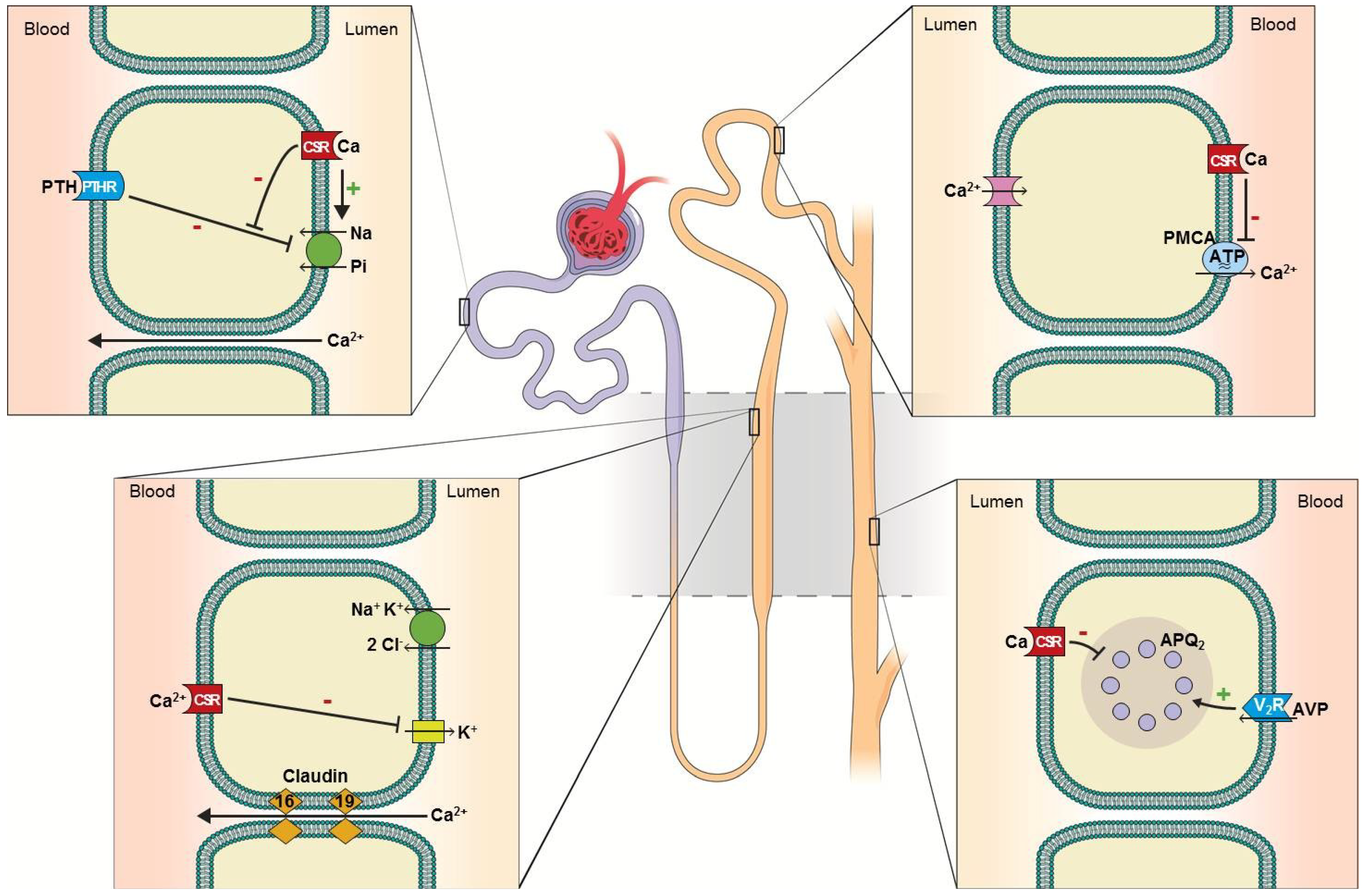
2. Calcium Homeostasis and Defects in Aging and CKD
Box 1. Klotho.
- Loss-of-function mutation in Klotho causes premature aging phenotype [19], which led to the initial discovery of the gene and appropriate naming of the gene: Klotho, the Goddess of life.
- Klotho encodes a single-pass transmembrane protein, also termed Klotho, that is expressed primarily in kidney and parathyroid gland [20].
- Klotho couples with FGF23 receptor to form a high-affinity receptor complex for FGF23 and mediates FGF23-induced phosphaturia [21].
- Klotho promotes Ca2+ conservation through stimulating TRPV-5 in the distal convoluted tubule [22].
- Extracellular domain of Klotho can be enzymatically cleaved and shed into the extracellular space, becoming a secreted form of Klotho.
- Klotho is regarded as an anti-aging molecule [23].

{kind=link}
{kind=link}
|
3. Phosphate
4. Vitamin D
5. Parathyroid Hormone
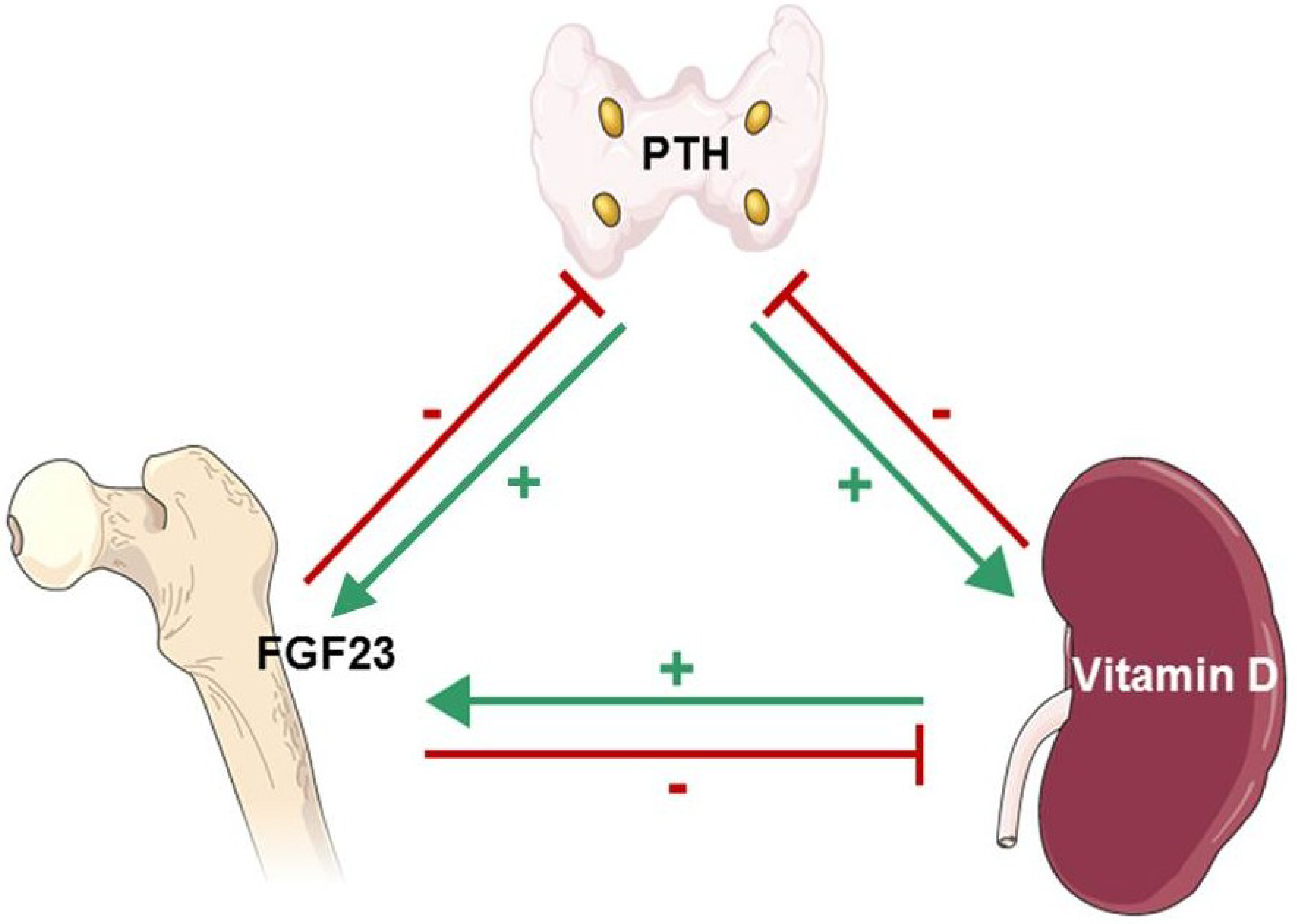
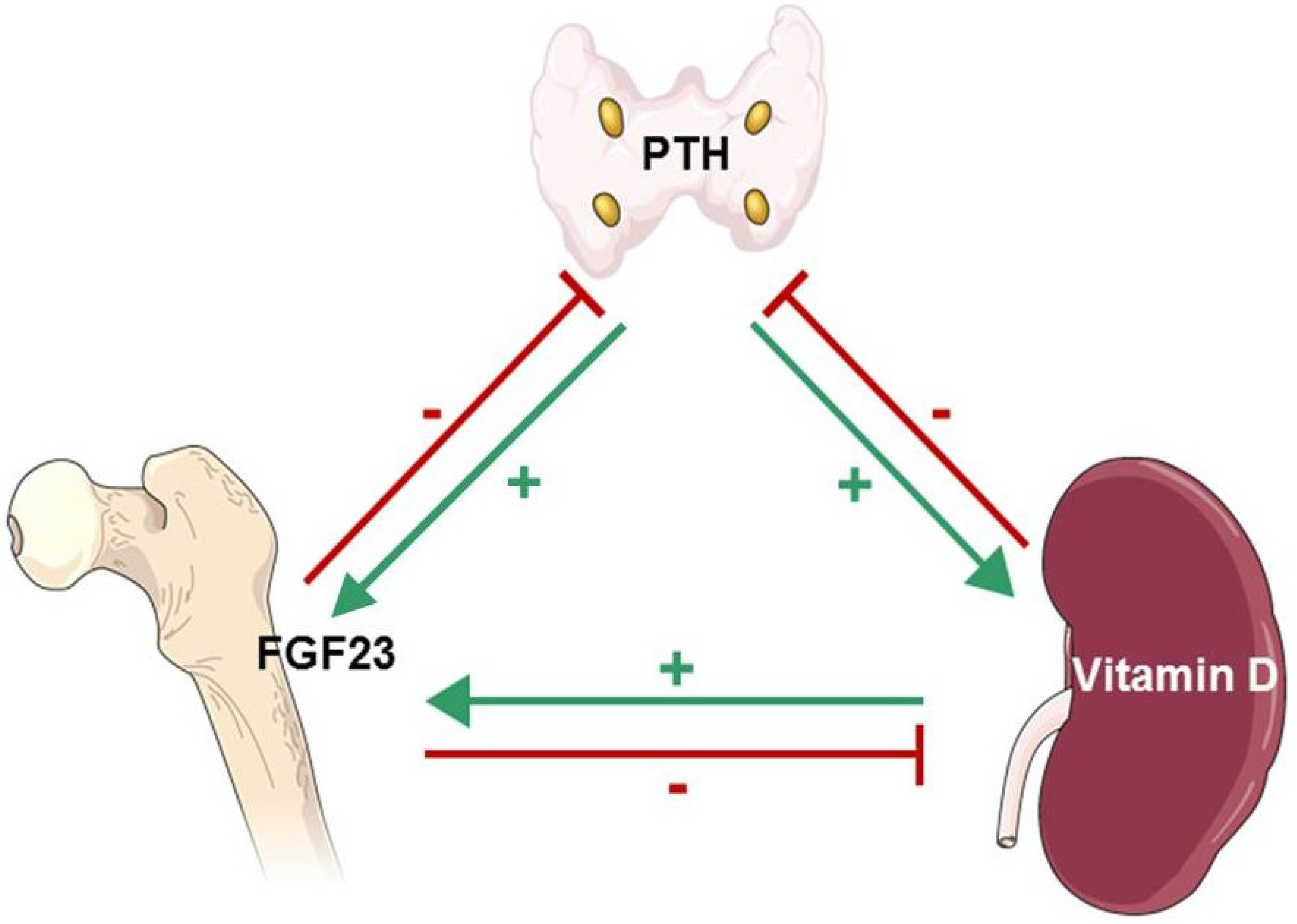
6. Fibroblast Growth Factor-23
7. Calcium Sensing Receptor
8. Mineral and Bone Disorder
- Laboratory abnormalities (i.e., serum Ca2+ and Pi, PTH and/or vitamin D).
- Abnormalities in bone turnover, mineralization or volume.
- Vascular or other soft tissue calcification.
9. Recommendations
10. Concluding Remarks
Conflict of Interest
References
- World Population Prospects: The 2010 Revision, Volume I: Comprehensive Tables; United Nations Department of Economic and Social Affairs Population Division: New York, NY, USA, 2011.
- Peacock, M. Calcium metabolism in health and disease. Clin. J. Am. Soc. Nephrol. 2010, 5, S23–S30. [Google Scholar] [CrossRef]
- Koeppen, B.M.; Stanton, B.A. Renal Physiology, 5th ed; Elsevier: Philadelphia, PA, USA; p. 240.
- Hebert, S.C. Calcium and salinity sensing by the thick ascending limb: A journey from mammals to fish and back again. Kidney Int. Suppl. 2004, 91, S28–S33. [Google Scholar]
- Hebert, S.C.; Brown, E.M.; Harris, H.W. Role of the Ca(2+)-sensing receptor in divalent mineral ion homeostasis. J. Exp. Biol. 1997, 200, 295–302. [Google Scholar]
- Hou, J.; Rajagopal, M.; Yu, A.S. Claudins and the Kidney. Annu. Rev. Physiol. 2013, 75, 479–501. [Google Scholar] [CrossRef]
- Vennekens, R.; Hoenderop, J.G.; Prenen, J.; Stuiver, M.; Willems, P.H.; Droogmans, G.; Nilius, B.; Bindels, R.J. Permeation and gating properties of the novel epithelial Ca(2+) channel. J. Biol. Chem. 2000, 275, 3963–3969. [Google Scholar]
- Chang, Q.; Hoefs, S.; van der Kemp, A.W.; Topala, C.N.; Bindels, R.J.; Hoenderop, J.G. The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 2005, 310, 490–493. [Google Scholar] [CrossRef]
- Cha, S.K.; Ortega, B.; Kurosu, H.; Rosenblatt, K.P.; Kuro-o, M.; Huang, C.L. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc. Natl. Acad. Sci. USA 2008, 105, 9805–9810. [Google Scholar]
- Blankenship, K.A.; Williams, J.J.; Lawrence, M.S.; McLeish, K.R.; Dean, W.L.; Arthur, J.M. The calcium-sensing receptor regulates calcium absorption in MDCK cells by inhibition of PMCA. Am. J. Physiol. Renal Physiol. 2001, 280, F815–F822. [Google Scholar]
- Ba, J.; Brown, D.; Friedman, P.A. Calcium-sensing receptor regulation of PTH-inhibitable proximal tubule phosphate transport. Am. J. Physiol. Renal Physiol. 2003, 285, F1233–F1243. [Google Scholar]
- Hebert, S.C. Bartter syndrome. Curr. Opin. Nephrol. Hypertens. 2003, 12, 527–532. [Google Scholar] [CrossRef]
- Earm, J.H.; Christensen, B.M.; Frøkiaer, J.; Marples, D.; Han, J.S.; Knepper, M.A.; Nielsen, S. Decreased aquaporin-2 expression and apical plasma membrane delivery in kidney collecting ducts of polyuric hypercalcemic rats. J. Am. Soc. Nephrol. 1998, 9, 2181–2193. [Google Scholar]
- Procino, G.; Mastrofrancesco, L.; Tamma, G.; Lasorsa, D.R.; Ranieri, M.; Stringini, G.; Emma, F.; Svelto, M.; Valenti, G. Calcium-sensing receptor and aquaporin 2 interplay in hypercalciuria-associated renal concentrating defect in humans. An in vivo and in vitro study. PLoS One 2012, 7, e33145. [Google Scholar]
- Renkema, K.Y.; Velic, A.; Dijkman, H.B.; Verkaart, S.; van der Kemp, A.W.; Nowik, M.; Timmermans, K.; Doucet, A.; Wagner, C.A.; Bindels, R.J.; Hoenderop, J.G. The calcium-sensing receptor promotes urinary acidification to prevent nephrolithiasis. J. Am. Soc. Nephrol. 2009, 20, 1705–1713. [Google Scholar]
- Nyengaard, J.R.; Bendtsen, T.F. Glomerular number and size in relation to age, kidney weight, and body surface in normal man. Anat. Rec. 1992, 232, 194–201. [Google Scholar] [CrossRef]
- Davies, D.F.; Shock, N.W. Age changes in glomerular filtration rate, effective renal plasma flow, and tubular excretory capacity in adult males. J. Clin. Invest. 1950, 29, 496–507. [Google Scholar] [CrossRef]
- CDC. Prevalence of chronic kidney disease and associated risk factors—United States, 1999–2004. MMWR Morb. Mortal. Wkly. Rep. 2007, 56, 161–165.
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef]
- Shiraki-Iida, T.; Aizawa, H.; Matsumura, Y.; Sekine, S.; Iida, A.; Anazawa, H.; Nagai, R.; Kuro-o, M.; Nabeshima, Y. Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Lett. 1998, 424, 6–10. [Google Scholar] [CrossRef]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.C.; Moe, O.W.; Kuro-o, M. Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef]
- Alexander, R.T.; Woudenberg-Vrenken, T.E.; Buurman, J.; Dijkman, H.; van der Eerden, B.C.; van Leeuwen, J.P.; Bindels, R.J.; Hoenderop, J.G. Klotho prevents renal calcium loss. J. Am. Soc. Nephrol. 2009, 20, 2371–2379. [Google Scholar] [CrossRef]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; Kuro-o, M. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef]
- Maekawa, Y.; Ishikawa, K.; Yasuda, O.; Oguro, R.; Hanasaki, H.; Kida, I.; Takemura, Y.; Ohishi, M.; Katsuya, T.; Rakugi, H. Klotho suppresses TNF-alpha-induced expression of adhesion molecules in the endothelium and attenuates NF-kappaB activation. Endocrine 2009, 35, 341–346. [Google Scholar] [CrossRef]
- Sugiura, H.; Yoshida, T.; Shiohira, S.; Kohei, J.; Mitobe, M.; Kurosu, H.; Kuro-o, M.; Nitta, K.; Tsuchiya, K. Reduced Klotho expression level in kidney aggravates renal interstitial fibrosis. Am. J. Physiol. Renal Physiol. 2012, 302, F1252–F1264. [Google Scholar]
- Kim, H.R.; Nam, B.Y.; Kim, D.W.; Kang, M.W.; Han, J.H.; Lee, M.J.; Shin, D.H.; Doh, F.M.; Koo, H.M.; Ko, K.I.; et al. Circulating alpha-Klotho Levels in CKD and Relationship to Progression. Am. J. Kidney Dis. 2013, 61, 899–909. [Google Scholar] [CrossRef]
- Stubbs, J.R.; He, N.; Idiculla, A.; Gillihan, R.; Liu, S.; David, V.; Hong, Y.; Quarles, L.D. Longitudinal evaluation of FGF23 changes and mineral metabolism abnormalities in a mouse model of chronic kidney disease. J. Bone Miner. Res. 2012, 27, 38–46. [Google Scholar] [CrossRef]
- Tsuruoka, S.; Nishiki, K.; Ioka, T.; Ando, H.; Saito, Y.; Kurabayashi, M.; Nagai, R.; Fujimura, A. Defect in parathyroid-hormone-induced luminal calcium absorption in connecting tubules of Klotho mice. Nephrol. Dial. Transpl. 2006, 21, 2762–2767. [Google Scholar] [CrossRef]
- Fliser, D.; Kollerits, B.; Neyer, U.; Ankerst, D.P.; Lhotta, K.; Lingenhel, A.; Ritz, E.; Kronenberg, F.; Kuen, E.; Konig, P.; et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: The Mild to Moderate Kidney Disease (MMKD) Study. J. Am. Soc. Nephrol. 2007, 18, 2600–2608. [Google Scholar] [CrossRef]
- Spiegel, D.M.; Brady, K. Calcium balance in normal individuals and in patients with chronic kidney disease on low- and high-calcium diets. Kidney Int. 2012, 81, 1116–1122. [Google Scholar]
- Foley, R.N.; Collins, A.J.; Ishani, A.; Kalra, P.A. Calcium-phosphate levels and cardiovascular disease in community-dwelling adults: The Atherosclerosis Risk in Communities (ARIC) Study. Am. Heart J. 2008, 156, 556–563. [Google Scholar] [CrossRef]
- Bolland, M.J.; Grey, A.; Avenell, A.; Gamble, G.D.; Reid, I.R. Calcium supplements with or without vitamin D and risk of cardiovascular events: Reanalysis of the Women’s Health Initiative limited access dataset and meta-analysis. BMJ 2011, 342, d2040. [Google Scholar]
- Xiao, Q.; Murphy, R.A.; Houston, D.K.; Harris, T.B.; Chow, W.H.; Park, Y. Dietary and Supplemental Calcium Intake and Cardiovascular Disease Mortality: The National Institutes of Health-AARP Diet and Health Study. JAMA Intern. Med. 2013, 173, 639–646. [Google Scholar]
- Reid, I.R.; Schooler, B.A.; Hannan, S.F.; Ibbertson, H.K. The acute biochemical effects of four proprietary calcium preparations. Aust. N. Z. J. Med. 1986, 16, 193–197. [Google Scholar]
- Michaëlsson, K.; Melhus, H.; Lemming, E.W.; Wolk, A.; Byberg, L. Long term calcium intake and rates of all cause and cardiovascular mortality: Community based prospective longitudinal cohort study. BMJ 2013, 346, f228. [Google Scholar] [CrossRef]
- Ginde, A.A.; Scragg, R.; Schwartz, R.S.; Camargo, C.A., Jr. Prospective study of serum 25-hydroxyvitamin D level, cardiovascular disease mortality, and all-cause mortality in older U.S. adults. J. Am. Geriatr. Soc. 2009, 57, 1595–1603. [Google Scholar] [CrossRef]
- Cauley, J.A.; Thompson, D.E.; Ensrud, K.C.; Scott, J.C.; Black, D. Risk of mortality following clinical fractures. Osteoporos Int. 2000, 11, 556–561. [Google Scholar] [CrossRef]
- Aishah, A.B.; Foo, Y.N. A retrospective study of serum calcium levels in a hospital population in Malaysia. Med. J. Malaysia 1995, 50, 246–249. [Google Scholar]
- Hastbacka, J.; Pettila, V. Prevalence and predictive value of ionized hypocalcemia among critically ill patients. Acta Anaesthesiol. Scand. 2003, 47, 1264–1269. [Google Scholar] [CrossRef]
- Kolb, J.P.; Schilling, A.F.; Bischoff, J.; Novo de Oliveira, A.; Spiro, A.; Hoffmann, M.; Amling, M.; Rueger, J.M.; Lehmann, W. Calcium homeostasis influences radiological fracture healing in postmenopausal women. Arch. Orthop. Trauma Surg. 2012, 133, 187–192. [Google Scholar]
- Fatayerji, D.; Mawer, E.B.; Eastell, R. The role of insulin-like growth factor I in age-related changes in calcium homeostasis in men. J. Clin. Endocrinol. Metab. 2000, 85, 4657–4662. [Google Scholar] [CrossRef]
- Levin, A.; Bakris, G.L.; Molitch, M.; Smulders, M.; Tian, J.; Williams, L.A.; Andress, D.L. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: Results of the study to evaluate early kidney disease. Kidney Int. 2007, 71, 31–38. [Google Scholar] [CrossRef]
- Vassalotti, J.A.; Uribarri, J.; Chen, S.C.; Li, S.; Wang, C.; Collins, A.J.; Calvo, M.S.; Whaley-Connell, A.T.; McCullough, P.A.; Norris, K.C. Trends in mineral metabolism: Kidney Early Evaluation Program (KEEP) and the National Health and Nutrition Examination Survey (NHANES) 1999–2004. Am. J. Kidney Dis. 2008, 51, S56–S68. [Google Scholar] [CrossRef]
- Markowitz, G.S.; Perazella, M.A. Acute phosphate nephropathy. Kidney Int. 2009, 76, 1027–1034. [Google Scholar] [CrossRef]
- Gutierrez, O.; Isakova, T.; Rhee, E.; Shah, A.; Holmes, J.; Collerone, G.; Juppner, H.; Wolf, M. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 2205–2215. [Google Scholar] [CrossRef]
- Moranne, O.; Froissart, M.; Rossert, J.; Gauci, C.; Boffa, J.J.; Haymann, J.P.; M’Rad, M.B.; Jacquot, C.; Houillier, P.; Stengel, B.; Fouqueray, B. Timing of onset of CKD-related metabolic complications. J. Am. Soc. Nephrol. 2009, 20, 164–171. [Google Scholar] [CrossRef]
- Cannata-Andia, J.B.; Naves-Diaz, M. Phosphorus and survival: Key questions that need answers. J. Am. Soc. Nephrol. 2009, 20, 234–236. [Google Scholar] [CrossRef]
- Mathew, S.; Tustison, K.S.; Sugatani, T.; Chaudhary, L.R.; Rifas, L.; Hruska, K.A. The mechanism of phosphorus as a cardiovascular risk factor in CKD. J. Am. Soc. Nephrol. 2008, 19, 1092–1105. [Google Scholar] [CrossRef]
- Kendrick, J.; Chonchol, M. The role of phosphorus in the development and progression of vascular calcification. Am. J. Kidney Dis. 2011, 58, 826–834. [Google Scholar] [CrossRef]
- Ix, J.H.; De Boer, I.H.; Peralta, C.A.; Adeney, K.L.; Duprez, D.A.; Jenny, N.S.; Siscovick, D.S.; Kestenbaum, B.R. Serum phosphorus concentrations and arterial stiffness among individuals with normal kidney function to moderate kidney disease in MESA. Clin. J. Am. Soc. Nephrol. 2009, 4, 609–615. [Google Scholar] [CrossRef]
- Dhingra, R.; Sullivan, L.M.; Fox, C.S.; Wang, T.J.; D’Agostino, R.B., Sr.; Gaziano, J.M.; Vasan, R.S. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch. Intern. Med. 2007, 167, 879–885. [Google Scholar] [CrossRef]
- Kestenbaum, B.; Sampson, J.N.; Rudser, K.D.; Patterson, D.J.; Seliger, S.L.; Young, B.; Sherrard, D.J.; Andress, D.L. Serum phosphate levels and mortality risk among people with chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 520–528. [Google Scholar] [CrossRef]
- Ohnishi, M.; Razzaque, M.S. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J. 2010, 24, 3562–3571. [Google Scholar] [CrossRef]
- Robinson, J.K. Sun exposure, sun protection, and vitamin D. JAMA 2005, 294, 1541–1543. [Google Scholar] [CrossRef]
- Holick, M.F. The vitamin D epidemic and its health consequences. J. Nutr. 2005, 135, 2739S–2748S. [Google Scholar]
- Bosworth, C.R.; Levin, G.; Robinson-Cohen, C.; Hoofnagle, A.N.; Ruzinski, J.; Young, B.; Schwartz, S.M.; Himmelfarb, J.; Kestenbaum, B.; de Boer, I.H. The serum 24,25-dihydroxyvitamin D concentration, a marker of vitamin D catabolism, is reduced in chronic kidney disease. Kidney Int. 2012, 82, 693–700. [Google Scholar] [CrossRef]
- Carlberg, C.; Bendik, I.; Wyss, A.; Meier, E.; Sturzenbecker, L.J.; Grippo, J.F.; Hunziker, W. Two nuclear signalling pathways for vitamin D. Nature 1993, 361, 657–660. [Google Scholar] [CrossRef]
- Haussler, M.R.; Jurutka, P.W.; Mizwicki, M.; Norman, A.W. Vitamin D receptor (VDR)-mediated actions of 1alpha,25(OH)(2)vitamin D(3): Genomic and non-genomic mechanisms. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 543–559. [Google Scholar] [CrossRef]
- Deb, D.K.; Sun, T.; Wong, K.E.; Zhang, Z.; Ning, G.; Zhang, Y.; Kong, J.; Shi, H.; Chang, A.; Li, Y.C. Combined vitamin D analog and AT1 receptor antagonist synergistically block the development of kidney disease in a model of type 2 diabetes. Kidney Int. 2010, 77, 1000–1009. [Google Scholar] [CrossRef]
- Vaidya, A.; Forman, J.P.; Williams, J.S. Vitamin D and the vascular sensitivity to angiotensin II in obese Caucasians with hypertension. J. Hum. Hypertens. 2011, 25, 672–678. [Google Scholar] [CrossRef]
- Ohara, I.; Tanimoto, M.; Gohda, T.; Yamazaki, T.; Hagiwara, S.; Murakoshi, M.; Aoki, T.; Toyoda, H.; Ishikawa, Y.; Funabiki, K.; et al. Effect of combination therapy with angiotensin receptor blocker and 1,25-dihydroxyvitamin D(3) in type 2 diabetic nephropathy in KK-A(y)/Ta mice. Nephron Exp. Nephrol. 2011, 117, e124–e132. [Google Scholar] [CrossRef]
- Vaidya, A.; Sun, B.; Larson, C.; Forman, J.P.; Williams, J.S. Vitamin D3 therapy corrects the tissue sensitivity to angiotensin II akin to the action of a converting enzyme inhibitor in obese hypertensives: An interventional study. J. Clin. Endocrinol. Metab. 2012, 97, 2456–2465. [Google Scholar] [CrossRef]
- Martini, L.A.; Wood, R.J. Milk intake and the risk of type 2 diabetes mellitus, hypertension and prostate cancer. Arq. Bras. Endocrinol. Metabol. 2009, 53, 688–694. [Google Scholar] [CrossRef]
- Gorham, E.D.; Garland, C.F.; Burgi, A.A.; Mohr, S.B.; Zeng, K.; Hofflich, H.; Kim, J.J.; Ricordi, C. Lower prediagnostic serum 25-hydroxyvitamin D concentration is associated with higher risk of insulin-requiring diabetes: A nested case-control study. Diabetologia 2012, 55, 3224–3227. [Google Scholar] [CrossRef]
- van der Rhee, H.; Coebergh, J.W.; de Vries, E. Is prevention of cancer by sun exposure more than just the effect of vitamin D? A systematic review of epidemiological studies. Eur. J. Cancer 2012, 49, 1422–1436. [Google Scholar] [CrossRef]
- Bergman, P.; Norlin, A.C.; Hansen, S.; Rekha, R.S.; Agerberth, B.; Bjorkhem-Bergman, L.; Ekstrom, L.; Lindh, J.D.; Andersson, J. Vitamin D3 supplementation in patients with frequent respiratory tract infections: A randomised and double-blind intervention study. BMJ Open 2012, 2, e001663. [Google Scholar] [CrossRef]
- Balion, C.; Griffith, L.E.; Strifler, L.; Henderson, M.; Patterson, C.; Heckman, G.; Llewellyn, D.J.; Raina, P. Vitamin D, cognition, and dementia: A systematic review and meta-analysis. Neurology 2012, 79, 1397–1405. [Google Scholar] [CrossRef]
- Mizwicki, M.T.; Menegaz, D.; Zhang, J.; Barrientos-Duran, A.; Tse, S.; Cashman, J.R.; Griffin, P.R.; Fiala, M. Genomic and nongenomic signaling induced by 1alpha,25(OH)2-vitamin D3 promotes the recovery of amyloid-beta phagocytosis by Alzheimer’s disease macrophages. J. Alzheimers Dis. 2012, 29, 51–62. [Google Scholar]
- Autier, P.; Gandini, S. Vitamin D supplementation and total mortality: A meta-analysis of randomized controlled trials. Arch. Intern. Med. 2007, 167, 1730–1737. [Google Scholar] [CrossRef]
- de Boer, I.H.; Levin, G.; Robinson-Cohen, C.; Biggs, M.L.; Hoofnagle, A.N.; Siscovick, D.S.; Kestenbaum, B. Serum 25-hydroxyvitamin D concentration and risk for major clinical disease events in a community-based population of older adults: A cohort study. Ann. Intern. Med. 2012, 156, 627–634. [Google Scholar] [CrossRef]
- Teng, M.; Wolf, M.; Lowrie, E.; Ofsthun, N.; Lazarus, J.M.; Thadhani, R. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N. Engl. J. Med. 2003, 349, 446–456. [Google Scholar] [CrossRef]
- Bischoff-Ferrari, H.A.; Shao, A.; Dawson-Hughes, B.; Hathcock, J.; Giovannucci, E.; Willett, W.C. Benefit-risk assessment of vitamin D supplementation. Osteoporos. Int. 2010, 21, 1121–1132. [Google Scholar] [CrossRef]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef]
- Wang, L.; Song, Y.; Manson, J.E.; Pilz, S.; Marz, W.; Michaelsson, K.; Lundqvist, A.; Jassal, S.K.; Barrett-Connor, E.; Zhang, C.; et al. Circulating 25-hydroxy-vitamin D and risk of cardiovascular disease: A meta-analysis of prospective studies. Circ. Cardiovasc. Qual. Outcomes 2012, 5, 819–829. [Google Scholar] [CrossRef]
- Holick, M.F. Vitamin D status: Measurement, interpretation, and clinical application. Ann. Epidemiol. 2009, 19, 73–78. [Google Scholar] [CrossRef]
- Forrest, K.Y.; Stuhldreher, W.L. Prevalence and correlates of vitamin D deficiency in US adults. Nutr. Res. 2011, 31, 48–54. [Google Scholar] [CrossRef]
- Janssen, H.C.; Emmelot-Vonk, M.H.; Verhaar, H.J.; van der Schouw, Y.T. Determinants of vitamin D status in healthy men and women aged 40–80 years. Maturitas 2013, 74, 79–83. [Google Scholar] [CrossRef]
- MacLaughlin, J.; Holick, M.F. Aging decreases the capacity of human skin to produce vitamin D3. J. Clin. Invest. 1985, 76, 1536–1538. [Google Scholar] [CrossRef]
- Ebeling, P.R.; Sandgren, M.E.; DiMagno, E.P.; Lane, A.W.; DeLuca, H.F.; Riggs, B.L. Evidence of an age-related decrease in intestinal responsiveness to vitamin D: Relationship between serum 1,25-dihydroxyvitamin D3 and intestinal vitamin D receptor concentrations in normal women. J. Clin. Endocrinol. Metab. 1992, 75, 176–182. [Google Scholar] [CrossRef]
- Tsiaras, W.G.; Weinstock, M.A. Factors influencing vitamin D status. Acta Derm. Venereol. 2011, 91, 115–124. [Google Scholar]
- Pilz, S.; Iodice, S.; Zittermann, A.; Grant, W.B.; Gandini, S. Vitamin D status and mortality risk in CKD: A meta-analysis of prospective studies. Am. J. Kidney Dis. 2011, 58, 374–382. [Google Scholar]
- Bischoff-Ferrari, H.A.; Willett, W.C.; Orav, E.J.; Lips, P.; Meunier, P.J.; Lyons, R.A.; Flicker, L.; Wark, J.; Jackson, R.D.; Cauley, J.A.; et al. A pooled analysis of vitamin D dose requirements for fracture prevention. N. Engl. J. Med. 2012, 367, 40–49. [Google Scholar] [CrossRef]
- de Zeeuw, D.; Agarwal, R.; Amdahl, M.; Audhya, P.; Coyne, D.; Garimella, T.; Parving, H.H.; Pritchett, Y.; Remuzzi, G.; Ritz, E.; Andress, D. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): A randomised controlled trial. Lancet 2010, 376, 1543–1551. [Google Scholar] [CrossRef]
- Verhave, G.; Siegert, C.E. Role of vitamin D in cardiovascular disease. Neth. J. Med. 2010, 68, 113–118. [Google Scholar]
- Rejnmark, L.; Avenell, A.; Masud, T.; Anderson, F.; Meyer, H.E.; Sanders, K.M.; Salovaara, K.; Cooper, C.; Smith, H.E.; Jacobs, E.T.; et al. Vitamin D with calcium reduces mortality: Patient level pooled analysis of 70,528 patients from eight major vitamin D trials. J. Clin. Endocrinol. Metab. 2012, 97, 2670–2681. [Google Scholar] [CrossRef]
- Shoben, A.B.; Rudser, K.D.; de Boer, I.H.; Young, B.; Kestenbaum, B. Association of oral calcitriol with improved survival in nondialyzed CKD. J. Am. Soc. Nephrol. 2008, 19, 1613–1619. [Google Scholar] [CrossRef]
- Teng, M.; Wolf, M.; Ofsthun, M.N.; Lazarus, J.M.; Hernan, M.A.; Camargo, C.A., Jr.; Thadhani, R. Activated injectable vitamin D and hemodialysis survival: A historical cohort study. J. Am. Soc. Nephrol. 2005, 16, 1115–1125. [Google Scholar] [CrossRef]
- Brondum-Jacobsen, P.; Benn, M.; Jensen, G.B.; Nordestgaard, B.G. 25-hydroxyvitamin D levels and risk of ischemic heart disease, myocardial infarction, and early death: Population-based study and meta-analyses of 18 and 17 studies. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2794–2802. [Google Scholar] [CrossRef]
- Lau, W.L.; Leaf, E.M.; Hu, M.C.; Takeno, M.M.; Kuro-o, M.; Moe, O.W.; Giachelli, C.M. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012, 82, 1261–1270. [Google Scholar] [CrossRef]
- Segre, G.V.; D’Amour, P.; Hultman, A.; Potts, J.T., Jr. Effects of hepatectomy, nephrectomy, and nephrectomy/uremia on the metabolism of parathyroid hormone in the rat. J. Clin. Invest. 1981, 67, 439–448. [Google Scholar] [CrossRef]
- D’Amour, P.; Lazure, C. Metabolism of radioiodinated carboxy-terminal fragments of bovine parathyroid hormone in normal and anephric rats. Endocrinology 1985, 117, 127–134. [Google Scholar] [CrossRef]
- Brown, E.M.; Gamba, G.; Riccardi, D.; Lombardi, M.; Butters, R.; Kifor, O.; Sun, A.; Hediger, M.A.; Lytton, J.; Hebert, S.C. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature 1993, 366, 575–580. [Google Scholar] [CrossRef]
- Egbuna, O.I.; Brown, E.M. Hypercalcaemic and hypocalcaemic conditions due to calcium-sensing receptor mutations. Best Pract. Res. Clin. Rheumatol. 2008, 22, 129–148. [Google Scholar] [CrossRef]
- Ritter, C.S.; Brown, A.J. Direct suppression of Pth gene expression by the vitamin D prohormones doxercalciferol and calcidiol requires the vitamin D receptor. J. Mol. Endocrinol. 2011, 46, 63–66. [Google Scholar]
- Lavi-Moshayoff, V.; Wasserman, G.; Meir, T.; Silver, J.; Naveh-Many, T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: A bone parathyroid feedback loop. Am. J. Physiol. Renal Physiol. 2010, 299, F882–F889. [Google Scholar] [CrossRef]
- Ganong, W.F.; Barrett, K.E. Ganong’s Review of Medical Physiology, 24th; Barrett, K.E., Boitano, S., Barman, S.M., Brooks, H.L., Eds.; McGraw-Hill: New York, NY, USA; p. 752.
- Ferrari, S.L.; Bonjour, J.P.; Rizzoli, R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J. Clin. Endocrinol. Metab. 2005, 90, 1519–1524. [Google Scholar] [CrossRef]
- Westerberg, P.A.; Ljunggren, O.; Larsson, T.E.; Wadstrom, J.; Linde, T. Fibroblast growth factor-23 and mineral metabolism after unilateral nephrectomy. Nephrol. Dial. Transpl. 2010, 25, 4068–4071. [Google Scholar] [CrossRef]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J. Bone Miner. Res. 2004, 19, 429–435. [Google Scholar]
- Heath, H.; Hodgson, S.F.; Kennedy, M.A. Primary Hyperparathyroidism. N. Engl. J. Med. 1980, 302, 189–193. [Google Scholar] [CrossRef]
- Block, G.A.; Martin, K.J.; de Francisco, A.L.; Turner, S.A.; Avram, M.M.; Suranyi, M.G.; Hercz, G.; Cunningham, J.; Abu-Alfa, A.K.; Messa, P.; et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N. Engl. J. Med. 2004, 350, 1516–1525. [Google Scholar] [CrossRef]
- Urena, P.; Bernard-Poenaru, O.; Cohen-Solal, M.; de Vernejoul, M.C. Plasma bone-specific alkaline phosphatase changes in hemodialysis patients treated by alfacalcidol. Clin. Nephrol. 2002, 57, 261–273. [Google Scholar]
- Goodman, W.G.; Ramirez, J.A.; Belin, T.R.; Chon, Y.; Gales, B.; Segre, G.V.; Salusky, I.B. Development of adynamic bone in patients with secondary hyperparathyroidism after intermittent calcitriol therapy. Kidney Int. 1994, 46, 1160–1166. [Google Scholar] [CrossRef]
- Yamashita, T.; Yoshioka, M.; Itoh, N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem. Biophys. Res. Commun. 2000, 277, 494–498. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, J.; Tang, W.; Jiang, X.; Rowe, D.W.; Quarles, L.D. Pathogenic role of Fgf23 in Hyp mice. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E38–E49. [Google Scholar] [CrossRef]
- Kumar, R. Phosphatonin—a new phosphaturetic hormone? (lessons from tumour-induced osteomalacia and X-linked hypophosphataemia). Nephrol. Dial. Transpl. 1997, 12, 11–13. [Google Scholar] [CrossRef]
- Canalejo, R.; Canalejo, A.; Martinez-Moreno, J.M.; Rodriguez-Ortiz, M.E.; Estepa, J.C.; Mendoza, F.J.; Munoz-Castaneda, J.R.; Shalhoub, V.; Almaden, Y.; Rodriguez, M. FGF23 fails to inhibit uremic parathyroid glands. J. Am. Soc. Nephrol. 2010, 21, 1125–1135. [Google Scholar] [CrossRef]
- Stubbs, J.R.; Egwuonwu, S. Is fibroblast growth factor 23 a harbinger of mortality in CKD? Pediatr. Nephrol. 2012, 27, 697–703. [Google Scholar] [CrossRef]
- Mak, M.P.; da Costa e Silva, V.T.; Martin, R.M.; Lerario, A.M.; Yu, L.; Hoff, P.M.; de Castro, G., Jr. Advanced prostate cancer as a cause of oncogenic osteomalacia: An underdiagnosed condition. Support Care Cancer 2012, 20, 2195–2197. [Google Scholar] [CrossRef]
- Folpe, A.L.; Fanburg-Smith, J.C.; Billings, S.D.; Bisceglia, M.; Bertoni, F.; Cho, J.Y.; Econs, M.J.; Inwards, C.Y.; Jan de Beur, S.M.; Mentzel, T.; et al. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: An analysis of 32 cases and a comprehensive review of the literature. Am. J. Surg. Pathol. 2004, 28, 1–30. [Google Scholar] [CrossRef]
- Bai, X.Y.; Miao, D.; Goltzman, D.; Karaplis, A.C. The autosomal dominant hypophosphatemic rickets R176Q mutation in fibroblast growth factor 23 resists proteolytic cleavage and enhances in vivo biological potency. J. Biol. Chem. 2003, 278, 9843–9849. [Google Scholar]
- Benet-Pages, A.; Orlik, P.; Strom, T.M.; Lorenz-Depiereux, B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum. Mol. Genet. 2005, 14, 385–390. [Google Scholar]
- Gutierrez, O.M.; Januzzi, J.L.; Isakova, T.; Laliberte, K.; Smith, K.; Collerone, G.; Sarwar, A.; Hoffmann, U.; Coglianese, E.; Christenson, R.; et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 2009, 119, 2545–2552. [Google Scholar] [CrossRef]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutierrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Invest. 2011, 121, 4393–4408. [Google Scholar] [CrossRef]
- Gutierrez, O.M.; Mannstadt, M.; Isakova, T.; Rauh-Hain, J.A.; Tamez, H.; Shah, A.; Smith, K.; Lee, H.; Thadhani, R.; Juppner, H.; Wolf, M. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N. Engl. J. Med. 2008, 359, 584–592. [Google Scholar] [CrossRef]
- Jean, G.; Terrat, J.C.; Vanel, T.; Hurot, J.M.; Lorriaux, C.; Mayor, B.; Chazot, C. High levels of serum fibroblast growth factor (FGF)-23 are associated with increased mortality in long haemodialysis patients. Nephrol. Dial. Transpl. 2009, 24, 2792–2796. [Google Scholar] [CrossRef]
- Hasegawa, H.; Nagano, N.; Urakawa, I.; Yamazaki, Y.; Iijima, K.; Fujita, T.; Yamashita, T.; Fukumoto, S.; Shimada, T. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int. 2010, 78, 975–980. [Google Scholar] [CrossRef]
- Shalhoub, V.; Shatzen, E.M.; Ward, S.C.; Davis, J.; Stevens, J.; Bi, V.; Renshaw, L.; Hawkins, N.; Wang, W.; Chen, C.; et al. FGF23 neutralization improves chronic kidney disease—associated hyperparathyroidism yet increases mortality. J. Clin. Invest. 2012, 122, 2543–2553. [Google Scholar] [CrossRef]
- Brown, A.J.; Zhong, M.; Finch, J.; Ritter, C.; McCracken, R.; Morrissey, J.; Slatopolsky, E. Rat calcium-sensing receptor is regulated by vitamin D but not by calcium. Am. J. Physiol. 1996, 270, F454–F460. [Google Scholar]
- Dvorak, M.M.; Chen, T.H.; Orwoll, B.; Garvey, C.; Chang, W.; Bikle, D.D.; Shoback, D.M. Constitutive activity of the osteoblast Ca2+-sensing receptor promotes loss of cancellous bone. Endocrinology 2007, 148, 3156–3163. [Google Scholar] [CrossRef]
- Pollak, M.R.; Brown, E.M.; Estep, H.L.; McLaine, P.N.; Kifor, O.; Park, J.; Hebert, S.C.; Seidman, C.E.; Seidman, J.G. Autosomal dominant hypocalcaemia caused by a Ca(2+)-sensing receptor gene mutation. Nat. Genet. 1994, 8, 303–307. [Google Scholar] [CrossRef]
- Theman, T.A.; Collins, M.T.; Dempster, D.W.; Zhou, H.; Reynolds, J.C.; Brahim, J.S.; Roschger, P.; Klaushofer, K.; Winer, K.K. PTH(1-34) replacement therapy in a child with hypoparathyroidism caused by a sporadic calcium receptor mutation. J. Bone Miner. Res. 2009, 24, 964–973. [Google Scholar] [CrossRef]
- Toke, J.; Czirjak, G.; Patocs, A.; Enyedi, B.; Gergics, P.; Csakvary, V.; Enyedi, P.; Toth, M. Neonatal severe hyperparathyroidism associated with a novel de novo heterozygous R551K inactivating mutation and a heterozygous A986S polymorphism of the calcium-sensing receptor gene. Clin. Endocrinol. (Oxf.) 2007, 67, 385–392. [Google Scholar] [CrossRef]
- Pollak, M.R.; Brown, E.M.; Chou, Y.H.; Hebert, S.C.; Marx, S.J.; Steinmann, B.; Levi, T.; Seidman, C.E.; Seidman, J.G. Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 1993, 75, 1297–1303. [Google Scholar] [CrossRef]
- Chang, W.; Rodriguez, L.; Chen, T.H.; Tu, C.; Shoback, D. Extracellular Ca2+-sensing in cartilage. J. Musculoskelet. Neuronal Interact. 2004, 4, 410–411. [Google Scholar]
- Moe, S.; Drueke, T.; Cunningham, J.; Goodman, W.; Martin, K.; Olgaard, K.; Ott, S.; Sprague, S.; Lameire, N.; Eknoyan, G. Definition, evaluation, and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006, 69, 1945–1953. [Google Scholar] [CrossRef]
- Joy, M.S.; Karagiannis, P.C.; Peyerl, F.W. Outcomes of secondary hyperparathyroidism in chronic kidney disease and the direct costs of treatment. J. Manag. Care Pharm. 2007, 13, 397–411. [Google Scholar]
- Suda, T.; Takahashi, N.; Udagawa, N.; Jimi, E.; Gillespie, M.T.; Martin, T.J. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 1999, 20, 345–357. [Google Scholar]
- Itoh, K.; Udagawa, N.; Matsuzaki, K.; Takami, M.; Amano, H.; Shinki, T.; Ueno, Y.; Takahashi, N.; Suda, T. Importance of membrane- or matrix-associated forms of M-CSF and RANKL/ODF in osteoclastogenesis supported by SaOS-4/3 cells expressing recombinant PTH/PTHrP receptors. J. Bone Miner. Res. 2000, 15, 1766–1775. [Google Scholar] [CrossRef]
- Kanzawa, M.; Sugimoto, T.; Kanatani, M.; Chihara, K. Involvement of osteoprotegerin/osteoclastogenesis inhibitory factor in the stimulation of osteoclast formation by parathyroid hormone in mouse bone cells. Eur. J. Endocrinol. 2000, 142, 661–664. [Google Scholar] [CrossRef]
- Jara, A.; Bover, J.; Felsenfeld, A.J. Development of secondary hyperparathyroidism and bone disease in diabetic rats with renal failure. Kidney Int. 1995, 47, 1746–1751. [Google Scholar]
- Couttenye, M.M.; D’Haese, P.C.; Deng, J.T.; van Hoof, V.O.; Verpooten, G.A.; de Broe, M.E. High prevalence of adynamic bone disease diagnosed by biochemical markers in a wide sample of the European CAPD population. Nephrol. Dial. Transpl. 1997, 12, 2144–2150. [Google Scholar] [CrossRef]
- Cannata-Andia, J.B.; Rodriguez Garcia, M.; Gomez Alonso, C. Osteoporosis and adynamic bone in chronic kidney disease. J. Nephrol. 2013, 26, 73–80. [Google Scholar] [CrossRef]
- Block, G.A.; Port, F.K. Re-evaluation of risks associated with hyperphosphatemia and hyperparathyroidism in dialysis patients: Recommendations for a change in management. Am. J. Kidney Dis. 2000, 35, 1226–1237. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int. Suppl. 2009, 113, S1–S130.
- Kazama, J.J.; Koda, R.; Yamamoto, S.; Narita, I.; Gejyo, F.; Tokumoto, A. Cancellous bone volume is an indicator for trabecular bone connectivity in dialysis patients. Clin. J. Am. Soc. Nephrol. 2010, 5, 292–298. [Google Scholar] [CrossRef]
- Malluche, H.H.; Monier-Faugere, M.C. Renal osteodystrophy: What’s in a name? Presentation of a clinically useful new model to interpret bone histologic findings. Clin. Nephrol. 2006, 65, 235–242. [Google Scholar]
- Mac Way, F.; Lessard, M.; Lafage-Proust, M.H. Pathophysiology of chronic kidney disease-mineral and bone disorder. Joint Bone Spine 2012, 79, 544–549. [Google Scholar] [CrossRef]
- Reeve, J.; Arlot, M.E.; Chavassieux, P.M.; Edouard, C.; Green, J.R.; Hesp, R.; Tellez, M.; Meunier, P.J. The assessment of bone formation and bone resorption in osteoporosis: A comparison between tetracycline-based iliac histomorphometry and whole body 85Sr kinetics. J. Bone Miner. Res. 1987, 2, 479–489. [Google Scholar]
- Kiel, D.P.; Kauppila, L.I.; Cupples, L.A.; Hannan, M.T.; O’Donnell, C.J.; Wilson, P.W. Bone loss and the progression of abdominal aortic calcification over a 25 year period: The Framingham Heart Study. Calcif. Tissue Int. 2001, 68, 271–276. [Google Scholar] [CrossRef]
- Naves, M.; Rodriguez-Garcia, M.; Diaz-Lopez, J.B.; Gomez-Alonso, C.; Cannata-Andia, J.B. Progression of vascular calcifications is associated with greater bone loss and increased bone fractures. Osteoporos. Int. 2008, 19, 1161–1166. [Google Scholar] [CrossRef]
- Rodriguez Garcia, M.; Naves Diaz, M.; Cannata Andia, J.B. Bone metabolism, vascular calcifications and mortality: Associations beyond mere coincidence. J. Nephrol. 2005, 18, 458–463. [Google Scholar]
- Sigrist, M.; Bungay, P.; Taal, M.W.; McIntyre, C.W. Vascular calcification and cardiovascular function in chronic kidney disease. Nephrol. Dial. Transpl. 2006, 21, 707–714. [Google Scholar] [CrossRef]
- Merjanian, R.; Budoff, M.; Adler, S.; Berman, N.; Mehrotra, R. Coronary artery, aortic wall, and valvular calcification in nondialyzed individuals with type 2 diabetes and renal disease. Kidney Int. 2003, 64, 263–271. [Google Scholar] [CrossRef]
- Goodman, W.G.; Goldin, J.; Kuizon, B.D.; Yoon, C.; Gales, B.; Sider, D.; Wang, Y.; Chung, J.; Emerick, A.; Greaser, L.; et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N. Engl. J. Med. 2000, 342, 1478–1483. [Google Scholar] [CrossRef]
- Stenvinkel, P. Chronic kidney disease: A public health priority and harbinger of premature cardiovascular disease. J. Intern. Med. 2010, 268, 456–467. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Larsson, T.E. Chronic Kidney Disease: A Clinical Model of Premature Aging. Am. J. Kidney Dis. 2013. in press. Available online: http://www.sciencedirect.com/science/article/pii/S0272638612015922 (accessed on 15 May 2013).
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: A potential mechanism for accelerated vascular calcification in ESRD. J. Am. Soc. Nephrol. 2004, 15, 2857–2867. [Google Scholar] [CrossRef]
- Lim, K.; Lu, T.S.; Molostvov, G.; Lee, C.; Lam, F.T.; Zehnder, D.; Hsiao, L.L. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 2012, 125, 2243–2255. [Google Scholar] [CrossRef]
- Sigrist, M.K.; Taal, M.W.; Bungay, P.; McIntyre, C.W. Progressive vascular calcification over 2 years is associated with arterial stiffening and increased mortality in patients with stages 4 and 5 chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2007, 2, 1241–1248. [Google Scholar] [CrossRef]
- Floege, J.; Kim, J.; Ireland, E.; Chazot, C.; Drueke, T.; de Francisco, A.; Kronenberg, F.; Marcelli, D.; Passlick-Deetjen, J.; Schernthaner, G.; et al. Serum iPTH, calcium and phosphate, and the risk of mortality in a European haemodialysis population. Nephrol. Dial. Transpl. 2011, 26, 1948–1955. [Google Scholar] [CrossRef]
- Block, G.A.; Wheeler, D.C.; Persky, M.S.; Kestenbaum, B.; Ketteler, M.; Spiegel, D.M.; Allison, M.A.; Asplin, J.; Smits, G.; Hoofnagle, A.N.; et al. Effects of phosphate binders in moderate CKD. J. Am. Soc. Nephrol. 2012, 23, 1407–1415. [Google Scholar] [CrossRef]
- Chue, C.D.; Townend, J.N.; Moody, W.E.; Zehnder, D.; Wall, N.A.; Harper, L.; Edwards, N.C.; Steeds, R.P.; Ferro, C.J. Cardiovascular Effects of Sevelamer in Stage 3 CKD. J. Am. Soc. Nephrol. 2013, 24, 842852. [Google Scholar]
- Andress, D.L.; Coyne, D.W.; Kalantar-Zadeh, K.; Molitch, M.E.; Zangeneh, F.; Sprague, S.M. Management of secondary hyperparathyroidism in stages 3 and 4 chronic kidney disease. Endocr. Pract. 2008, 14, 18–27. [Google Scholar] [CrossRef]
- Cunningham, J.; Danese, M.; Olson, K.; Klassen, P.; Chertow, G.M. Effects of the calcimimetic cinacalcet HCl on cardiovascular disease, fracture, and health-related quality of life in secondary hyperparathyroidism. Kidney Int. 2005, 68, 1793–1800. [Google Scholar] [CrossRef]
- Chertow, G.M.; Block, G.A.; Correa-Rotter, R.; Drueke, T.B.; Floege, J.; Goodman, W.G.; Herzog, C.A.; Kubo, Y.; London, G.M.; Mahaffey, K.W.; et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N. Engl. J. Med. 2012, 367, 2482–2494. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tejwani, V.; Qian, Q. Calcium Regulation and Bone Mineral Metabolism in Elderly Patients with Chronic Kidney Disease. Nutrients 2013, 5, 1913-1936. https://doi.org/10.3390/nu5061913
Tejwani V, Qian Q. Calcium Regulation and Bone Mineral Metabolism in Elderly Patients with Chronic Kidney Disease. Nutrients. 2013; 5(6):1913-1936. https://doi.org/10.3390/nu5061913
Chicago/Turabian StyleTejwani, Vickram, and Qi Qian. 2013. "Calcium Regulation and Bone Mineral Metabolism in Elderly Patients with Chronic Kidney Disease" Nutrients 5, no. 6: 1913-1936. https://doi.org/10.3390/nu5061913
APA StyleTejwani, V., & Qian, Q. (2013). Calcium Regulation and Bone Mineral Metabolism in Elderly Patients with Chronic Kidney Disease. Nutrients, 5(6), 1913-1936. https://doi.org/10.3390/nu5061913