Genomic and Epigenomic Insights into Nutrition and Brain Disorders

Abstract
:1. Introduction
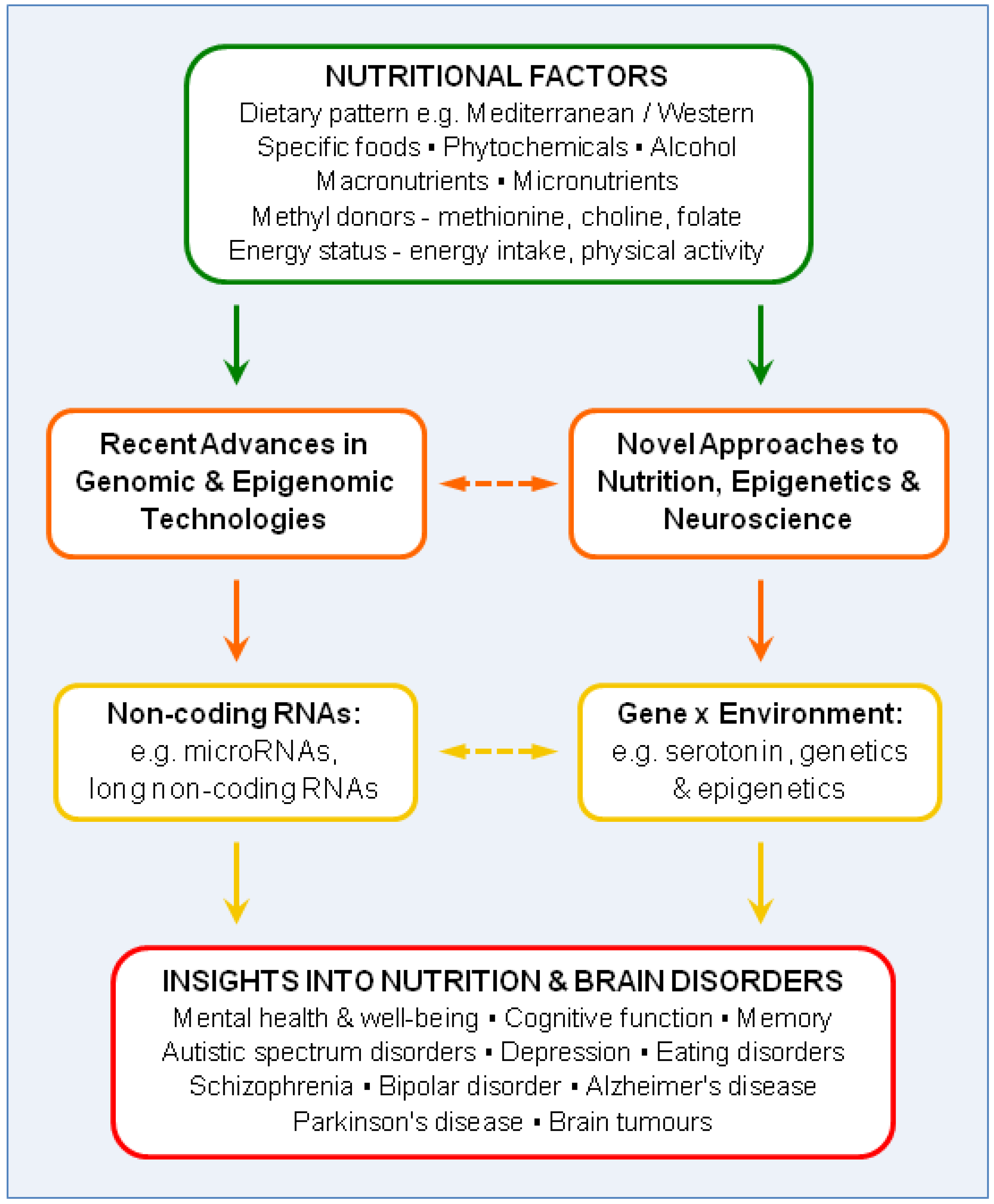
2. Recent Advances in Genomic and Epigenomic Technologies
2.1. Whole Genome Strategies, Next-Generation DNA Sequencing and Brain Disorders
2.2. Genetic Variation and the 1000 Genomes Project
2.3. ENCODE: An Encyclopedia of DNA Elements in the Human Genome
2.4. Epigenomics and New Technologies
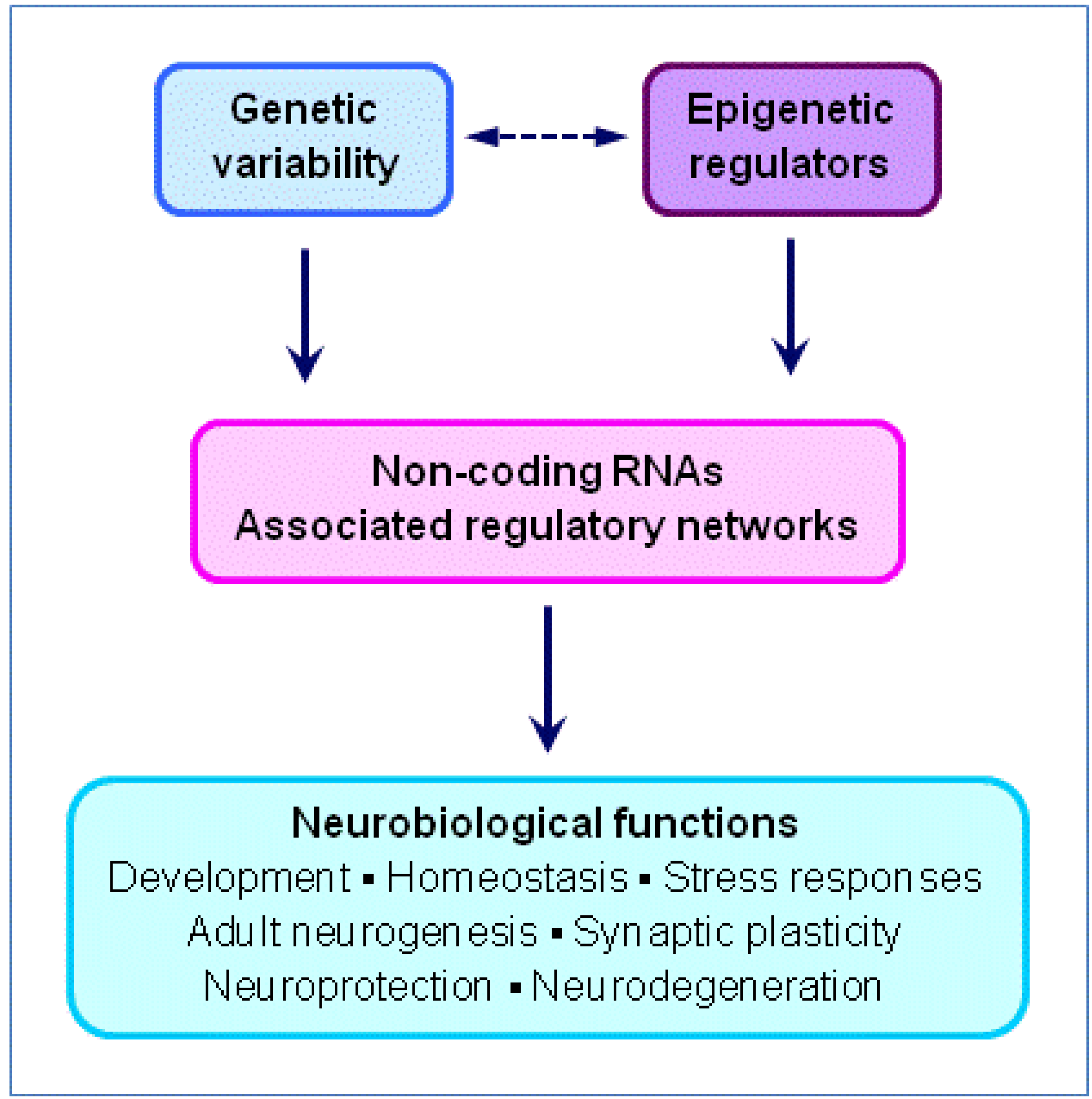
3. Non-Coding RNAs (ncRNAs), Gene Regulation and Neuroscience
3.1. Genomic and Epigenomic Regulation by ncRNAs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Small ncRNAs (<200/400 nucleotides) |
|
| Long ncRNAs (>200/400 nucleotides, sometimes >100,000 nucleotides) |
|
3.2. NcRNAs and Brain Disorders
3.3. NcRNAs, Nutrition and the Brain
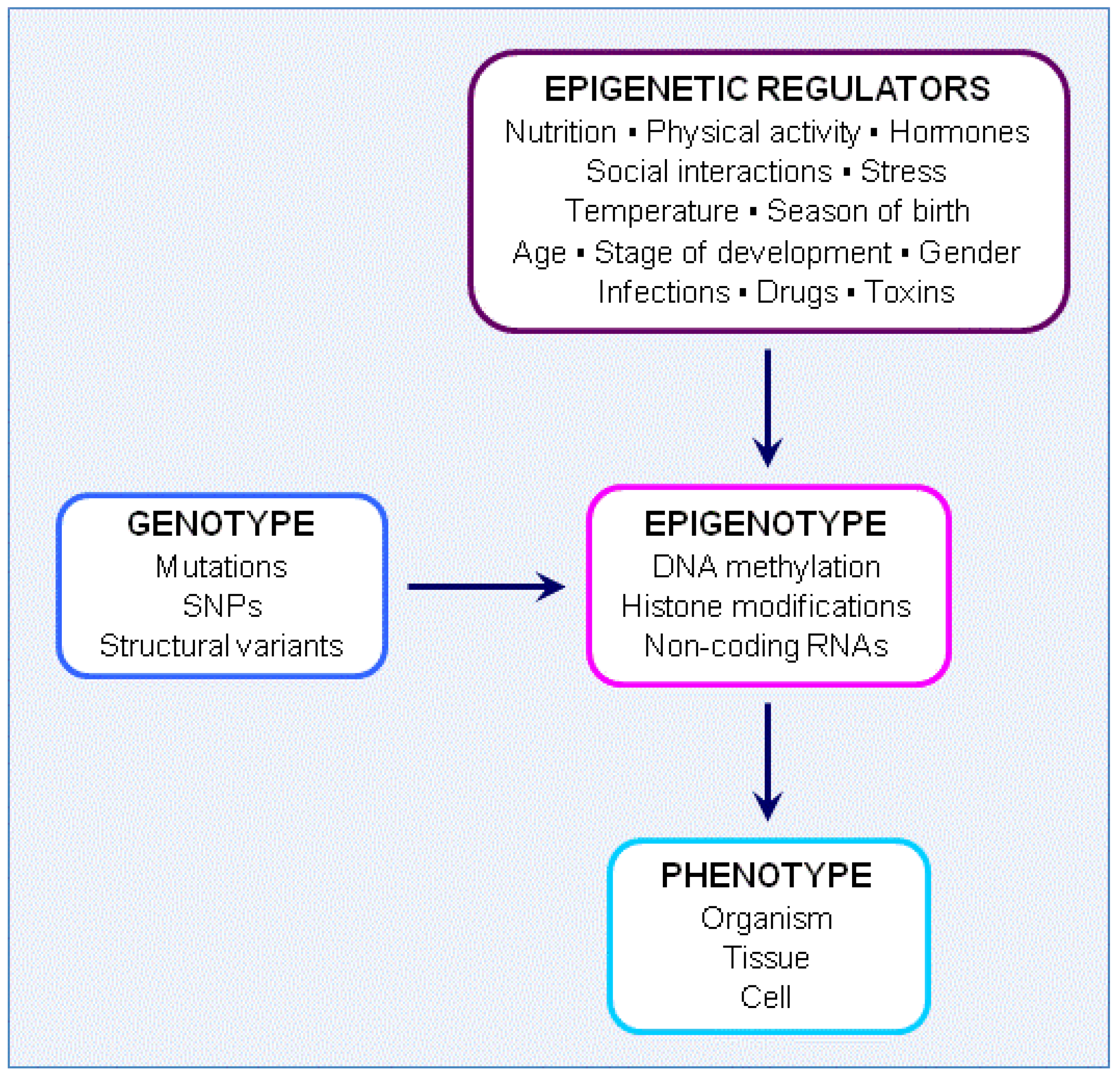
4. Novel Approaches to Nutrition, Epigenetics and Neuroscience
4.1. Genotype, Epigenotype and Phenotype
4.2. Nutrition, Honeybees and Epigenetics
4.3. Phenotypic Plasticity, Locusts and Neurological Function
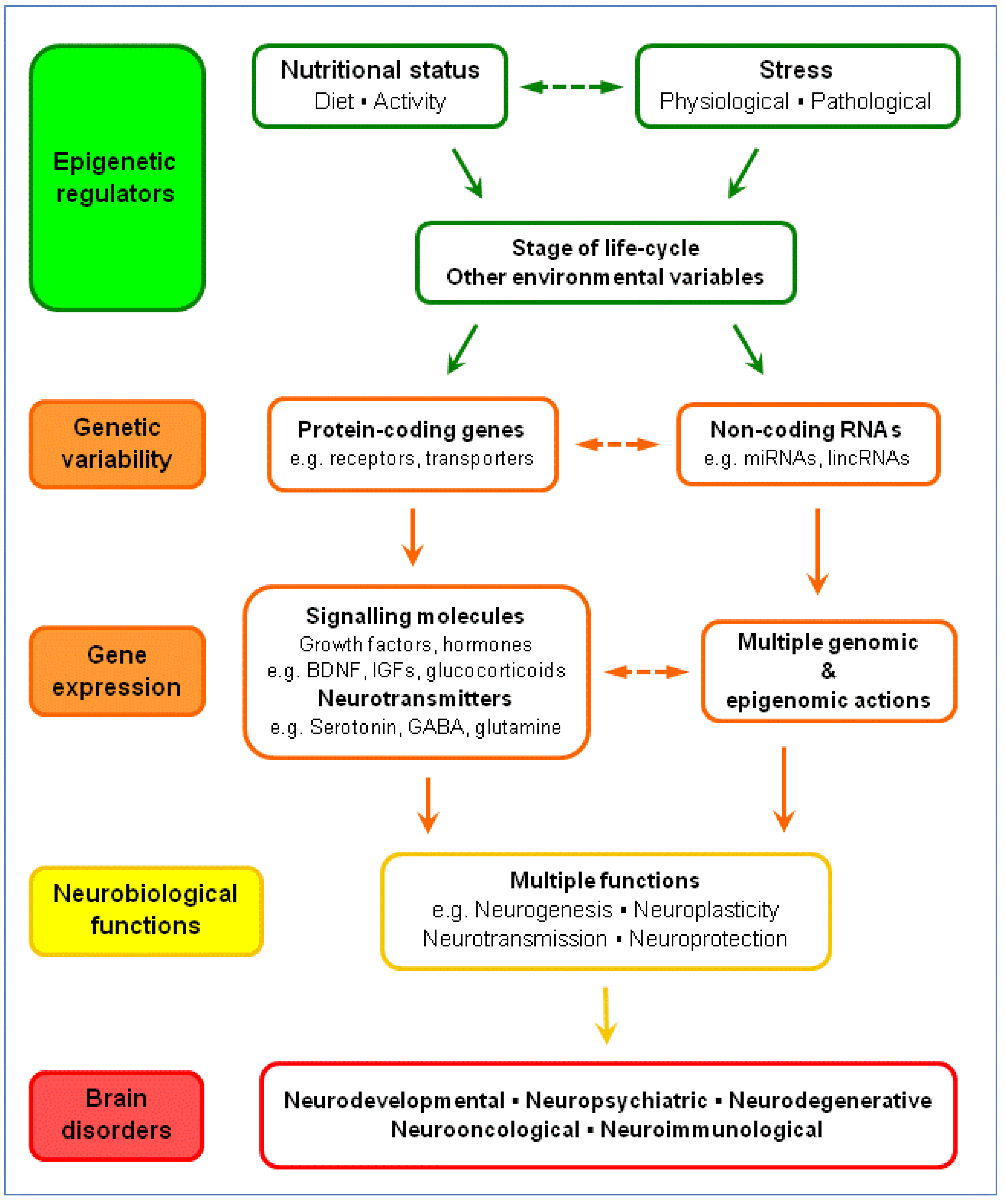
5. Gene-Environment Interactions: The Serotonergic System and Brain Disorders
5.1. Dietary Tryptophan and Brain Serotonin
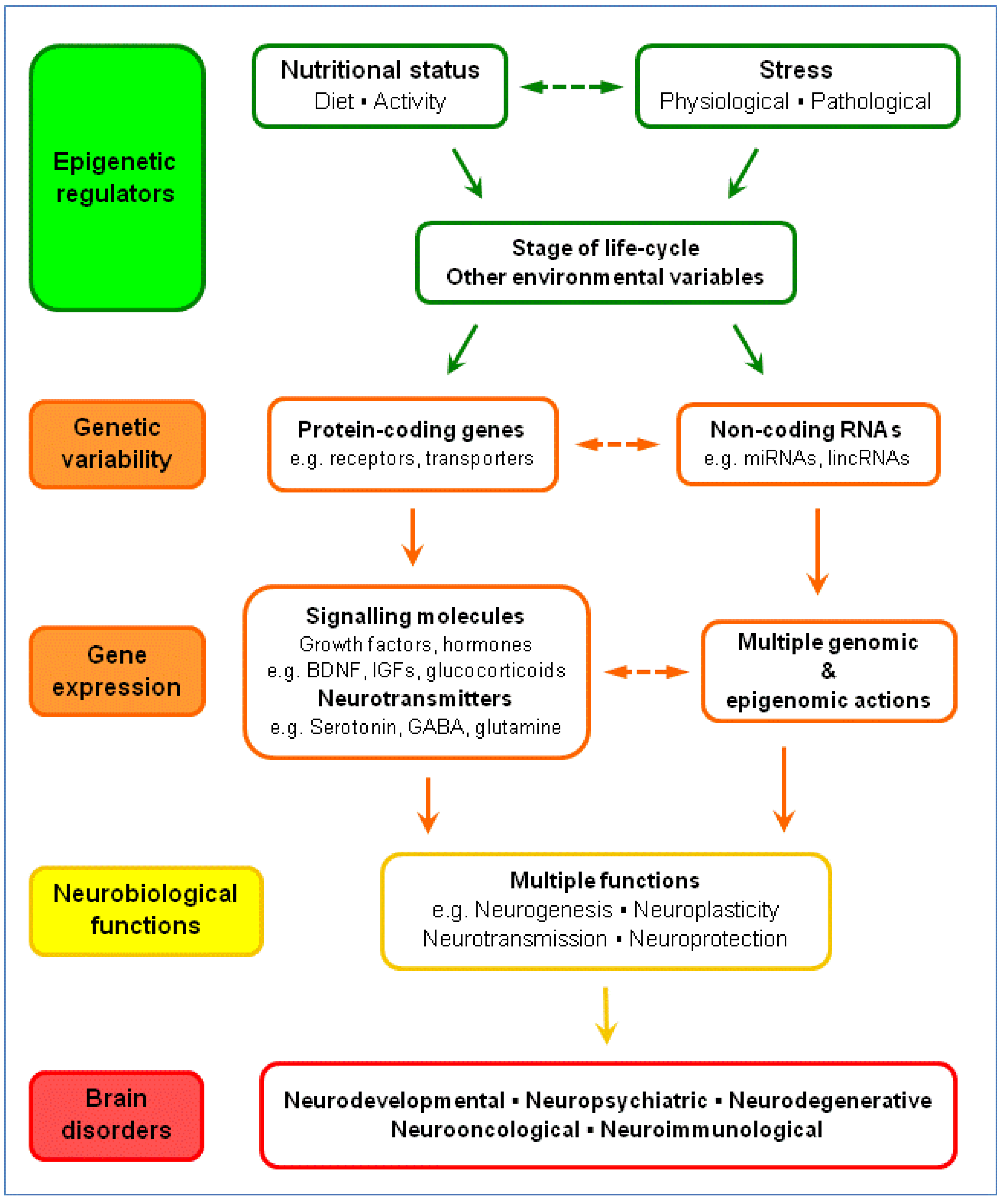
5.2. Brain Plasticity: BDNF, Glucocorticoids and Serotonin
5.3. Serotonergic System: Gene Variability, Epigenetics and Brain Disorders
5.4. Nutrition-Stress-Gene Interactions and Brain Disorders
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Bras, J.; Guerreiro, R.; Hardy, J. Use of next-generation sequencing and other whole-genome strategies to dissect neurological disease. Nat. Rev. Neurosci. 2012, 13, 453–464. [Google Scholar] [CrossRef]
- Sullivan, P.F.; Daly, M.J.; O’Donovan, M. Genetic architectures of psychiatric disorders: The emerging picture and its implications. Nat. Rev. Genet. 2012, 13, 537–551. [Google Scholar] [CrossRef]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Qureshi, I.A.; Mehler, M.F. Epigenetic mechanisms governing the process of neurodegeneration. Mol. Aspects Med. 2012, in press. [Google Scholar]
- Dauncey, M.J. Recent advances in nutrition, genes and brain health. Proc. Nutr. Soc. 2012, 71, 581–591. [Google Scholar] [CrossRef]
- Dauncey, M.J. Novos conhecimentos sobre nutrição, genes e saúde do cérebro. Nutr. Pauta 2012, 115, 3–10. [Google Scholar]
- Dauncey, M.J. Novos conhecimentos sobre nutrição, genes e doenças do cérebro. Nutr. Pauta 2012, 116, 3–12. [Google Scholar]
- Gomez-Pinilla, F. Brain foods: The effects of nutrients on brain function. Nat. Rev. Neurosci. 2008, 9, 568–578. [Google Scholar] [CrossRef]
- Nurk, E.; Refsum, H.; Drevon, C.A.; Tell, G.S.; Nygaard, H.A.; Engedal, K.; Smith, A.D. Intake of flavonoid-rich wine, tea, and chocolate by elderly men and women is associated with better cognitive test performance. J. Nutr. 2009, 139, 120–127. [Google Scholar]
- Dauncey, M.J. New insights into nutrition and cognitive neuroscience. Proc. Nutr. Soc. 2009, 68, 408–415. [Google Scholar] [CrossRef]
- Dauncey, M.J. Recentes avanços em nutrição e neurociência cognitiva. Nutr. Pauta 2009, 97, 4–13. [Google Scholar]
- Gomez-Pinilla, F.; Nguyen, T.T. Natural mood foods: The actions of polyphenols against psychiatric and cognitive disorders. Nutr. Neurosci. 2012, 15, 127–133. [Google Scholar]
- Milte, C.M.; Parletta, N.; Buckley, J.D.; Coates, A.M.; Young, R.M.; Howe, P.R. Eicosapentaenoic and docosahexaenoic acids, cognition, and behavior in children with attention-deficit/hyperactivity disorder: A randomized controlled trial. Nutrition 2012, 28, 670–677. [Google Scholar] [CrossRef]
- Morris, M.S. The role of B vitamins in preventing and treating cognitive impairment and decline. Adv. Nutr. 2012, 3, 801–812. [Google Scholar] [CrossRef]
- Sinn, N.; Milte, C.M.; Street, S.J.; Buckley, J.D.; Coates, A.M.; Petkov, J.; Howe, P.R. Effects of n-3 fatty acids, EPA v. DHA, on depressive symptoms, quality of life, memory and executive function in older adults with mild cognitive impairment: A 6-month randomised controlled trial. Br. J. Nutr. 2012, 107, 1682–1693. [Google Scholar] [CrossRef]
- Susser, E.; Kirkbride, J.B.; Heijmans, B.T.; Kresovich, J.K.; Lumey, L.H.; Stein, A.D. Maternal prenatal nutrition and health in grandchildren and subsequent generations. Ann. Rev. Anthropol. 2012, 41, 577–610. [Google Scholar] [CrossRef]
- Dauncey, M.J.; Bicknell, R.J. Nutrition and neurodevelopment: Mechanisms of developmental dysfunction and disease in later life. Nutr. Res. Rev. 1999, 12, 231–253. [Google Scholar] [CrossRef]
- Dauncey, M.J.; Astley, S. Genômica nutricional: Novos estudos sobre as interações entre nutrição e o genoma humano. Nutr. Pauta 2006, 77, 4–9. [Google Scholar]
- Hackett, J.A.; Zylicz, J.J.; Surani, M.A. Parallel mechanisms of epigenetic reprogramming in the germline. Trends Genet. 2012, 28, 164–174. [Google Scholar] [CrossRef]
- Babenko, O.; Kovalchuk, I.; Metz, G.A. Epigenetic programming of neurodegenerative diseases by an adverse environment. Brain Res. 2012, 1444, 96–111. [Google Scholar] [CrossRef]
- Dauncey, M.J.; White, P.; Burton, K.A.; Katsumata, M. Nutrition-hormone receptor-gene interactions: Implications for development and disease. Proc. Nutr. Soc. 2001, 60, 63–72. [Google Scholar] [CrossRef]
- Dauncey, M.J.; White, P. Nutrition and cell communication: Insulin signalling in development, health and disease. Rec. Res. Dev. Nutr. 2004, 6, 49–81. [Google Scholar]
- Park, L.K.; Friso, S.; Choi, S.W. Nutritional influences on epigenetics and age-related disease. Proc. Nutr. Soc. 2012, 71, 75–83. [Google Scholar] [CrossRef]
- Dauncey, M.J. From early nutrition and later development to underlying mechanisms and optimal health. Br. J. Nutr. 1997, 78, S113–S123. [Google Scholar] [CrossRef]
- Dauncey, M.J. Interações precoces nutrição-hormônios: Implicações nas doenças degenerativas de adultos. Nutr. Pauta 2004, 66, 30–35. [Google Scholar]
- Langie, S.A.; Lara, J.; Mathers, J.C. Early determinants of the ageing trajectory. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 613–626. [Google Scholar] [CrossRef]
- Robinson, S.; Fall, C. Infant nutrition and later health: A review of current evidence. Nutrients 2012, 4, 859–874. [Google Scholar] [CrossRef]
- Liu, G.E. Recent applications of DNA sequencing technologies in food, nutrition and agriculture. Rec. Pat. Food Nutr. Agric. 2011, 3, 187–195. [Google Scholar] [CrossRef]
- Kilpinen, H.; Barrett, J.C. How next-generation sequencing is transforming complex disease genetics. Trends Genet. 2013, 29, 23–30. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar]
- Steinberg, S.; de Jong, S.; Mattheisen, M.; Costas, J.; Demontis, D.; Jamain, S.; Pietilainen, O.P.; Lin, K.; Papiol, S.; Huttenlocher, J.; et al. Common variant at 16p11.2 conferring risk of psychosis. Mol. Psychiatry 2012. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, H.; Bloss, C.S.; Duvvuri, V.; Kaye, W.; Schork, N.J.; Berrettini, W.; Hakonarson, H. A genome-wide association study on common SNPs and rare CNVs in anorexia nervosa. Mol. Psychiatry 2011, 16, 949–959. [Google Scholar] [CrossRef]
- Freilinger, T.; Anttila, V.; de Vries, B.; Malik, R.; Kallela, M.; Terwindt, G.M.; Pozo-Rosich, P.; Winsvold, B.; Nyholt, D.R.; van Oosterhout, W.P.; et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat. Genet. 2012, 44, 777–782. [Google Scholar] [CrossRef]
- Boraska, V.; Davis, O.S.; Cherkas, L.F.; Helder, S.G.; Harris, J.; Krug, I.; Liao, T.P.; Treasure, J.; Ntalla, I.; Karhunen, L.; et al. Genome-wide association analysis of eating disorder-related symptoms, behaviors, and personality traits. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 803–811. [Google Scholar]
- Palotie, A.; Widen, E.; Ripatti, S. From genetic discovery to future personalized health research. N. Biotechnol. 2012, in press. [Google Scholar]
- Nadeau, J.H.; Dudley, A.M. Genetics. Systems genetics. Science 2011, 331, 1015–1016. [Google Scholar] [CrossRef]
- Kalupahana, N.S.; Moustaid-Moussa, N. Overview of symposium “Systems genetics in nutrition and obesity research”. J. Nutr. 2011, 141, 512–514. [Google Scholar] [CrossRef]
- Voy, B.H. Systems genetics: A powerful approach for gene-environment interactions. J. Nutr. 2011, 141, 515–519. [Google Scholar] [CrossRef]
- Vidal, M.; Cusick, M.E.; Barabasi, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef]
- Human Genome Far More Active Than Thought. GENCODE Consortium Discovers Far More Genes Than Previously Thought. Available online: http://www.sanger.ac.uk/about/press/2012/120905.html (accessed on 10 December 2012).
- Attar, N. The allure of the epigenome. Genome Biol. 2012, 13, 419. [Google Scholar] [CrossRef]
- Meissner, A. What can epigenomics do for you? Genome Biol. 2012, 13, 420. [Google Scholar] [CrossRef]
- Clark, C.; Palta, P.; Joyce, C.J.; Scott, C.; Grundberg, E.; Deloukas, P.; Palotie, A.; Coffey, A.J. A comparison of the whole genome approach of MeDIP-Seq to the targeted approach of the Infinium HumanMethylation450 BeadChip® for methylome profiling. PLoS One 2012, 7, e50233. [Google Scholar]
- Davies, M.N.; Volta, M.; Pidsley, R.; Lunnon, K.; Dixit, A.; Lovestone, S.; Coarfa, C.; Harris, R.A.; Milosavljevic, A.; Troakes, C.; et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012, 13, R43. [Google Scholar] [CrossRef]
- Decock, A.; Ongenaert, M.; Hoebeeck, J.; De Preter, K.; Van Peer, G.; Van Criekinge, W.; Ladenstein, R.; Schulte, J.H.; Noguera, R.; Stallings, R.L.; et al. Genome-wide promoter methylation analysis in neuroblastoma identifies prognostic methylation biomarkers. Genome Biol. 2012, 13, R95. [Google Scholar] [CrossRef]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473, 43–49. [Google Scholar]
- Huttenhofer, A.; Schattner, P.; Polacek, N. Non-coding RNAs: Hope or hype? Trends Genet. 2005, 21, 289–297. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef]
- Magistri, M.; Faghihi, M.A.; St Laurent, G., III; Wahlestedt, C. Regulation of chromatin structure by long noncoding RNAs: Focus on natural antisense transcripts. Trends Genet. 2012, 28, 389–396. [Google Scholar] [CrossRef]
- Qureshi, I.A.; Mehler, M.F. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat. Rev. Neurosci. 2012, 13, 528–541. [Google Scholar] [CrossRef]
- Salta, E.; de Strooper, B. Non-coding RNAs with essential roles in neurodegenerative disorders. Lancet Neurol. 2012, 11, 189–200. [Google Scholar] [CrossRef]
- Qureshi, I.A.; Mattick, J.S.; Mehler, M.F. Long non-coding RNAs in nervous system function and disease. Brain Res. 2010, 1338, 20–35. [Google Scholar] [CrossRef]
- Gascon, E.; Gao, F.B. Cause or effect: Misregulation of microRNA pathways in neurodegeneration. Front. Neurosci. 2012, 6, 48. [Google Scholar]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef]
- Spadaro, P.A.; Bredy, T.W. Emerging role of non-coding RNA in neural plasticity, cognitive function, and neuropsychiatric disorders. Front. Genet. 2012, 3, 132. [Google Scholar]
- Tan, L.; Yu, J.T.; Hu, N.; Tan, L. Non-coding RNAs in Alzheimer’s Disease. Mol. Neurobiol. 2013, 47, 382–393. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Jung, K.H.; Kim, J.H.; Huh, J.Y.; Yoon, H.; Park, D.K.; Lim, J.Y.; Kim, J.M.; Jeon, D.; et al. miR-206 regulates brain-derived neurotrophic factor in Alzheimer disease model. Ann. Neurol. 2012, 72, 269–277. [Google Scholar] [CrossRef]
- Gomez-Pinilla, F. The combined effects of exercise and foods in preventing neurological and cognitive disorders. Prev. Med. 2011, 52, S75–S80. [Google Scholar] [CrossRef]
- Harraz, M.M.; Dawson, T.M.; Dawson, V.L. MicroRNAs in Parkinson’s disease. J. Chem. Neuroanat. 2011, 42, 127–130. [Google Scholar] [CrossRef]
- Wang, G.; van der Walt, J.M.; Mayhew, G.; Li, Y.J.; Zuchner, S.; Scott, W.K.; Martin, E.R.; Vance, J.M. Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of alpha-synuclein. Am. J. Hum. Genet. 2008, 82, 283–289. [Google Scholar] [CrossRef]
- Foley, N.H.; Bray, I.; Watters, K.M.; Das, S.; Bryan, K.; Bernas, T.; Prehn, J.H.; Stallings, R.L. MicroRNAs 10a and 10b are potent inducers of neuroblastoma cell differentiation through targeting of nuclear receptor corepressor 2. Cell. Death Differ. 2011, 18, 1089–1098. [Google Scholar] [CrossRef]
- Setty, M.; Helmy, K.; Khan, A.A.; Silber, J.; Arvey, A.; Neezen, F.; Agius, P.; Huse, J.T.; Holland, E.C.; Leslie, C.S. Inferring transcriptional and microRNA-mediated regulatory programs in glioblastoma. Mol. Syst. Biol. 2012, 8, 605. [Google Scholar]
- Ross, S.A.; Davis, C.D. MicroRNA, nutrition, and cancer preventio. Adv. Nutr. 2011, 2, 472–485. [Google Scholar] [CrossRef]
- Kesse-Guyot, E.; Fezeu, L.; Andreeva, V.A.; Touvier, M.; Scalbert, A.; Hercberg, S.; Galan, P. Total and specific polyphenol intakes in midlife are associated with cognitive function measured 13 years later. J. Nutr. 2012, 142, 76–83. [Google Scholar] [CrossRef]
- Milenkovic, D.; Deval, C.; Gouranton, E.; Landrier, J.F.; Scalbert, A.; Morand, C.; Mazur, A. Modulation of miRNA expression by dietary polyphenols in apoE deficient mice: A new mechanism of the action of polyphenols. PLoS One 2012, 7, e29837. [Google Scholar]
- Baggish, A.L.; Hale, A.; Weiner, R.B.; Lewis, G.D.; Systrom, D.; Wang, F.; Wang, T.J.; Chan, S.Y. Dynamic regulation of circulating microRNA during acute exhaustive exercise and sustained aerobic exercise training. J. Physiol. 2011, 589, 3983–3994. [Google Scholar] [CrossRef]
- Fernandes-Silva, M.M.; Carvalho, V.O.; Guimarães, G.V.; Bacal, F. Physical exercise and microRNAs: New frontiers in heart failure. Arq. Bras. Cardiol. 2012, 98, 459–466. [Google Scholar] [CrossRef]
- Barrès, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell. Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef]
- Habibi, E.; Masoudi-Nejad, A.; Abdolmaleky, H.M.; Haggarty, S.J. Emerging roles of epigenetic mechanisms in Parkinson’s disease. Funct. Integr. Genomics 2011, 11, 523–537. [Google Scholar] [CrossRef]
- Labrie, V.; Pai, S.; Petronis, A. Epigenetics of major psychosis: Progress, problems and perspectives. Trends Genet. 2012, 28, 427–435. [Google Scholar] [CrossRef]
- Jimenez-Chillaron, J.C.; Diaz, R.; Martinez, D.; Pentinat, T.; Ramon-Krauel, M.; Ribo, S.; Plosch, T. The role of nutrition on epigenetic modifications and their implications on health. Biochimie 2012, 94, 2242–2263. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Zimbron, J.; Lewis, G.; Jones, P.B. Prenatal maternal infection, neurodevelopment and adult schizophrenia: A systematic review of population-based studies. Psychol. Med. 2013, 43, 239–257. [Google Scholar] [CrossRef]
- Johannsen, W. The genotype conception of heredity. Am. Nat. 1911, 45, 129–159. [Google Scholar]
- Dauncey, M.J.; Ingram, D.L. Acclimatization to warm or cold temperatures and the role of food intake. J. Therm. Biol. 1986, 11, 89–93. [Google Scholar] [CrossRef]
- Dauncey, M.J. From whole body to molecule: An integrated approach to the regulation of metabolism and growth. Thermochim. Acta 1995, 250, 305–318. [Google Scholar] [CrossRef]
- Anstey, M.L.; Rogers, S.M.; Ott, S.R.; Burrows, M.; Simpson, S.J. Serotonin mediates behavioral gregarization underlying swarm formation in desert locusts. Science 2009, 323, 627–630. [Google Scholar] [CrossRef]
- Menzel, R. The honeybee as a model for understanding the basis of cognition. Nat. Rev. Neurosci. 2012, 13, 758–768. [Google Scholar] [CrossRef]
- Wakeling, L.A.; Ions, L.J.; Ford, D. Could Sirt1-mediated epigenetic effects contribute to the longevity response to dietary restriction and be mimicked by other dietary interventions? Age (Dordr.) 2009, 31, 327–341. [Google Scholar] [CrossRef]
- McKay, J.A.; Mathers, J.C. Diet induced epigenetic changes and their implications for health. Acta Physiol. (Oxf.) 2011, 202, 103–118. [Google Scholar] [CrossRef]
- Singh, K.; Molenaar, A.J.; Swanson, K.M.; Gudex, B.; Arias, J.A.; Erdman, R.A.; Stelwagen, K. Epigenetics: A possible role in acute and transgenerational regulation of dairy cow milk production. Animal 2012, 6, 375–381. [Google Scholar]
- Weiner, S.A.; Toth, A.L. Epigenetics in social insects: a new direction for understanding the evolution of castes. Genet. Res. Int. 2012, 2012, 609810. [Google Scholar]
- Gerhauser, C. Cancer chemoprevention and nutri-epigenetics: State of the art and future challenges. Top. Curr. Chem. 2013, 329, 73–132. [Google Scholar] [CrossRef]
- Kucharski, R.; Maleszka, J.; Foret, S.; Maleszka, R. Nutritional control of reproductive status in honeybees via DNA methylation. Science 2008, 319, 1827–1830. [Google Scholar] [CrossRef]
- Herb, B.R.; Wolschin, F.; Hansen, K.D.; Aryee, M.J.; Langmead, B.; Irizarry, R.; Amdam, G.V.; Feinberg, A.P. Reversible switching between epigenetic states in honeybee behavioral subcastes. Nat. Neurosci. 2012, 15, 1371–1373. [Google Scholar] [CrossRef]
- Ford, D. Honeybees and cell lines as models of DNA methylation and aging in response to diet. Exp. Gerontol. 2012. [Google Scholar] [CrossRef]
- Burrows, M.; Rogers, S.M.; Ott, S.R. Epigenetic remodelling of brain, body and behaviour during phase change in locusts. Neural. Syst. Circuits 2011, 1, 11. [Google Scholar] [CrossRef]
- Guo, W.; Wang, X.; Ma, Z.; Xue, L.; Han, J.; Yu, D.; Kang, L. CSP and takeout genes modulate the switch between attraction and repulsion during behavioral phase change in the migratory locust. PLoS Genet. 2011, 7, e1001291. [Google Scholar] [CrossRef]
- Ma, Z.; Guo, W.; Guo, X.; Wang, X.; Kang, L. Modulation of behavioral phase changes of the migratory locust by the catecholamine metabolic pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 3882–3887. [Google Scholar]
- Robinson, K.L.; Tohidi-Esfahani, D.; Lo, N.; Simpson, S.J.; Sword, G.A. Evidence for widespread genomic methylation in the migratory locust, Locusta migrat oria (Orthoptera: Acrididae). PLoS One 2011, 6, e28167. [Google Scholar]
- Geyer, M.A.; Vollenweider, F.X. Serotonin research: Contributions to understanding psychoses. Trends Pharmacol. Sci. 2008, 29, 445–453. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Noristani, H.N.; Verkhratsky, A. The serotonergic system in ageing and Alzheimer’s disease. Prog. Neurobiol. 2012, 99, 15–41. [Google Scholar] [CrossRef]
- Wurtman, R.J.; Wurtman, J.J.; Regan, M.M.; McDermott, J.M.; Tsay, R.H.; Breu, J.J. Effects of normal meals rich in carbohydrates or proteins on plasma tryptophan and tyrosine ratios. Am. J. Clin. Nutr. 2003, 77, 128–132. [Google Scholar]
- Le Floc’h, N.; Otten, W.; Merlot, E. Tryptophan metabolism, from nutrition to potential therapeutic applications. Amino Acids 2011, 41, 1195–1205. [Google Scholar] [CrossRef]
- Doron, R.; Lotan, D.; Rak-Rabl, A.; Raskin-Ramot, A.; Lavi, K.; Rehavi, M. Anxiolytic effects of a novel herbal treatment in mice models of anxiety. Life Sci. 2012, 90, 995–1000. [Google Scholar] [CrossRef]
- Gray, J.D.; Milner, T.A.; McEwen, B.S. Dynamic plasticity: The role of glucocorticoids, brain-derived neurotrophic factor and other trophic factors. Neuroscience 2012, in press. [Google Scholar]
- Numakawa, T.; Adachi, N.; Richards, M.; Chiba, S.; Kunugi, H. Brain-derived neurotrophic factor and glucocorticoids: Reciprocal influence on the central nervous system. Neuroscience 2012, in press. [Google Scholar]
- Suri, D.; Vaidya, V.A. Glucocorticoid regulation of brain-derived neurotrophic factor: Relevance to hippocampal structural and functional plasticity. Neuroscience 2012, in press. [Google Scholar]
- Hashimoto, K. Understanding depression: Linking brain-derived neurotrophic factor, transglutaminase 2 and serotonin. Expert Rev. Neurother. 2013, 13, 5–7. [Google Scholar] [CrossRef]
- Yoshida, T.; Ishikawa, M.; Niitsu, T.; Nakazato, M.; Watanabe, H.; Shiraishi, T.; Shiina, A.; Hashimoto, T.; Kanahara, N.; Hasegawa, T.; et al. Decreased serum levels of mature brain-derived neurotrophic factor (BDNF), but not its precursor proBDNF, in patients with major depressive disorder. PLoS One 2012, 7, e42676. [Google Scholar]
- Kutiyanawalla, A.; Terry, A.V., Jr.; Pillai, A. Cysteamine attenuates the decreases in TrkB protein levels and the anxiety/depression-like behaviors in mice induced by corticosterone treatment. PLoS One 2011, 6, e26153. [Google Scholar] [CrossRef]
- Pidsley, R.; Mill, J. Research highlights: Epigenetic changes to serotonin receptor gene expression in schizophrenia and bipolar disorder. Epigenomics 2011, 3, 537–538. [Google Scholar] [CrossRef]
- Carrard, A.; Salzmann, A.; Malafosse, A.; Karege, F. Increased DNA methylation status of the serotonin receptor 5HTR1A gene promoter in schizophrenia and bipolar disorder. J. Affect. Disord. 2011, 132, 450–453. [Google Scholar] [CrossRef]
- Abdolmaleky, H.M.; Yaqubi, S.; Papageorgis, P.; Lambert, A.W.; Ozturk, S.; Sivaraman, V.; Thiagalingam, S. Epigenetic dysregulation of HTR2A in the brain of patients with schizophrenia and bipolar disorder. Schizophr. Res. 2011, 129, 183–190. [Google Scholar] [CrossRef]
- Kaufman, J.; Yang, B.Z.; Douglas-Palumberi, H.; Grasso, D.; Lipschitz, D.; Houshyar, S.; Krystal, J.H.; Gelernter, J. Brain-derived neurotrophic factor-5-HTTLPR gene interactions and environmental modifiers of depression in children. Biol. Psychiatry 2006, 59, 673–680. [Google Scholar] [CrossRef]
- Kalueff, A.V.; Wheaton, M.; Ren-Patterson, R.; Murphy, D.L. Brain-derived neurotrophic factor, serotonin transporter, and depression: Comment on Kaufman et al. Biol. Psychiatry 2007, 61, 1112–1113; author reply 1113–1115. [Google Scholar]
- Van Den Hove, D.L.; Jakob, S.B.; Schraut, K.G.; Kenis, G.; Schmitt, A.G.; Kneitz, S.; Scholz, C.J.; Wiescholleck, V.; Ortega, G.; Prickaerts, J.; et al. Differential effects of prenatal stress in 5-Htt deficient mice: Towards molecular mechanisms of gene × environment interactions. PLoS One 2011, 6, e22715. [Google Scholar]
- Bellani, M.; Nobile, M.; Bianchi, V.; van Os, J.; Brambilla, P. G × E interaction and neurodevelopment II. Focus on adversities in paediatric depression: the moderating role of serotonin transporter. Epidemiol. Psychiatr. Sci. 2012, 22, 21–28. [Google Scholar]
- Owens, M.; Goodyer, I.M.; Wilkinson, P.; Bhardwaj, A.; Abbott, R.; Croudace, T.; Dunn, V.; Jones, P.B.; Walsh, N.D.; Ban, M.; Sahakian, B.J. 5-HTTLPR and early childhood adversities moderate cognitive and emotional processing in adolescence. PLoS One 2012, 7, e48482. [Google Scholar] [CrossRef]
- Jasinska, A.J.; Lowry, C.A.; Burmeister, M. Serotonin transporter gene, stress and raphe-raphe interactions: A molecular mechanism of depression. Trends Neurosci. 2012, 35, 395–402. [Google Scholar] [CrossRef]
- Heim, C.; Binder, E.B. Current research trends in early life stress and depression: Review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp. Neurol. 2012, 233, 102–111. [Google Scholar] [CrossRef]
- Kagias, K.; Nehammer, C.; Pocock, R. Neuronal responses to physiological stress. Front. Genet. 2012, 3, 222. [Google Scholar]
- Vedhara, K.; Metcalfe, C.; Brant, H.; Crown, A.; Northstone, K.; Dawe, K.; Lightman, S.; Smith, G.D. Maternal mood and neuroendocrine programming: Effects of time of exposure and sex. J. Neuroendocrinol. 2012, 24, 999–1011. [Google Scholar] [CrossRef]
- Sinclair, D.; Fullerton, J.M.; Webster, M.J.; Shannon Weickert, C. Glucocorticoid receptor 1B and 1C mRNA transcript alterations in schizophrenia and bipolar disorder, and their possible regulation by GR gene variants. PLoS One 2012, 7, e31720. [Google Scholar]
- Carrard, A.; Salzmann, A.; Perroud, N.; Gafner, J.; Malafosse, A.; Karege, F. Genetic association of the phosphoinositide-3 kinase in schizophrenia and bipolar disorder and interaction with a BDNF gene polymorphism. Brain Behav. 2011, 1, 119–124. [Google Scholar] [CrossRef]
- Nestler, E.J. Epigenetics: Stress makes its molecular mark. Nature 2012, 490, 171–172. [Google Scholar] [CrossRef]
- Aas, M.; Navari, S.; Gibbs, A.; Mondelli, V.; Fisher, H.L.; Morgan, C.; Morgan, K.; MacCabe, J.; Reichenberg, A.; Zanelli, J.; et al. Is there a link between childhood trauma, cognition, and amygdala and hippocampus volume in first-episode psychosis? Schizophr. Res. 2012, 137, 73–79. [Google Scholar] [CrossRef]
- Nosarti, C.; Reichenberg, A.; Murray, R.M.; Cnattingius, S.; Lambe, M.P.; Yin, L.; MacCabe, J.; Rifkin, L.; Hultman, C.M. Preterm birth and psychiatric disorders in young adult life. Arch. Gen. Psychiatry 2012, 69, E1–E8. [Google Scholar]
- Meredith, R.M.; Dawitz, J.; Kramvis, I. Sensitive time-windows for susceptibility in neurodevelopmental disorders. Trends Neurosci. 2012, 35, 335–344. [Google Scholar] [CrossRef]
- Kirkbride, J.B.; Susser, E.; Kundakovic, M.; Kresovich, J.K.; Davey Smith, G.; Relton, C.L. Prenatal nutrition, epigenetics and schizophrenia risk: Can we test causal effects? Epigenomics 2012, 4, 303–315. [Google Scholar] [CrossRef]
- Myers, M.G., Jr.; Olson, D.P. Central nervous system control of metabolism. Nature 2012, 491, 357–363. [Google Scholar] [CrossRef]
- Hunter, R.G. Epigenetic effects of stress and corticosteroids in the brain. Front. Cell. Neurosci. 2012, 6, 18. [Google Scholar] [CrossRef]
- Kino, T.; Hurt, D.E.; Ichijo, T.; Nader, N.; Chrousos, G.P. Noncoding RNA Gas5 is a growth arrest and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal. 2010, 3, ra8. [Google Scholar] [CrossRef]
- Jiang, X.; Yan, J.; West, A.A.; Perry, C.A.; Malysheva, O.V.; Devapatla, S.; Pressman, E.; Vermeylen, F.; Caudill, M.A. Maternal choline intake alters the epigenetic state of fetal cortisol-regulating genes in humans. FASEB J. 2012, 26, 3563–3574. [Google Scholar] [CrossRef]
- Nakaoka, H.; Cui, T.; Tajima, A.; Oka, A.; Mitsunaga, S.; Kashiwase, K.; Homma, Y.; Sato, S.; Suzuki, Y.; Inoko, H.; Inoue, I. A systems genetics approach provides a bridge from discovered genetic variants to biological pathways in rheumatoid arthritis. PLoS One 2011, 6, e25389. [Google Scholar] [CrossRef]
- Harris, J.R.; Burton, P.; Knoppers, B.M.; Lindpaintner, K.; Bledsoe, M.; Brookes, A.J.; Budin-Ljosne, I.; Chisholm, R.; Cox, D.; Deschenes, M.; et al. Toward a roadmap in global biobanking for health. Eur. J. Hum. Genet. 2012, 20, 1105–1111. [Google Scholar] [CrossRef]
- Groenen, M.A.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gaillard, C.; Park, C.; Milan, D.; Megens, H.J.; et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012, 491, 393–398. [Google Scholar]
- Al-Harthi, L. Wnt/beta-catenin and its diverse physiological cell signaling pathways in neurodegenerative and neuropsychiatric disorders. J. Neuroimmune Pharmacol. 2012, 7, 725–730. [Google Scholar] [CrossRef]
- Van Alphen, B.; van Swinderen, B. Drosophila strategies to study psychiatric disorders. Brain Res. Bull. 2013, 92, 1–11. [Google Scholar] [CrossRef]
- Jin, K.; Xue, C.; Wu, X.; Qian, J.; Zhu, Y.; Yang, Z.; Yonezawa, T.; Crabbe, M.J.; Cao, Y.; Hasegawa, M.; et al. Why does the giant panda eat bamboo? A comparative analysis of appetite-reward-related genes among mammals. PLoS One 2011, 6, e22602. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dauncey, M.J. Genomic and Epigenomic Insights into Nutrition and Brain Disorders. Nutrients 2013, 5, 887-914. https://doi.org/10.3390/nu5030887
Dauncey MJ. Genomic and Epigenomic Insights into Nutrition and Brain Disorders. Nutrients. 2013; 5(3):887-914. https://doi.org/10.3390/nu5030887
Chicago/Turabian StyleDauncey, Margaret Joy. 2013. "Genomic and Epigenomic Insights into Nutrition and Brain Disorders" Nutrients 5, no. 3: 887-914. https://doi.org/10.3390/nu5030887
APA StyleDauncey, M. J. (2013). Genomic and Epigenomic Insights into Nutrition and Brain Disorders. Nutrients, 5(3), 887-914. https://doi.org/10.3390/nu5030887