Is Selenium a Potential Treatment for Cancer Metastasis?

Abstract
:Abbreviations
| Se | selenium |
| Sec | selenocysteine |
| SeMet | selenomethionine |
| MSC | Se-methyl-selenocysteine |
| SECIS | selenocysteine-insertion sequence |
| MeCN | methylselenocyanate |
| SELECT | the Selenium and Vitamin E Cancer Prevention Trial |
| GPX | glutathione peroxidase |
| TXR | thioredoxin reductase |
| SBP | selenium-binding protein |
| MSA | methylseleninic acid |
| VEGF | vascular endothelial growth factor |
| HGF | hepatocyte growth factor |
| IL-1 | interlukin-1 |
| IL-8 | interlukin-8 |
| SDF-1 | stromal cell-derived factor 1 |
| GRO-α | growth-regulated peptide-alpha/growth-regulated oncogene-1 |
| OPN | osteopontin |
| FGF | fibroblast growth factors |
| MMP | matrix metalloproteinase |
| HUVEC | human umbilical vein endothelial cells |
| HIF-1α | hypoxia-induced factor 1alpha |
| SCID mice | severe combined immunodeficient mice |
| Sep15 | the 15-KDa selenoprotein |
| uPA | urokinase-type plasminogen activator |
| ECM | extracellular matrix |
| TIMP1 | tissue inhibitor of metalloproteinase 1 |
| TIMP2 | tissue inhibitor of metalloproteinase 2 |
| PMA | 12-o-tetradecanoylphorbol-13-acetate |
| MT1-MMP | membrane-type 1 matrix metalloproteinase |
| IL-18 | interlukin-18 |
| PAI-1 | plasminogen activator inhibitor-1 |
| TNFα | tumor necrosis factor alpha |
| IGF II | insulin-like growth factor II |
| COX-2 | cyclooxygenase-2 |
| iNOS | inducible nitric oxide synthase |
1. Introduction
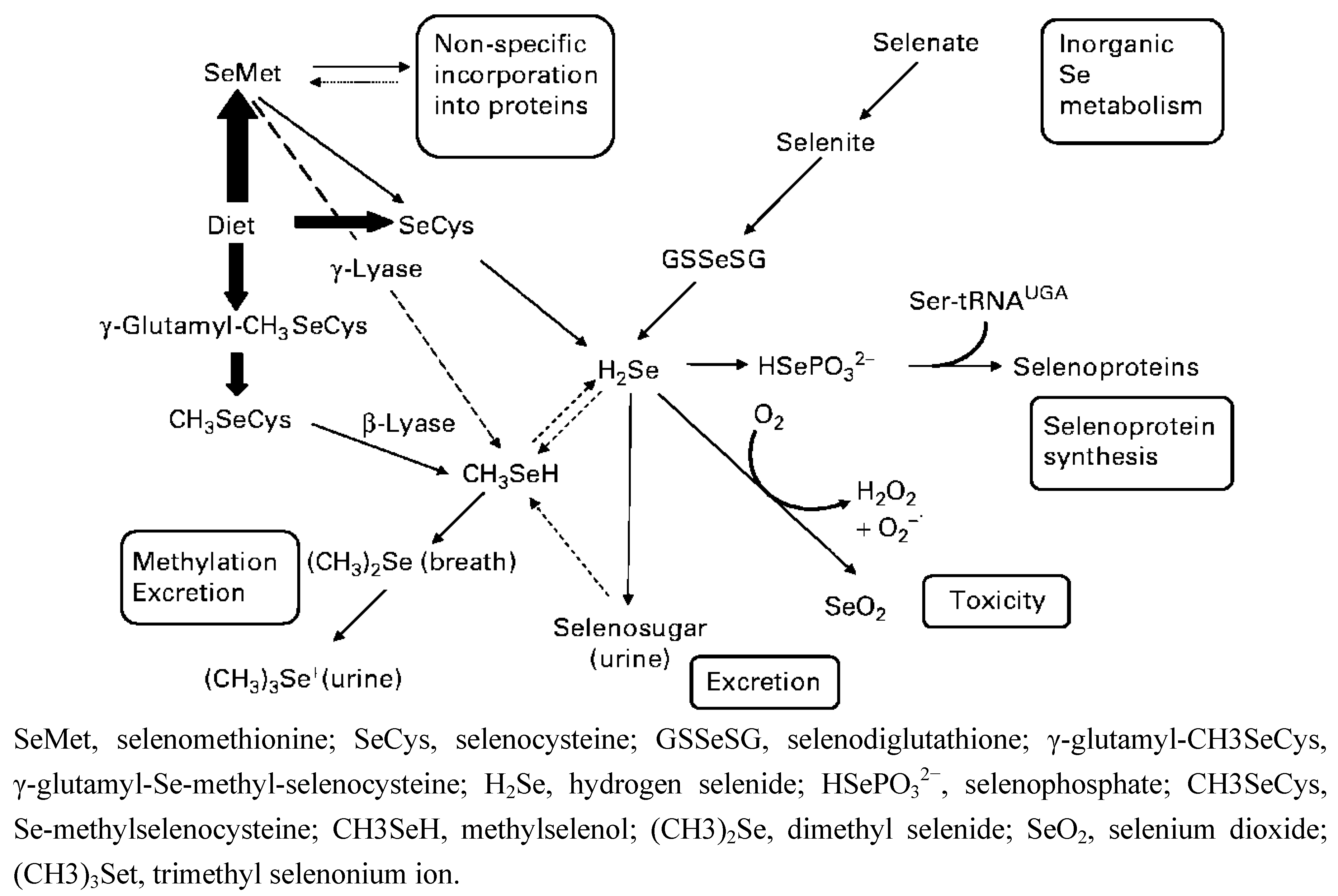

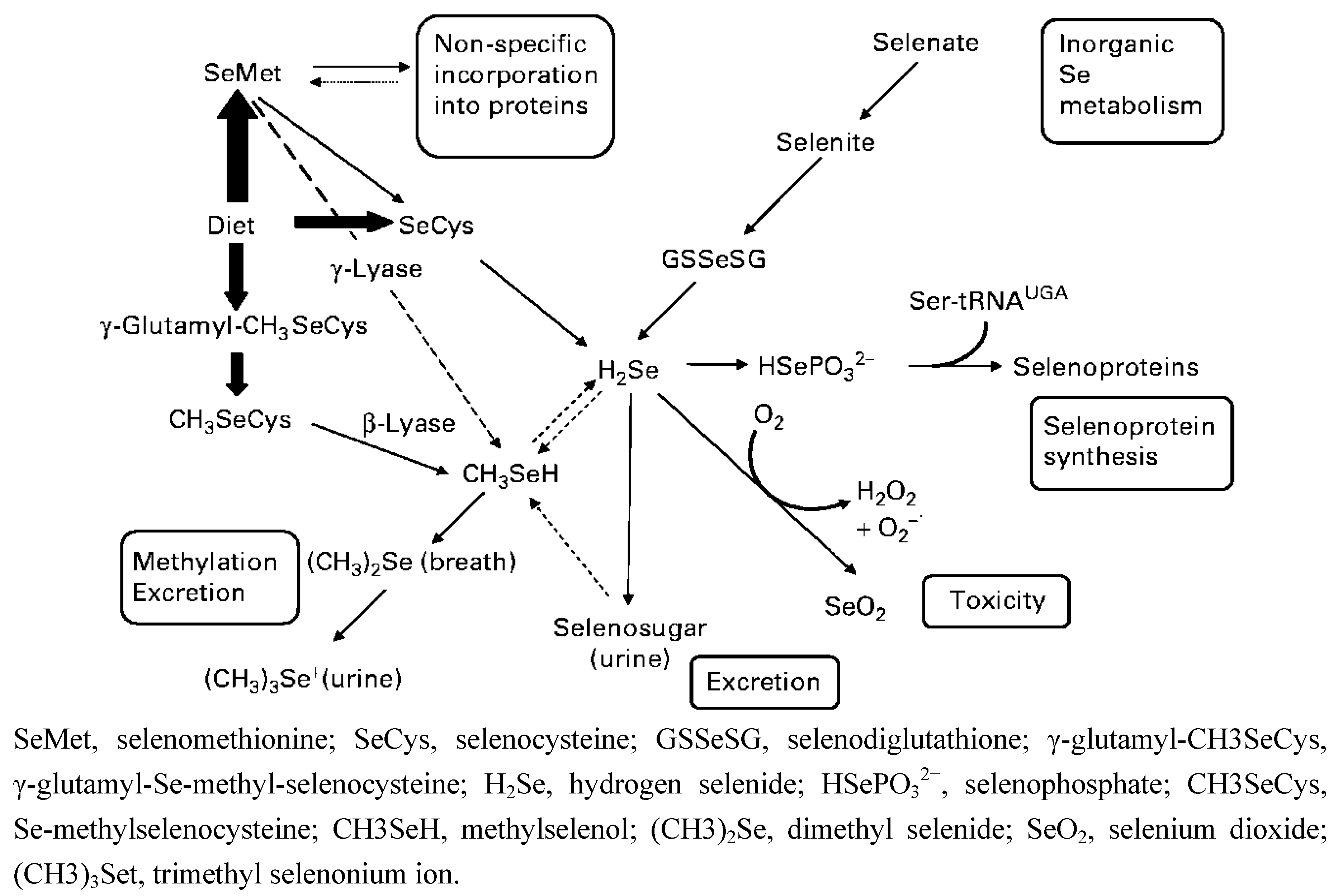{kind=link}
| Se compounds | Studies | Cancer/cells | Function | Reference |
|---|---|---|---|---|
| MSA | in vitro | HUVEC | reduce MMP-2, apoptosis | [9,30] |
| in vitro | human breast cancer cells, MDA-MB-468 and MCF-7 | reduce VEGF | [8] | |
| in vitro | human prostate cancer cells, DU145 | reduce VEGF | [8] | |
| mice | human prostate cancer cells, DU145 | reduce tumor growth and angiogenesis | [31] | |
| rat | rat prostate cancer cells, PAIII | reduce HIF-1α, and VEGF, reduce metastatic lung foci | [32] | |
| in vitro | human fibrosarcoma cell, HT1080 | inhibit cell invasion, inhibit MMP-2 activation, reduce MT1-MMP and increase TIMP-2 | [9] | |
| mice | Lewis lung carcinoma cell | reduce lung metastasis, reduce plasma uPAand PAI-1 | [33] | |
| in vitro | human clear cell renal cell carcinoma, RC2 | reduce HIF-1α, and VEGF | [34] | |
| in vitro | human head and neck squamous cell carcinoma, FaDu | reduce HIF-1α, and VEGF, increase prolyl hroxylases | [35] | |
| MSC | in vitro | murine breast cancer cells, TM6 | inhibit migration | [36] |
| rat | carcinogen-induced breast cancer | reduce angiogenesis | [37] | |
| mice | human breast cancer cells, MCF-7 | reduce angiogenesis | [38] | |
| mice | human colon cancer cells, HCT-8, HT-29 and GEO | reduce angiogenesis | [39,40] | |
| mice | human small cell lung cancer, H69 | reduce microvessel density, increase vascular maturation | [41] | |
| mice | human nonsmall epithelial lung carcinimo, A549 | increase vascular maturation | [41] | |
| mice | human head and neck squamous cell carcinoma, FaDu | reduce COX-2, iNOS, HIF-1α, and VEGF, reduce microvessel density, increase vascular maturation, drug delivery and distribution | [35,42,43] | |
| mice | human head and neck squamous cell carcinoma, A253 | reduce microvessel density, increase vascular maturation, drug delivery and distribution | [41] | |
| SeM | mice | murine breast cancer cells, 4T1.2 | most protection against metastasis | [44] |
| mice | melanoma | reduce lung metastasis | [45] | |
| MeCN | in vitro | HUVEC | reduce MMP-2 | [9] |
| methylselenol | in vitro | human fibrosarcoma cell, HT1080 | reduce cell migration and invasion, decrease expression and activity of MMP-2 and MMP-9, increase TIMP1 and TIMP2 | [46] |
| selenite | in vitro | HUVEC | apoptosis | [30] |
| in vitro | mammaery endothelial cells | reduce VEGF | [47] | |
| rat | carcinogen-induced breast cancer | inhibit VEGF, reduce angiogenesis | [37] | |
| in vitro | human fibrosarcoma cell, HT1080 | reduce cell migration, reduce cell-ECM attachment, reduce MMP-2, MMP-9 and uPA, increase TIMP-1 | [48] | |
| mice | murine melanoma cell, B16BL6 | reduce lung metastasis | [49] | |
| mice | murine melanoma cell, B16F10 | reduce lung metastasis | [50] | |
| in vitro | murine melanoma cell, B16F10 | inhibit cell migration decrease HIF-1α, VEGF, and IL-18 | [51] | |
| rat | carcinogen-indeced liver cancer | reduce angiogenesis, inhibit angiogenic factors | [41] | |
| in vitro | human astrocytoma cell, IPSB-18 | reduce MMPs amd EGFR, increase MMP inhibitors | [52] | |
| selenate | in vitro | human breast cancer cells, MDA-MB-231 and MCF-7 | enhance epithelial tight junction, inhibit motility and trans-endothelial invasion | [53] |
| Se-enriched garlic | rat | carcinogen-induced breast cancer | inhibit VEGF, reduce angiogenesis | [37] |
| high Se isolated soy proteins | mice | murine melanoma cell, B16BL6 | reduce lung metastasis | [54] |
| Se-enriched malt | rat | carcinogen-indeced liver cancer | reduce angiogenesis, inhibit angiogenic factors | [41,55] |
| Selenoproteins | Studies | Cancer/cell | Function | Reference |
|---|---|---|---|---|
| TXR1 | in vitro | mammary endothelial cells | TXR1 inhibition reduces VEGF, cell migration, proliferation and tube formation | [47] |
| mice | Lewis lung carcinoma cell | TXR1 inhibition reduces lung metastasis | [56] | |
| in vitro | Lewis lung carcinoma cell | TXR1 inhibition reduces HGF and OPN | [56] | |
| clinically | human oral squamous cell carcinoma | correlated with lymph node metastasis and with the clinical stage | [57] | |
| GPX3 | in vitro | human prostate cancer cells, PC3, DU145 and LNCaP | GPX3 overexpression reduces cell invasion | [58] |
| mice | human prostate cancer cells, PC3 with GPX3 overexpression | GPX3 overexpression reduces primary tumor sizes, eliminates metastasis, and promotes survival | [58] | |
| GPX2 | in vitro | human colon adenocarcinoma cell, HT29 | inhibit cell migration and invasion | [59] |
| SBP1 | in vitro | human colon cancer cell, HCT116 | SBP1 overexpression inhibits cell migration | [60] |
| in vitro | human liver cancer cell, SMMC7721 | SBP1 reduction increases cell migration and GPX1 activity | [61] | |
| Sep15 | mice | murine colon cancer cells, CT26 | Sep15 inhibition reduces lung metastasis | [62] |
2. The Effects of Se on Endothelial Cells
3. Se in Breast Cancer
4. Prostate Cancer
5. Colorectal Cancer
6. Fibrosarcoma
7. Melanoma
8. Lung Cancer
9. Other Cancers
10. Conclusion
The Future Role of Se in Cancer Metastasis
Acknowledgements
References
- Papp, L.V.; Lu, J.; Holmgren, A.; Khanna, K.K. From selenium to selenoproteins: Synthesis, identity, and their role in human health. Antioxid. Redox Signal. 2007, 9, 775–806. [Google Scholar] [CrossRef]
- Davis, C.; Tsuji, P.A.; Milner, J.A. Selenoproteins and cancer prevention. Ann. Rev. Nutr. 2012, 32, 73–95. [Google Scholar] [CrossRef]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar]
- Fang, W.; Goldberg, M.L.; Pohl, N.M.; Bi, X.; Tong, C.; Xiong, B.; Koh, T.J.; Diamond, A.M.; Yang, W. Functional and physical interaction between the selenium-binding protein 1 (SBP1) and the glutathione peroxidase 1 selenoprotein. Carcinogenesis 2010, 31, 1360–1366. [Google Scholar] [CrossRef]
- Rayman, M.P.; Infante, H.G.; Sargent, M. Food-chain selenium and human health: Spotlight on speciation. Br. J. Nutr. 2008, 100, 238–253. [Google Scholar]
- Chen, Y.C.; Sosnoski, D.M.; Gandhi, U.H.; Novinger, L.J.; Prabhu, K.S.; Mastro, A.M. Selenium modifies the osteoblast inflammatory stress response to bone metastatic breast cancer. Carcinogenesis 2009, 30, 1941–1948. [Google Scholar] [CrossRef]
- Suzuki, K.T.; Kurasaki, K.; Ogawa, S.; Suzuki, N. Metabolic transformation of methylseleninic acid through key selenium intermediate selenide. Toxicol. Appl. Pharmacol. 2006, 215, 189–197. [Google Scholar] [CrossRef]
- Brozmanova, J.; Manikova, D.; Vlckova, V.; Chovanec, M. Selenium: A double-edged sword for defense and offence in cancer. Arch. Toxicol. 2010, 84, 919–938. [Google Scholar]
- Boudreau, R.T.; Conrad, D.M.; Hoskin, D.W. Differential involvement of reactive oxygen species in apoptosis caused by the inhibition of protein phosphatase 2A in Jurkat and CCRF-CEM human T-leukemia cells. Exp. Mol. Pathol. 2007, 83, 347–356. [Google Scholar] [CrossRef]
- Jiang, C.; Ganther, H.; Lu, J. Monomethyl selenium—specific inhibition of MMP-2 and VEGF expression: Implications for angiogenic switch regulation. Mol. Carcinog. 2000, 29, 236–250. [Google Scholar] [CrossRef]
- Park, J.M.; Kim, A.; Oh, J.H.; Chung, A.S. Methylseleninic acid inhibits PMA-stimulated pro-MMP-2 activation mediated by Mt1-MMP expression and further tumor invasion through suppression of NF-kappaB activation. Carcinogenesis 2007, 28, 837–847. [Google Scholar] [CrossRef]
- Li, G.X.; Hu, H.; Jiang, C.; Schuster, T.; Lu, J. Differential involvement of reactive oxygen species in apoptosis induced by two classes of selenium compounds in human prostate cancer cells. Int. J. Cancer 2007, 120, 2034–2043. [Google Scholar] [CrossRef]
- Bhattacharya, A. Methylselenocysteine: A promising antiangiogenic agent for overcoming drug delivery barriers in solid malignancies for therapeutic synergy with anticancer drugs. Expert Opin. Drug Deliv. 2011, 8, 749–763. [Google Scholar] [CrossRef]
- Patrick, L. Selenium biochemistry and cancer: A review of the literature. Altern. Med. Rev. 2004, 9, 239–258. [Google Scholar]
- Clark, L.C.; Dalkin, B.; Krongrad, A.; Combs, G.F., Jr.; Turnbull, B.W.; Slate, E.H.; Witherington, R.; Herlong, J.H.; Janosko, E.; Carpenter, D.; et al. Decreased incidence of prostate cancer with selenium supplementation: Results of a double-blind cancer prevention trial. Br. J. Urol. 1998, 81, 730–734. [Google Scholar]
- Duffield-Lillico, A.J.; Reid, M.E.; Turnbull, B.W.; Combs, G.F., Jr.; Slate, E.H.; Fischbach, L.A.; Marshall, J.R.; Clark, L.C. Baseline characteristics and the effect of selenium supplementation on cancer incidence in a randomized clinical trial: A summary report of the nutritional prevention of cancer trial. Cancer Epidemiol. Biomarkers Prev. 2002, 11, 630–639. [Google Scholar]
- Ledesma, M.C.; Jung-Hynes, B.; Schmit, T.L.; Kumar, R.; Mukhtar, H.; Ahmad, N. Selenium and vitamin E for prostate cancer: Post-SELECT (selenium and vitamin E cancer prevention trial) status. Mol. Med. 2011, 17, 134–143. [Google Scholar]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The selenium and vitamin E cancer prevention trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Amaral, A.F.; Cantor, K.P.; Silverman, D.T.; Malats, N. Selenium and bladder cancer risk: A meta-analysis. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 2407–2415. [Google Scholar] [CrossRef]
- Glattre, E.; Thomassen, Y.; Thoresen, S.O.; Haldorsen, T.; Lund-Larsen, P.G.; Theodorsen, L.; Aaseth, J. Prediagnostic serum selenium in a case-control study of thyroid cancer. Int. J. Epidemiol. 1989, 18, 45–49. [Google Scholar] [CrossRef]
- Peters, U.; Takata, Y. Selenium and the prevention of prostate and colorectal cancer. Mol. Nutr. Food Res. 2008, 52, 1261–1272. [Google Scholar] [CrossRef]
- Wei, W.Q.; Abnet, C.C.; Qiao, Y.L.; Dawsey, S.M.; Dong, Z.W.; Sun, X.D.; Fan, J.H.; Gunter, E.W.; Taylor, P.R.; Mark, S.D. Prospective study of serum selenium concentrations and esophageal and gastric cardia cancer, heart disease, stroke, and total death. Am. J. Clin. Nutr. 2004, 79, 80–85. [Google Scholar]
- Zhuo, H.; Smith, A.H.; Steinmaus, C. Selenium and lung cancer: A quantitative analysis of heterogeneity in the current epidemiological literature. Cancer Epidemiol. Biomarkers Prev. 2004, 13, 771–778. [Google Scholar]
- Ip, C.; Thompson, H.J.; Zhu, Z.; Ganther, H.E. in vitro and in vivo studies of methylseleninic acid: Evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000, 60, 2882–2886. [Google Scholar]
- Li, G.X.; Lee, H.J.; Wang, Z.; Hu, H.; Liao, J.D.; Watts, J.C.; Combs, G.F., Jr.; Lu, J. Superior in vivo inhibitory efficacy of methylseleninic acid against human prostate cancer over selenomethionine or selenite. Carcinogenesis 2008, 29, 1005–1012. [Google Scholar] [CrossRef]
- Whanger, P.D. Selenium and its relationship to cancer: An update. Br. J. Nutr. 2004, 91, 11–28. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R. Selenium compounds and selenoproteins in cancer. Chem. Biodivers. 2008, 5, 389–395. [Google Scholar] [CrossRef]
- Rayman, M. Selenium; Humana Press/Springer: New York, NY, USA, 2010; pp. 411–448. [Google Scholar]
- van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef]
- Neufeld, G.; Kessler, O. Pro-angiogenic cytokines and their role in tumor angiogenesis. Cancer Metastasis Rev. 2006, 25, 373–385. [Google Scholar] [CrossRef]
- Eckhardt, B.L.; Parker, B.S.; van Laar, R.K.; Restall, C.M.; Natoli, A.L.; Tavaria, M.D.; Stanley, K.L.; Sloan, E.K.; Moseley, J.M.; Anderson, R.L. Genomic analysis of a spontaneous model of breast cancer metastasis to bone reveals a role for the extracellular matrix. Mol. Cancer Res. 2005, 3, 1–13. [Google Scholar]
- Hurst, R.; Hooper, L.; Norat, T.; Lau, R.; Aune, D.; Greenwood, D.C.; Vieira, R.; Collings, R.; Harvey, L.J.; Sterne, J.A.; et al. Selenium and prostate cancer: Systematic review and meta-analysis. Am. J. Clin. Nutr. 2012, 96, 111–122. [Google Scholar] [CrossRef]
- Li, D.; Graef, G.L.; Yee, J.A.; Yan, L. Dietary supplementation with high-selenium soy protein reduces pulmonary metastasis of melanoma cells in mice. J. Nutr. 2004, 134, 1536–1540. [Google Scholar]
- Huang, C.; Ding, G.; Gu, C.; Zhou, J.; Kuang, M.; Ji, Y.; He, Y.; Kondo, T.; Fan, J. Decreased selenium-binding protein 1 enhances glutathione peroxidase 1 activity and downregulates HIF-1alpha to promote hepatocellular carcinoma invasiveness. Clin. Cancer Res. 2012, 18, 3042–3053. [Google Scholar] [CrossRef]
- Chintala, S.; Najrana, T.; Toth, K.; Cao, S.; Durrani, F.A.; Pili, R.; Rustum, Y.M. Prolyl hydroxylase 2 dependent and Von-Hippel-Lindau independent degradation of hypoxia-inducible factor 1 and 2 alpha by selenium in clear cell renal cell carcinoma leads to tumor growth inhibition. BMC Cancer 2012, 12, 293. [Google Scholar] [CrossRef]
- Jiang, C.; Kim, K.H.; Wang, Z.; Lu, J. Methyl selenium-induced vascular endothelial apoptosis is executed by caspases and principally mediated by p38 MAPK pathway. Nutr. Cancer 2004, 49, 174–183. [Google Scholar] [CrossRef]
- Martin, T.A.; Jiang, W.G. Loss of tight junction barrier function and its role in cancer metastasis. Biochim. Biophys. Acta 2009, 1788, 872–891. [Google Scholar] [CrossRef]
- Martin, T.A.; Das, T.; Mansel, R.E.; Jiang, W.G. Enhanced tight junction function in human breast cancer cells by antioxidant, selenium and polyunsaturated lipid. J. Cell. Biochem. 2007, 101, 155–166. [Google Scholar]
- Banning, A.; Kipp, A.; Schmitmeier, S.; Lowinger, M.; Florian, S.; Krehl, S.; Thalmann, S.; Thierbach, R.; Steinberg, P.; Brigelius-Flohe, R. Glutathione peroxidase 2 inhibits cyclooxygenase-2-mediated migration and invasion of HT-29 adenocarcinoma cells but supports their growth as tumors in nude mice. Cancer Res. 2008, 68, 9746–9753. [Google Scholar] [CrossRef]
- Murawaki, Y.; Tsuchiya, H.; Kanbe, T.; Harada, K.; Yashima, K.; Nozaka, K.; Tanida, O.; Kohno, M.; Mukoyama, T.; Nishimuki, E.; et al. Aberrant expression of selenoproteins in the progression of colorectal cancer. Cancer Lett. 2008, 259, 218–230. [Google Scholar] [CrossRef]
- Yan, L.; DeMars, L.C. Dietary supplementation with methylseleninic acid, but not selenomethionine, reduces spontaneous metastasis of lewis lung carcinoma in mice. Int. J. Cancer 2012, 131, 1260–1266. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Seshadri, M.; Oven, S.D.; Toth, K.; Vaughan, M.M.; Rustum, Y.M. Tumor vascular maturation and improved drug delivery induced by methylselenocysteine leads to therapeutic synergy with anticancer drugs. Clin. Cancer Res. 2008, 14, 3926–3932. [Google Scholar] [CrossRef]
- Chintala, S.; Toth, K.; Cao, S.; Durrani, F.A.; Vaughan, M.M.; Jensen, R.L.; Rustum, Y.M. Se-methylselenocysteine sensitizes hypoxic tumor cells to irinotecan by targeting hypoxia-inducible factor 1alpha. Cancer Chemother. Pharmacol. 2010, 66, 899–911. [Google Scholar] [CrossRef]
- Chen, Y.C.; Prabhu, K.S.; Das, A.; Mastro, A.M. Dietary selenium supplementation modifies breast tumor growth and metastasis. Int. J. Cancer 2013, in press. [Google Scholar]
- Bhattacharya, A.; Turowski, S.G.; San Martin, I.D.; Rajput, A.; Rustum, Y.M.; Hoffman, R.M.; Seshadri, M. Magnetic resonance and fluorescence-protein imaging of the anti-angiogenic and anti-tumor efficacy of selenium in an orthotopic model of human colon cancer. Anticancer Res. 2011, 31, 387–393. [Google Scholar]
- Bhattacharya, A.; Toth, K.; Sen, A.; Seshadri, M.; Cao, S.; Durrani, F.A.; Faber, E.; Repasky, E.A.; Rustum, Y.M. Inhibition of colon cancer growth by methylselenocysteine-induced angiogenic chemomodulation is influenced by histologic characteristics of the tumor. Clin. Colorectal Cancer 2009, 8, 155–162. [Google Scholar] [CrossRef]
- Lu, J.; Jiang, C. Antiangiogenic activity of selenium in cancer chemoprevention: Metabolite-specific effects. Nutr. Cancer 2001, 40, 64–73. [Google Scholar] [CrossRef]
- Irons, R.; Tsuji, P.A.; Carlson, B.A.; Ouyang, P.; Yoo, M.H.; Xu, X.M.; Hatfield, D.L.; Gladyshev, V.N.; Davis, C.D. Deficiency in the 15-kDa selenoprotein inhibits tumorigenicity and metastasis of colon cancer cells. Cancer Prev. Res. (Phila.) 2010, 3, 630–639. [Google Scholar] [CrossRef]
- Yoon, S.O.; Kim, M.M.; Chung, A.S. Inhibitory effect of selenite on invasion of HT1080 tumor cells. J. Biol. Chem. 2001, 276, 20085–20092. [Google Scholar] [CrossRef]
- Yan, L.; Yee, J.A.; Li, D.; McGuire, M.H.; Graef, G.L. Dietary supplementation of selenomethionine reduces metastasis of melanoma cells in mice. Anticancer Res. 1999, 19, 1337–1342. [Google Scholar]
- Yan, L.; Yee, J.A.; McGuire, M.H.; Graef, G.L. Effect of dietary supplementation of selenite on pulmonary metastasis of melanoma cells in mice. Nutr. Cancer 1997, 28, 165–169. [Google Scholar] [CrossRef]
- Liu, J.G.; Zhao, H.J.; Liu, Y.J.; Liu, Y.W.; Wang, X.L. Effect of two selenium sources on hepatocarcinogenesis and several angiogenic cytokines in diethylnitrosamine-induced hepatocarcinoma rats. J. Trace Elem. Med. Biol. 2012, 26, 255–261. [Google Scholar] [CrossRef]
- Streicher, K.L.; Sylte, M.J.; Johnson, S.E.; Sordillo, L.M. Thioredoxin reductase regulates angiogenesis by increasing endothelial cell-derived vascular endothelial growth factor. Nutr. Cancer 2004, 50, 221–231. [Google Scholar] [CrossRef]
- Zeng, H.; Briske-Anderson, M.; Idso, J.P.; Hunt, C.D. The selenium metabolite methylselenol inhibits the migration and invasion potential of HT1080 tumor cells. J. Nutr. 2006, 136, 1528–1532. [Google Scholar]
- Rustum, Y.M.; Toth, K.; Seshadri, M.; Sen, A.; Durrani, F.A.; Stott, E.; Morrison, C.D.; Cao, S.; Bhattacharya, A. Architectural heterogeneity in tumors caused by differentiation alters intratumoral drug distribution and affects therapeutic synergy of antiangiogenic organoselenium compound. J. Oncol. 2010, 2010, 396286. [Google Scholar]
- Liu, Y.H.; Tian, H.S.; Wang, D.X. Inhibitory effect of selenium yeast on the metastasis of lewis lung carcinoma in C57BL mice. Studies with reference of histochemistry and ultrastructure. Chin. Med. J. (Engl.) 1987, 100, 549–554. [Google Scholar]
- Schrauzer, G.N. Selenium yeast: Composition, quality, analysis, and safety. Pure Appl. Chem. 2006, 78, 105–109. [Google Scholar] [CrossRef]
- Richman, E.L.; Chan, J.M. Selenium and prostate cancer: The puzzle isn’t finished yet. Am. J. Clin. Nutr. 2012, 96, 1–2. [Google Scholar] [CrossRef]
- Pohl, N.M.; Tong, C.; Fang, W.; Bi, X.; Li, T.; Yang, W. Transcriptional regulation and biological functions of selenium-binding protein 1 in colorectal cancer in vitro and in nude mouse xenografts. PLoS One 2009, 4, e7774. [Google Scholar]
- Yu, Y.P.; Yu, G.; Tseng, G.; Cieply, K.; Nelson, J.; Defrances, M.; Zarnegar, R.; Michalopoulos, G.; Luo, J.H. Glutathione peroxidase 3, deleted or methylated in prostate cancer, suppresses prostate cancer growth and metastasis. Cancer Res. 2007, 67, 8043–8050. [Google Scholar] [CrossRef]
- Yoo, M.H.; Xu, X.M.; Carlson, B.A.; Gladyshev, V.N.; Hatfield, D.L. Thioredoxin reductase 1 deficiency reverses tumor phenotype and tumorigenicity of lung carcinoma cells. J. Biol. Chem. 2006, 281, 13005–13008. [Google Scholar]
- Florian, S.; Wingler, K.; Schmehl, K.; Jacobasch, G.; Kreuzer, O.J.; Meyerhof, W.; Brigelius-Flohe, R. Cellular and subcellular localization of gastrointestinal glutathione peroxidase in normal and malignant human intestinal tissue. Free Radic. Res. 2001, 35, 655–663. [Google Scholar] [CrossRef]
- Roomi, M.W.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Anti-angiogenic effects of a nutrient mixture on human umbilical vein endothelial cells. Oncol. Rep. 2005, 14, 1399–1404. [Google Scholar]
- Unni, E.; Kittrell, F.S.; Singh, U.; Sinha, R. Osteopontin is a potential target gene in mouse mammary cancer chemoprevention by Se-methylselenocysteine. Breast Cancer Res. 2004, 6, R586–R592. [Google Scholar]
- Jiang, C.; Jiang, W.; Ip, C.; Ganther, H.; Lu, J. Selenium-induced inhibition of angiogenesis in mammary cancer at chemopreventive levels of intake. Mol. Carcinog. 1999, 26, 213–225. [Google Scholar] [CrossRef]
- Li, Z.; Carrier, L.; Belame, A.; Thiyagarajah, A.; Salvo, V.A.; Burow, M.E.; Rowan, B.G. Combination of methylselenocysteine with tamoxifen inhibits MCF-7 breast cancer xenografts in nude mice through elevated apoptosis and reduced angiogenesis. Breast Cancer Res. Treat. 2009, 118, 33–43. [Google Scholar]
- Wang, Z.; Hu, H.; Li, G.; Lee, H.J.; Jiang, C.; Kim, S.H.; Lu, J. Methylseleninic acid inhibits microvascular endothelial G1 cell cycle progression and decreases tumor microvessel density. Int. J. Cancer 2008, 122, 15–24. [Google Scholar] [CrossRef]
- Sinha, I.; Null, K.; Wolter, W.; Suckow, M.A.; King, T.; Pinto, J.T.; Sinha, R. Methylseleninic acid downregulates hypoxia-inducible factor-1alpha in invasive prostate cancer. Int. J. Cancer 2012, 130, 1430–1439. [Google Scholar] [CrossRef]
- Kim, H.; Kang, H.J.; You, K.T.; Kim, S.H.; Lee, K.Y.; Kim, T.I.; Kim, C.; Song, S.Y.; Kim, H.J.; Lee, C.; et al. Suppression of human selenium-binding protein 1 is a late event in colorectal carcinogenesis and is associated with poor survival. Proteomics 2006, 6, 3466–3476. [Google Scholar] [CrossRef]
- Li, T.; Yang, W.; Li, M.; Byun, D.S.; Tong, C.; Nasser, S.; Zhuang, M.; Arango, D.; Mariadason, J.M.; Augenlicht, L.H. Expression of selenium-binding protein 1 characterizes intestinal cell maturation and predicts survival for patients with colorectal cancer. Mol. Nutr. Food Res. 2008, 52, 1289–1299. [Google Scholar] [CrossRef]
- Song, H.; Hur, I.; Park, H.J.; Nam, J.; Park, G.B.; Kong, K.H.; Hwang, Y.M.; Kim, Y.S.; Cho, D.H.; Lee, W.J.; et al. Selenium inhibits metastasis of murine melanoma cells through the induction of cell cycle arrest and cell death. Immune Netw. 2009, 9, 236–242. [Google Scholar] [CrossRef]
- Song, H.; Kim, J.; Lee, H.K.; Park, H.J.; Nam, J.; Park, G.B.; Kim, Y.S.; Cho, D.; Hur, D.Y. Selenium inhibits migration of murine melanoma cells via down-modulation of IL-18 expression. Int. Immunopharmacol. 2011, 11, 2208–2213. [Google Scholar] [CrossRef]
- Iwasawa, S.; Yamano, Y.; Takiguchi, Y.; Tanzawa, H.; Tatsumi, K.; Uzawa, K. Upregulation of thioredoxin reductase 1 in human oral squamous cell carcinoma. Oncol. Rep. 2011, 25, 637–644. [Google Scholar]
- Liu, J.G.; Zhao, H.J.; Liu, Y.J.; Wang, X.L. Effect of selenium-enriched malt on VEGF and several relevant angiogenic cytokines in diethylnitrosamine-induced hepatocarcinoma rats. J. Trace Elem. Med. Biol. 2010, 24, 52–57. [Google Scholar] [CrossRef]
- Yin, M.B.; Li, Z.R.; Toth, K.; Cao, S.; Durrani, F.A.; Hapke, G.; Bhattacharya, A.; Azrak, R.G.; Frank, C.; Rustum, Y.M. Potentiation of irinotecan sensitivity by Se-methylselenocysteine in an in vivo tumor model is associated with downregulation of cyclooxygenase-2, inducible nitric oxide synthase, and hypoxia-inducible factor 1alpha expression, resulting in reduced angiogenesis. Oncogene 2006, 25, 2509–2519. [Google Scholar]
- Whanger, P.D. Selenium and the brain: A review. Nutr. Neurosci. 2001, 4, 81–97. [Google Scholar]
- Rooprai, H.K.; Kyriazis, I.; Nuttall, R.K.; Edwards, D.R.; Zicha, D.; Aubyn, D.; Davies, D.; Gullan, R.; Pilkington, G.J. Inhibition of invasion and induction of apoptosis by selenium in human malignant brain tumour cells in vitro. Int. J. Oncol. 2007, 30, 1263–1271. [Google Scholar]
- Ramis, G.; Thomas-Moya, E.; Fernandez de Mattos, S.; Rodriguez, J.; Villalonga, P. EGFR inhibition in glioma cells modulates Rho signaling to inhibit cell motility and invasion and cooperates with temozolomide to reduce cell growth. PLoS One 2012, 7, e38770. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, Y.-C.; Prabhu, K.S.; Mastro, A.M. Is Selenium a Potential Treatment for Cancer Metastasis? Nutrients 2013, 5, 1149-1168. https://doi.org/10.3390/nu5041149
Chen Y-C, Prabhu KS, Mastro AM. Is Selenium a Potential Treatment for Cancer Metastasis? Nutrients. 2013; 5(4):1149-1168. https://doi.org/10.3390/nu5041149
Chicago/Turabian StyleChen, Yu-Chi, K. Sandeep Prabhu, and Andrea M. Mastro. 2013. "Is Selenium a Potential Treatment for Cancer Metastasis?" Nutrients 5, no. 4: 1149-1168. https://doi.org/10.3390/nu5041149
APA StyleChen, Y.-C., Prabhu, K. S., & Mastro, A. M. (2013). Is Selenium a Potential Treatment for Cancer Metastasis? Nutrients, 5(4), 1149-1168. https://doi.org/10.3390/nu5041149