1. History of the Milk-Alkali Syndrome
In 1915, Bertram Welton Sippy introduced a cocktail to treat peptic ulcer disease with hourly administration of milk and cream with Sippy powders, which were a mixture of sodium bicarbonate with calcinated magnesia or bismuth subcarbonate [
1]. With the Sippy diet, about 32 g of sodium bicarbonate and 10–32 g of calcium carbonate (4 to 13 g of elemental calcium) were administered daily [
2]. As Sippy’s treatment became prevalent, some patients who were treated with high doses of calcium and sodium bicarbonate developed a constellation of symptoms including headache, nausea, vomiting, light-headedness, anorexia, distaste for milk, musculoskeletal pains, weakness, dizziness, mental clouding, and renal failure [
2,
3]. Later, in 1949, Burnett
et al. [
4] described these symptoms as part of “milk and alkali syndrome”. The most common findings were hypercalcemia, metabolic alkalosis, renal insufficiency, and soft tissue calcium deposits.
Three different progressive stages have been described. First, there is an acute form, referred to as “toxemia”, which occurs 2–30 days after calcium ingestion. This stage has been characterized by irritability, vertigo, apathy, headaches, weakness, myalgias, and vomiting. Second, the intermediate phase, called “Cope’s syndrome,” includes symptoms of the acute form plus conjunctivitis. Last is the chronic form, or Burnett’s syndrome, which is characterized by soft tissue calcification including conjunctivitis, band keratopathy of the cornea, musculoskeletal deposits, and nephrocalcinosis [
5].
3. Populations at Risk
Whereas the traditional milk-alkali syndrome affected younger male patients with peptic ulcer disease [
12,
13], the demographics have changed to post-menopausal women, solid organ transplant recipients, pregnant women, bulimic patients, and those on dialysis [
6,
11,
12,
13,
14]. Post-menopausal women and solid organ transplant recipients are encouraged to take calcium supplementation along with vitamin D for the prevention and treatment of osteoporosis. Post-menopausal women also have decreased estrogen which results in bone loss and decreased calcium absorption. Estrogen may also upregulate the transient receptor potential vanilloid member 5 (TRPV5), a channel that is critical in calcium uptake in the kidney, resulting in enhanced calcium-reabsorption [
15]. Thus, lack of estrogen in and following the menopausal transition may result in a rise in obligatory urinary calcium loss, reduced intestinal calcium absorption, negative calcium balance, and increased risk of osteoporosis [
16].
Elderly patients are at risk of developing calcium-alkali syndrome relating to aging bone metabolism and reduced renal function, leading to impaired bone mineralization and reduced calcium excretion, respectively. Pregnant women are susceptible to calcium-alkali syndrome due to hyperemesis, which causes volume depletion and metabolic alkalosis, and increased calcium absorption through the gastrointestinal tract, which is thought to be mediated by prolactin, placental lactogen, and increased 1,25-dihydroxyvitamin D [
17]. Bulimic patients are a high-risk group for developing calcium-alkali syndrome related to metabolic alkalosis that is associated with vomiting and diuretic abuse, as well as poor eating habits. In the Far East, Asia, and South Pacific, calcium-alkali syndrome has been reported in betel nut chewers. Worldwide, betel nut is used by an estimated 600 million people [
18]. The lime paste added to the nut is made from ground oyster shells which consist of calcium oxide and calcium hydroxide [
19].
4. Physiology
The integral features of calcium-alkali syndrome are excess ingestion of calcium and often absorbable alkali leading to the classic triad of hypercalcemia, metabolic alkalosis, and varying degrees of renal insufficiency [
10,
13,
20]. Initially, the traditional milk-alkali syndrome was characterized by elevated serum phosphate concentration, probably secondary to the ingestion of phosphate-rich milk and cream in the Sippy diet [
21]. In contrast, calcium-alkali syndrome is associated with hypophosphatemia or low-normal serum phosphorus level as a result of the phosphate-binding capacity of calcium carbonate and other calcium containing supplements resulting in decreased phosphate absorption. These effects are more pronounced in elderly patients and patients with eating disorders who have reduced dietary phosphate and protein intake [
6]. The vast majority of cases are associated with calcium carbonate supplements [
10].
The pathogenesis of calcium-alkali syndrome is intricate and involves the interplay of multiple systems including intestine, kidney, and bone [
6,
22]. An average healthy adult contains about 1 kg of calcium (or 25,000 mmol) with over 99% located in the bone and less than 1% (20 mmol) in the extracellular fluid [
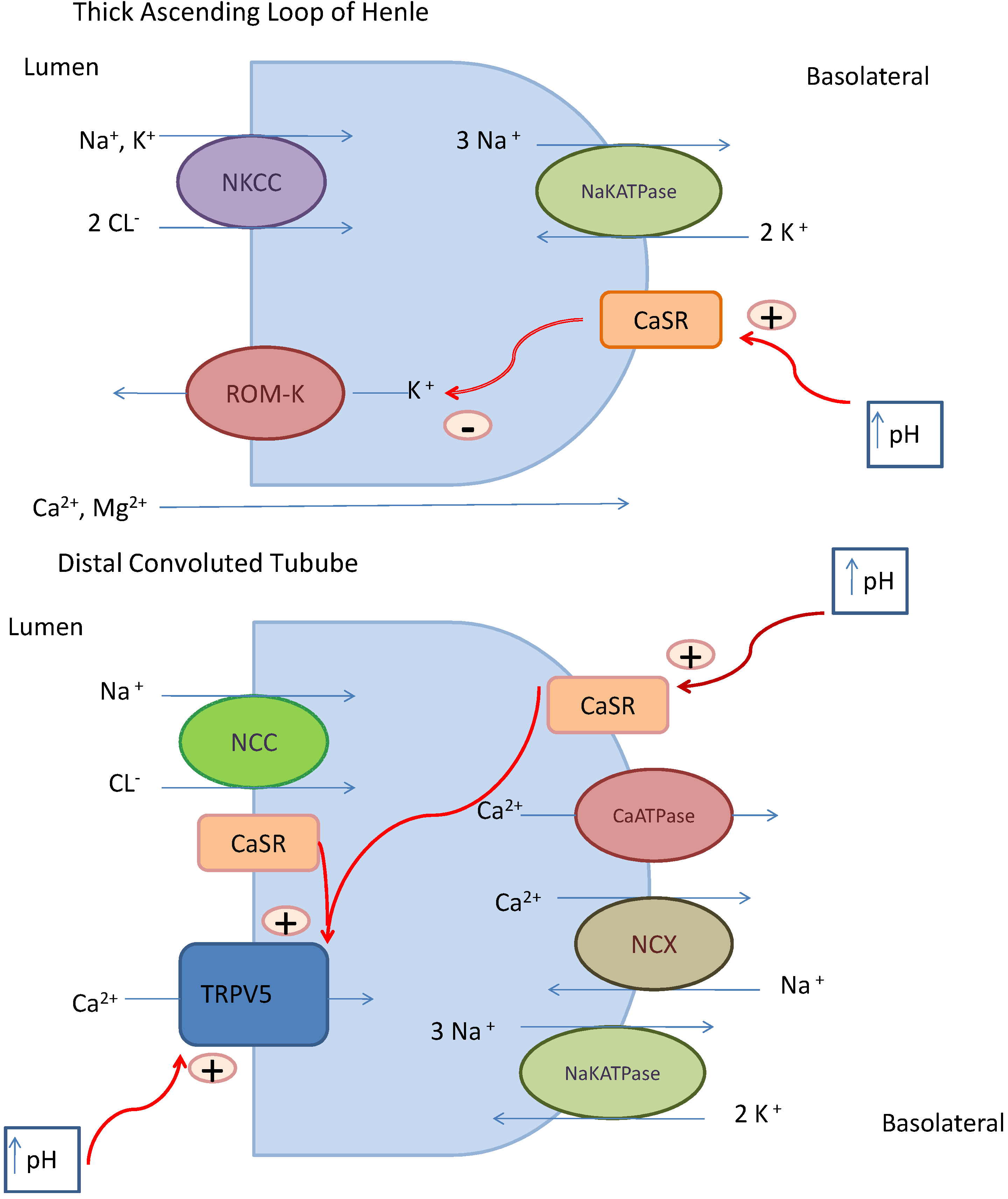
22]. The kidney plays an important role in calcium balance. Fine-tuning of calcium reabsorption occurs in the distal convoluted tubule of the nephron under the influence of several factors, including parathyroid hormone, calcium, calcitriol, and calcitonin. Nevertheless, the rate-limiting step in renal calcium reabsorption is the TRPV5 calcium channel activity and to a lesser extent, TRPV6, both located on the luminal side of the distal tubule. Through TRPV5, calcium enters the cell, is carried across the cell cytoplasm by calbindin D23. Calcium ions then exits through a sodium-calcium exchanger or calcium ATPase pump located on the basolateral side (
Figure 1) [
6].
Figure 1.
Mechanisms of renal calcium transport [
9]. In the thick ascending limb of Henle’s loop, the creation of a lumen positive voltage tends to drive Ca
2+ and Mg
2+ through a paracellular path and thereby produces a high rate of reabsorption of the divalent ions. The CaSR modulates this transport activity by altering rates of K efflux and thereby contributing to the lumen positive voltage. In the distal convoluted tubule, a three-step process facilitates active and transcellular Ca
2+ transport. The first step is entry of luminal Ca
2+ at the apical side of the cell through the TRPV5 channel. Subsequently, calbindin, a calcium binding protein buffers Ca
2+ and the Ca
2+ diffuses to the basolateral membrane. At the basolateral membrane, Ca
2+ is extruded by across the basolateral membrane by NCX. This process is controlled by calciotropic hormones, including parathyroid hormone and 1,25(OH)2D3. Abbreviations: NKCC, sodium potassium-2-chloride co-transporter; ROM-K, renal outer medullary potassium channel; CaSR, calcium sensing receptor; NaKATPase, sodium potassium ATPase; +, stimulates; −, inhibits; NCC, sodium chloride co-transporter; NCX, sodium calcium exchanger; TRPV5, transient receptor potential vanilloid membrane calcium channel. (Note: Permission obtained from [
9], Copyright American Society of Nephrology.)
Figure 1.
Mechanisms of renal calcium transport [
9]. In the thick ascending limb of Henle’s loop, the creation of a lumen positive voltage tends to drive Ca
2+ and Mg
2+ through a paracellular path and thereby produces a high rate of reabsorption of the divalent ions. The CaSR modulates this transport activity by altering rates of K efflux and thereby contributing to the lumen positive voltage. In the distal convoluted tubule, a three-step process facilitates active and transcellular Ca
2+ transport. The first step is entry of luminal Ca
2+ at the apical side of the cell through the TRPV5 channel. Subsequently, calbindin, a calcium binding protein buffers Ca
2+ and the Ca
2+ diffuses to the basolateral membrane. At the basolateral membrane, Ca
2+ is extruded by across the basolateral membrane by NCX. This process is controlled by calciotropic hormones, including parathyroid hormone and 1,25(OH)2D3. Abbreviations: NKCC, sodium potassium-2-chloride co-transporter; ROM-K, renal outer medullary potassium channel; CaSR, calcium sensing receptor; NaKATPase, sodium potassium ATPase; +, stimulates; −, inhibits; NCC, sodium chloride co-transporter; NCX, sodium calcium exchanger; TRPV5, transient receptor potential vanilloid membrane calcium channel. (Note: Permission obtained from [
9], Copyright American Society of Nephrology.)
![]()
Hypercalcemia occurs when the influx of calcium into extracellular fluid exceeds the excretion by the kidney [
22]. Calcium-alkali syndrome can be divided into generation and maintenance phases. In the generation phase, the intestinal component is the most important since it is the site of calcium absorption. Factors affecting the amount of calcium absorption include dietary intake, vitamin D level, and acidity. An acidic environment increases the availability of free calcium for absorption in the gut, predominantly in the proximal small intestine through a combination of active vitamin D-dependent transport (saturable) and passive unregulated paracellular diffusion [
6,
16]. Therefore, meals that raise gastric acidity such as animal protein and carbohydrate-based meals are associated with increased calcium absorption [
6]. The threshold amount of calcium generally considered to predispose to calcium-alkali is above 4 g; however, there are reports of calcium-alkali with as little as 1–1.5 g of calcium supplementation, a dose that is consistent with many dietary guidelines [
11,
13,
23].
Once calcium is absorbed into the extracellular fluid, uptake occurs in the bone envelope but more slowly in structural bone [
24]. However, following post-epiphyseal closure, the ability of bone to buffer calcium loads diminishes. In the young adult and child, calcium preferentially deposits in the bone with high intake and renal calcium excretion is minimal to allow a positive calcium balance for growth. However, in the aging individual, the net flux of calcium is out of bone, making bone less available for buffering excessive amounts of calcium [
22]. Thus, the elderly are more susceptible to the calcium-alkali syndrome. Natural protective mechanisms against the development of calcium-alkali syndrome also operate in elderly individuals, including decreased intestinal absorption of calcium, decreased sensitivity to vitamin D supplementation, and reduced gastric acidity [
6].
In the event that hypercalcemia occurs, counter-regulatory mechanisms re-establish normocalcemia. First, the high calcium level suppresses parathyroid hormone (PTH) secretion, which reduces bone efflux of calcium. A decreased PTH concentration also functions to suppress calcitriol production, resulting in decreased intestinal absorption of calcium. Despite these modifying mechanisms, other features of the calcium-alkali syndrome may further perpetuate the syndrome. Hypercalcemia-mediated renal vasoconstriction causes a decline in glomerular filtration rate, filtered load of calcium, and ultimately, reduced calcium excretion in a self-propagating cycle [
13]. Furthermore, an enhanced reabsorption of any filtered calcium in the proximal tubule operates via a paracellular pathway through solvent drag in addition to enhanced reabsorption of bicarbonate. Alkaline tubular pH also sensitizes the TRPV5 channel, resulting in enhanced calcium absorption in the distal tubule (see
Figure 1) [
6].
Hypercalcemia affects water and salt conservation. Hypercalcemia causes a furosemide-like effect by inhibiting secretory potassium channel activity via actions initiated by the calcium-sensing receptor (CaSR) in the thick ascending limb of Henle’s loop (
Figure 1). Without the luminal positive voltage caused by potassium efflux, the voltage driving force is obliterated and there is less sodium chloride reabsorption through the sodium potassium-2-chloride co-transporter (NKCC) channel [
18]. Metabolic alkalosis enhances the sensitivity of the CaSR to hypercalcemia. Moreover, hypercalcemia causes nephrogenic diabetes insipidus by inhibiting vasopressin action at the V2 receptor in the collecting duct [
25]. These mechanisms lead to salt and water losses and, therefore, they contribute to volume depletion which further stimulates proximal tubular calcium reabsorption. Over time, chronic renal ischemia induced by the calcium-alkali syndrome can result in irreversible renal dysfunction. In addition, renal dysfunction can occur as a result of calcium precipitation in the kidney, manifested as nephrocalcinosis. Calcium deposits may also develop in other tissues, such as the vasculature, cardiac valves, and the cornea [
13].
Commonly used medications can influence calcium retention, predisposing some individuals to the development of calcium alkali syndrome. Thiazide diuretics directly enhance calcium absorption in the proximal tubule of the nephron by promoting volume depletion and subsequent metabolic alkalosis. Metabolic alkalosis favors calcium absorption, as described previously. Agents that decrease glomerular filtration rate can predispose to calcium-alkali by decreasing calcium excretion. Inhibitors of the renin-angiotensin system, widely used for the treatment of hypertension, cardiovascular disease, and renal disease, can result in a decline in glomerular filtration rate. Non-steroidal anti-inflammatory drugs such as ibuprofen and naproxen, can also decrease glomerular filtration rate and subsequent calcium excretion via their inhibitory effects on prostaglandins [
6]. Consumption of absorbable alkali such as aluminum hydroxide and magnesium hydroxide, components of over-the-counter dyspepsia remedies, can also increase tubular calcium reabsorption in the kidney via their effects on pH. With an increase in pH, the affinity of the calcium-sensing receptor for calcium is increased which initiates a cascade of events that promotes calcium reabsorption in both the proximal and distal tubule of the nephron (see
Figure 1) [
9].
6. Role of Supplemental Calcium Intake
On average, men and women above the age of 50 years consume about 750–950 mg of elemental calcium per day in their diet. About 60% of the ingested calcium comes from dairy products [
27]. Around 40% of the U.S. population, and almost 70% of older women, use calcium-containing dietary supplements [
27]. Due to the widespread use of calcium supplementation for osteoporosis prevention and treatment, about 5% of women older than 50 years have an estimated calcium intake that exceeds the Tolerable Upper Intake Level for calcium (established by the Institute of Medicine, Washington, DC, USA [
28]) by a substantial amount each day,
i.e., total calcium intake may exceed 2300 mg per day, according to NHANES (National Health and Nutrition Examination Survey, Centers for Disease Control and Prevention, Atlanta, GA, USA) data from 2003 to 2006 [
27,
29]. Chronic exposure to high calcium intake can lead to a positive calcium balance, especially in the elderly, because of reduced renal function with resultant hypocalciuria and aging bone metabolism with a decreased capacity to take up the calcium load.
The beneficial effect of calcium supplements on the prevention of osteoporotic fracture is modest. In a meta-analysis by Bischoff-Ferrari
et al. [
30], hip fracture risk was not reduced significantly with increased calcium intake from 800 to 1600 mg (RR 0.92 with 95% CI 0.81–1.05). Furthermore, calcium carbonate and citrate can reduce phosphate absorption, which may be detrimental for bone mineralization. Therefore, the benefits of calcium supplementation on osteoporosis should be weighed against the potential risks. In most cases, calcium intake is not rate-limiting for bone formation and modest improvements in bone mineral density is not accompanied by reduction in rates of bone fracture. According to the Institute of Medicine, risk of harm increases when calcium intake surpasses 2000 mg per day and the vitamin D intake is greater than 4000 IUs per day [
28]. As per the U.S. Preventive Services Task Force (Rockville, MD, USA), evidence is insufficient for the benefit beyond 400 IU of vitamin D3 and more than 1000 mg of calcium for the primary prevention of fractures in non-institutionalized postmenopausal women [
31]. According to the National Osteoporosis Foundation (Washington, DC, USA), dietary calcium intake in excess of 1200–1500 mg may increase the risk for cardiovascular disease or kidney stones [
8].
Table 1 shows the Recommended Dietary Allowances for calcium and vitamin D intake by age groups [
28].
Table 1.
Recommended dietary allowances (RDAs) for calcium and vitamin D [
28].
Table 1.
Recommended dietary allowances (RDAs) for calcium and vitamin D [28].
| Age | Calcium (mg) | Vitamin D (IU) |
|---|
| Infants |
| 0–6 months | 200 | 400 |
| 6–12 months | 260 | 400 |
| 1–3 years | 700 | 600 |
| 4–8 years | 1000 | 600 |
| 9–18 years | 1300 | 600 |
| 19–50 years | 1000 | 600 |
| Pregnancy | 1000 | 600 |
| 51–70 Years |
| Male | 1000 | 600 |
| Female | 1200 | 600 |
| Greater than 70 years | 1200 | 800 |
The quantity of elemental calcium contained in various popular calcium supplements is summarized in
Table 2 [
9]. Calcium carbonate is more commonly available and is inexpensive. It contains about 40% of elemental calcium and requires stomach acid for absorption; therefore, calcium carbonate should be taken with meals. On the other hand, calcium citrate is considered the better absorbed calcium supplement because it does not require extra acid for absorption and can be taken on empty or full stomach [
32]. However, it contains 21% elemental calcium [
29].
Table 2.
Amount of elemental calcium in various supplements [
9]. Caltrate
® (Pfizer, Kings Mountain, USA); Centrum
® (Pfizer, Kings Mountain, NC, USA); Rolaids
® (Chattem Inc., Chattanooga, TN, USA); Os-Cal
® (GlaxoSmithKline, Brentford, UK); Tums
® (GlaxoSmithKline, Brentford, UK); Tums Ultra
® (GlaxoSmithKline, Brentford, UK); Viactiv
® (McNeil Nutritionals, Fort Washington, DC, USA); Citracal
® (Bayer, Pittsburg, PN, USA); Phoslo
® (Fresenius Medical Care North America, Waltham, MA, USA). (Note: Permission obtained from [
9], Copyright American Society of Nephrology).
Table 2.
Amount of elemental calcium in various supplements [9]. Caltrate® (Pfizer, Kings Mountain, USA); Centrum® (Pfizer, Kings Mountain, NC, USA); Rolaids® (Chattem Inc., Chattanooga, TN, USA); Os-Cal® (GlaxoSmithKline, Brentford, UK); Tums® (GlaxoSmithKline, Brentford, UK); Tums Ultra® (GlaxoSmithKline, Brentford, UK); Viactiv® (McNeil Nutritionals, Fort Washington, DC, USA); Citracal® (Bayer, Pittsburg, PN, USA); Phoslo® (Fresenius Medical Care North America, Waltham, MA, USA). (Note: Permission obtained from [9], Copyright American Society of Nephrology).
| Type | Trade Name | Elemental Calcium (mg) | Vitamin D (IU) |
|---|
| Calcium carbonate | Caltrate® | 600 | 400
400
800 |
| Centrum® | 200 |
| Centrum® Ultra Women’s | 500 |
| Rolaids® Extra Strength | 471 |
| Os-Cal® | 500 |
| Tums® | 200 |
| Tums Ultra® | 400 |
| Viactiv® | 500 |
| Calcium citrate | Citracal® Regular with Vit D | 250 | 200 |
| Calcium acetate | Phoslo® | 167 | - |
Calcium supplements can raise serum calcium levels to a modest degree. Changes in serum calcium level are often reflected by opposite but magnified and sustained responses in the PTH level [
32]. An acute load of calcium above 400 mg has been shown to suppress serum PTH secretion for as long as 8–10 h [
33]. PTH changes sometimes occur before a calcemic response is detected [
32]. On a 1300 mg calcium diet for one week, fasting serum calcium level increased by 3% in one small study by Martini
et al. [
34]. 1 g of calcium as either citrate or as lactate-gluconate raised the mean ionized calcium level from 1.22 to 1.30 mmol/L [
35]. The less absorbable calcium carbonate increased ionized calcium by approximately 0.05 mmol/L, and the same calcium load as bone meal only raised it by 0.03 mmol/L. In comparison, dairy products increased ionized calcium by only one-sixth of the level seen following consumption of soluble calcium supplement with comparable calcium content [
35]. Ingesting 1200 mg calcium as fortified skim milk increased the total (not ionized) serum calcium level by only 0.03 mmol/L [
35]. However, in another study by Martini
et al. [
34] involving 12 subjects, no difference was observed in the postprandial total calcium level and the degree of PTH reduction between orange juice fortified with calcium-citrate malate, skim milk, or calcium carbonate. Equal doses of calcium carbonate and calcium citrate produced a similar increment in total serum calcium and reduction in the PTH level [
36]. In a study by Hanzlik
et al. [
32], 1200 mg calcium carbonate produced a serum calcium rise of about 4% and decreased the PTH level by about 20%–40%; nevertheless, these changes did not differ significantly from placebo. Administration of similar dose of calcium citrate caused the serum calcium to rise about 9% (
p = 0.001 [
32]. In summary, varying effects on the serum calcium and PTH concentrations result from calcium supplementation compared to dietary calcium intake from food, and certain calcium supplements, such as calcium formate, have even a more pronounced elevation of serum calcium than other supplements [
36].
In general, ingestion of equivalent doses of calcium from dairy products has a smaller effect on serum calcium level than calcium supplements, which may explain the lack of detrimental vascular effect by dietary calcium intake as found in observational studies [
35,
37,
38]. Calcium in food is less bioavailable because of the presence of calcium-binding agents, such as oxalic acid, phytates, fiber, and phosphate. Oxalic acid, which exists in some plant foods, binds to calcium directly, making calcium less available for absorption. Foods with high amounts of oxalic acid such as spinach, collard greens, sweet potatoes, rhubarb, and beans actually permit little calcium absorption. Phytates can also bind to the calcium, decreasing its absorption. Foods high in phytates include whole-grain products, wheat bran, beans, seeds, nuts, and soy isolates. Unlike oxalic acid, phytates will bind the calcium from other food sources consumed at the same meal. Furthermore, fibers such as in wheat bran, can also bind to the calcium in the intestine and decrease its absorption [
16,
29]. Individuals may also ingest medications with phosphate binding properties such as aluminum hydroxide, increasing the bioavailability of calcium for absorption [
39].
Due to stimulation of active enteric absorption of calcium by 1,25-dihydroxyvitamin D, a higher percentage of calcium is absorbed from smaller doses of calcium (below 500 mg) than from larger calcium loads. Increases in serum ionized calcium were similar after loads of 1000 and 2000 mg calcium, which results from saturation of the vitamin-D mediated active mechanism of calcium absorption in the intestinal tract [
33]. As calcium intake increases, net absorbed calcium increases steeply until active transport becomes saturated, after which the percentage of calcium absorbed drops. Therefore, the peak absorption of calcium occurs at around a 400 mg calcium load and then it levels off. Absorption of calcium may be as high as 60% at very low intake and as low as 20% at a high intake [
40]. 1,25-Dihydroxyvitamin D (hormonal form) may also mediate enteric calcium absorption [
41], and according to the NHANES 2003–2006 data, 37% of the U.S. population used a vitamin D supplement [
27].
7. Long-Term Sequelae of Calcium Supplementation
Chronic ingestion of moderate to high doses of calcium supplementation has been found to have variable effects on cardiovascular morbidity and mortality. In a study by Wang
et al. [
42], 1471 healthy post-menopausal women were randomized to calcium 600 mg or 1200 mg per day. Although increased serum calcium and phosphate levels were associated with aortic calcification in elderly women, neither dietary calcium intake nor calcium supplementation was associated with changes in aortic calcification in this trial [
42]. In the Women’s Health Initiative study involving nearly 36,000 postmenopausal women followed for seven years randomized to 1 g of calcium carbonate supplementation with 400 IU vitamin D, there was no increase in cardiovascular events (HR 1.04, 95% CI 0.92–1.18) [
43]. On the other hand, a meta-analysis of 15 trials by Bolland
et al. [
38] found that calcium supplementation of 500 mg or greater was associated with hazard ratio of 1.31 for myocardial infarction (95% CI 1.02–1.67,
p = 0.035). The authors of this meta-analysis concluded that the number needed to treat with calcium for five years to cause one event was 69 for myocardial infraction, 100 for stroke, and 77 for death while the number needed to treat to prevent one fracture was 39 [
38]. Interestingly, supplemental calcium treatment was associated with increased risk of myocardial infarction in people with dietary intake above the median of 805 mg/day (HR 1.85, 95% CI 1.28–2.67), but there was no increased risk in those with dietary intake intakes below the median (HR 0.98, 95% CI 0.69–1.38). This finding suggests that a threshold may exist above which combined dietary and supplemental calcium may result in increased cardiovascular risk and that the higher risk may be due to higher calcium from supplements rather than from dietary calcium. This meta-analysis did not examine studies with vitamin D supplementation [
38]. In a recent prospective study by Li
et al. [
44] of the Heidelberg cohort of 23,980 participants, dietary calcium did not raise the risk for cardiovascular disease; however, calcium supplementation was significantly associated with increased risk for myocardial infarction (HR 1.86, 95% CI 1.12–5.12). Again, this article indicates that calcium supplements rather than dietary calcium might confer increased cardiovascular risk.
Calcium supplements may contribute to vascular calcification by causing a positive calcium balance as evidenced by a reduction in PTH and a modest rise in serum calcium level. Serum calcium is positively correlated with carotid artery plaque thickness, aortic calcification, incidence of myocardial infarction, and mortality [
45,
46]. It is possible that an increase in serum calcium level from supplements induces vascular calcification by altering inhibitors such as fetuin-A, matrix Gla protein, pyrophosphate, osteoprotegerin, and bone morphogenic protein-7. It is important to recognize that 29 million Americans have stage 3 chronic kidney disease. In these individuals fetuin-A and matrix Gla protein are frequently reduced, thus, promoting the development and progression of vascular calcification [
47]. In patients with renal failure, calcium supplements have been well established to worsen vascular calcification and increase mortality in both dialysis and pre-dialysis populations. Serum calcium can also directly bind to the calcium-sensing receptor that is expressed on vascular smooth muscle cells [
35]. In a study by Alam
et al. [
39], exposing vascular smooth muscles cells
in vitro to high calcium level caused increased mineralization.


{kind=link}