Cobalamin Deficiency: Clinical Picture and Radiological Findings

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Vitamin B12: Physiology and Causes of Deficiency
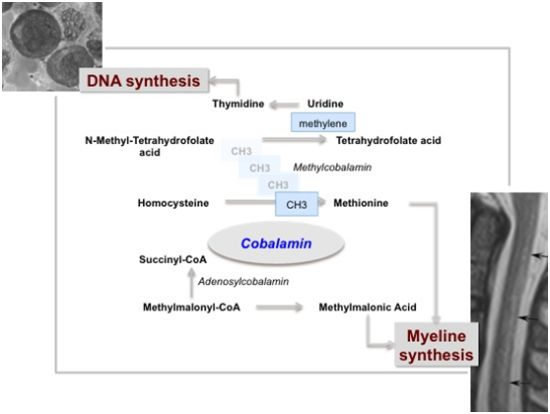
2.1. Cobalamin-Folate Relationships
2.2. Causes of Cobalamin Deficiency
2.3. Diseases Leading to Insufficient Cobalamin Absorption
2.4. Clinical Tests to Evaluate Cobalamin Absorption
2.4.1. The Schilling Test
2.4.2. Study of Gastric Functions
2.5. Biochemical Indicators of Cobalamin Deficiency
3. The Multifaceted Clinical Presentation of Cobalamin Deficiency
3.1. Biological and Morphological Expressions of Cobalamin Deficiency
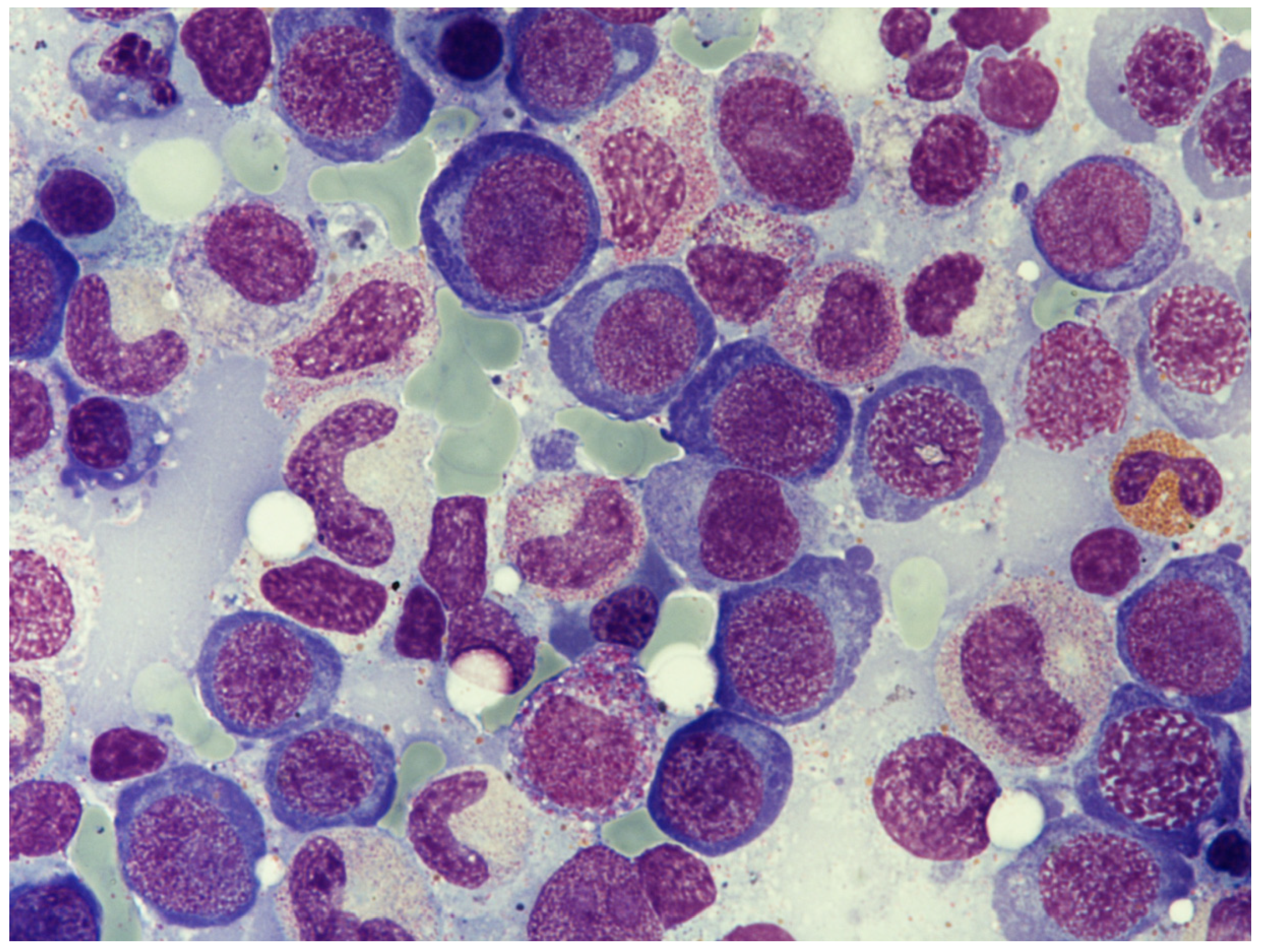
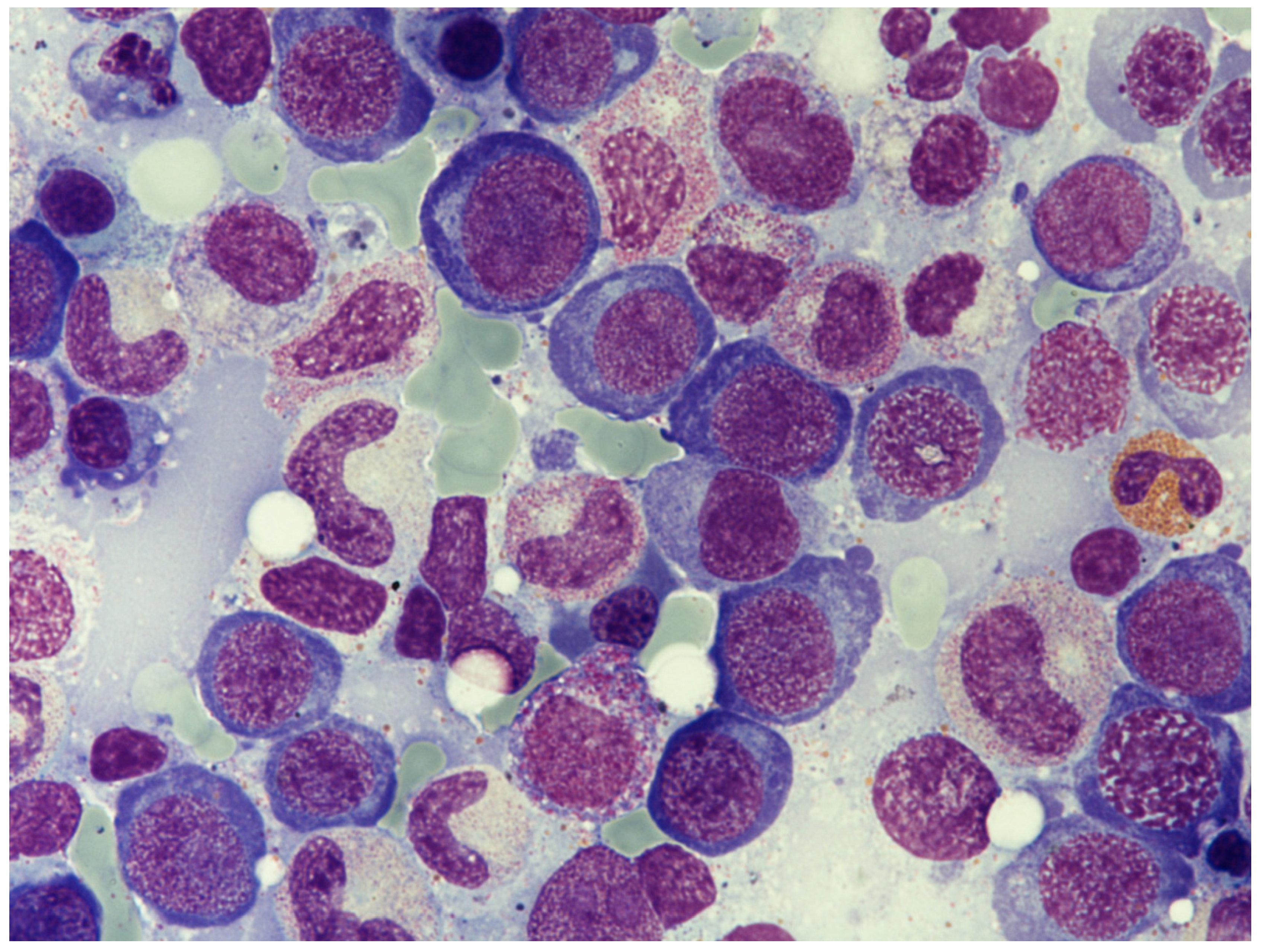
3.2. Hematological Manifestation of Cobalamin Deficiency
3.3. Neurological Manifestation of Cobalamin Deficiency
3.4. Other Effects of Cobalamin Deficiency
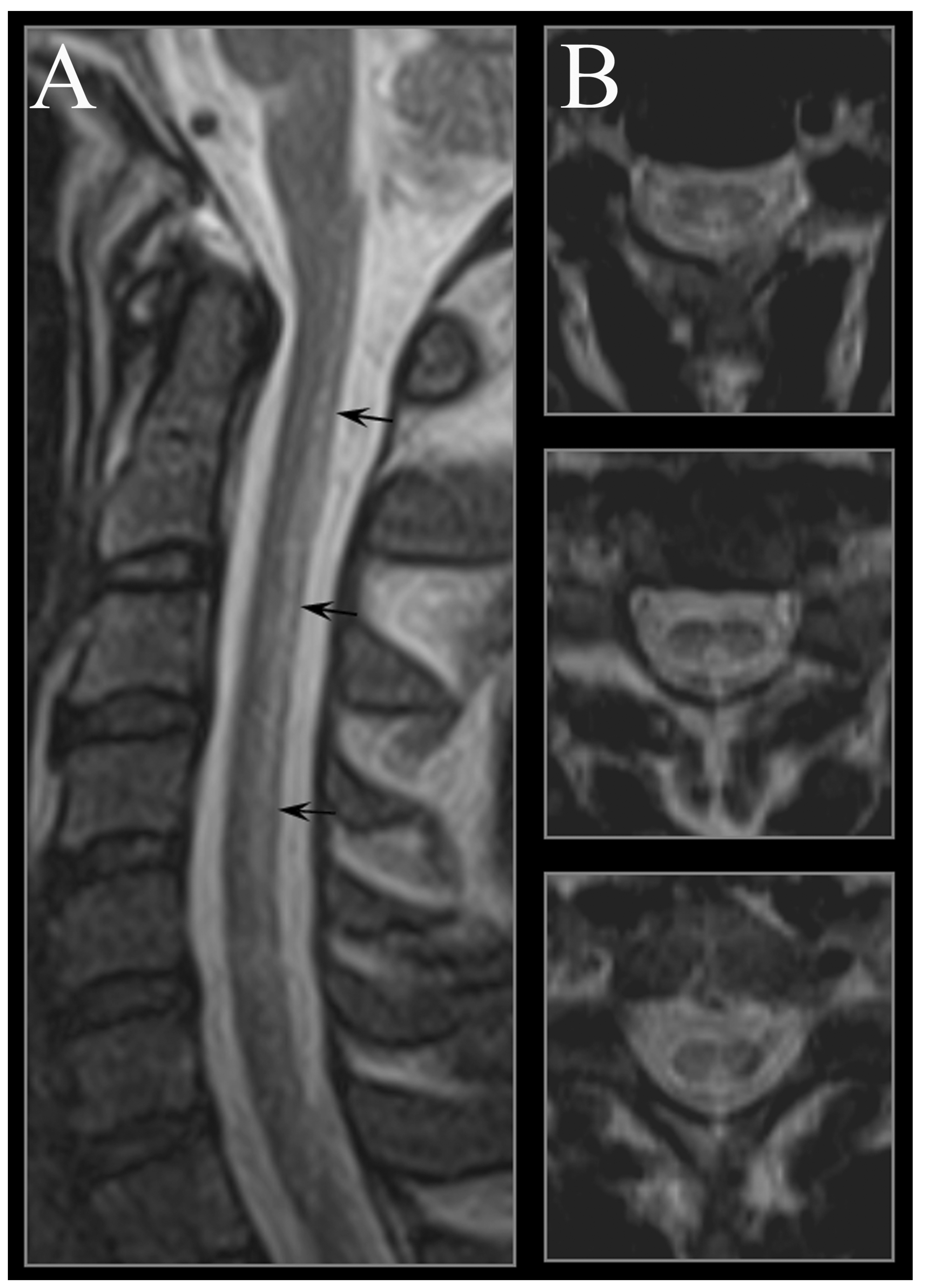
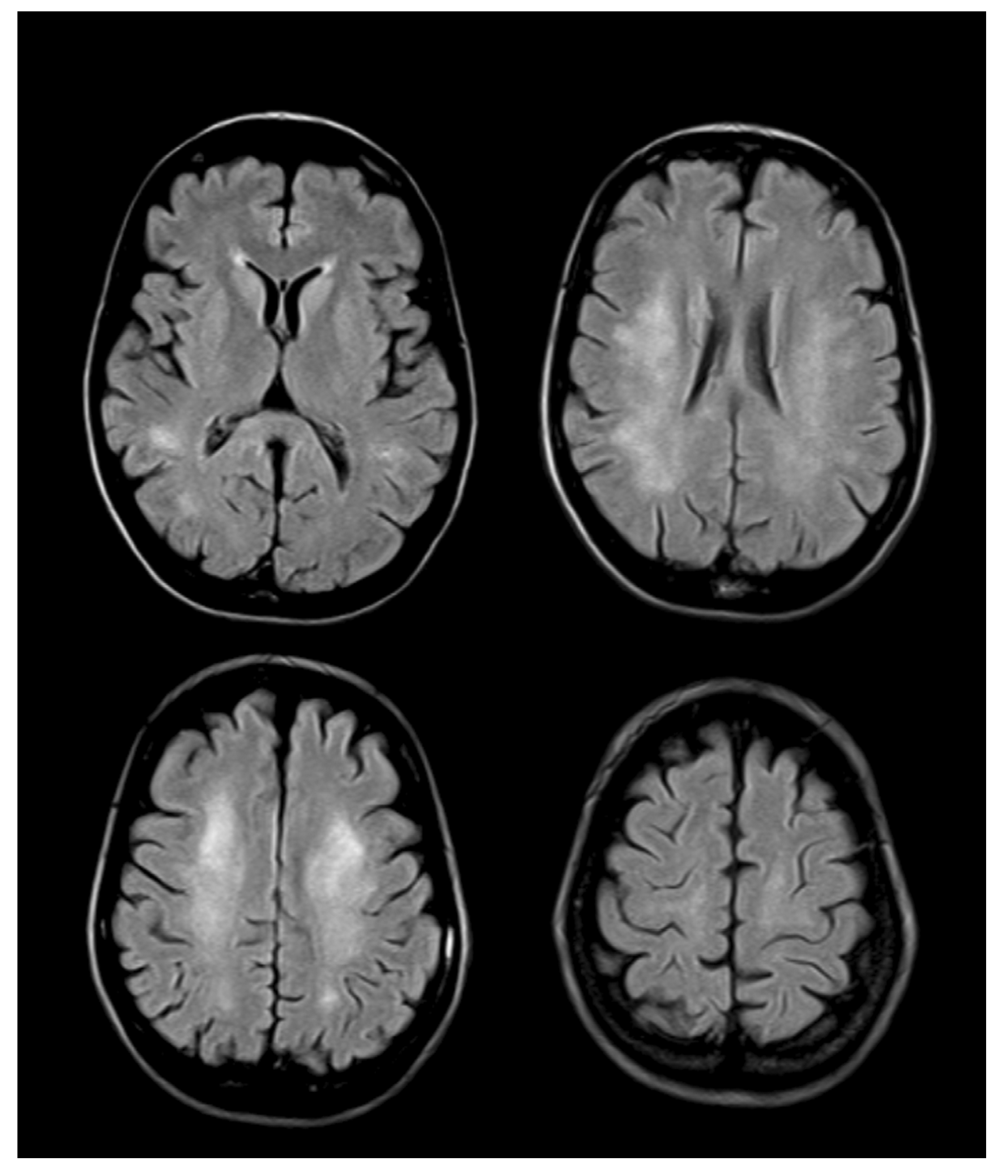
4. Neuroimaging

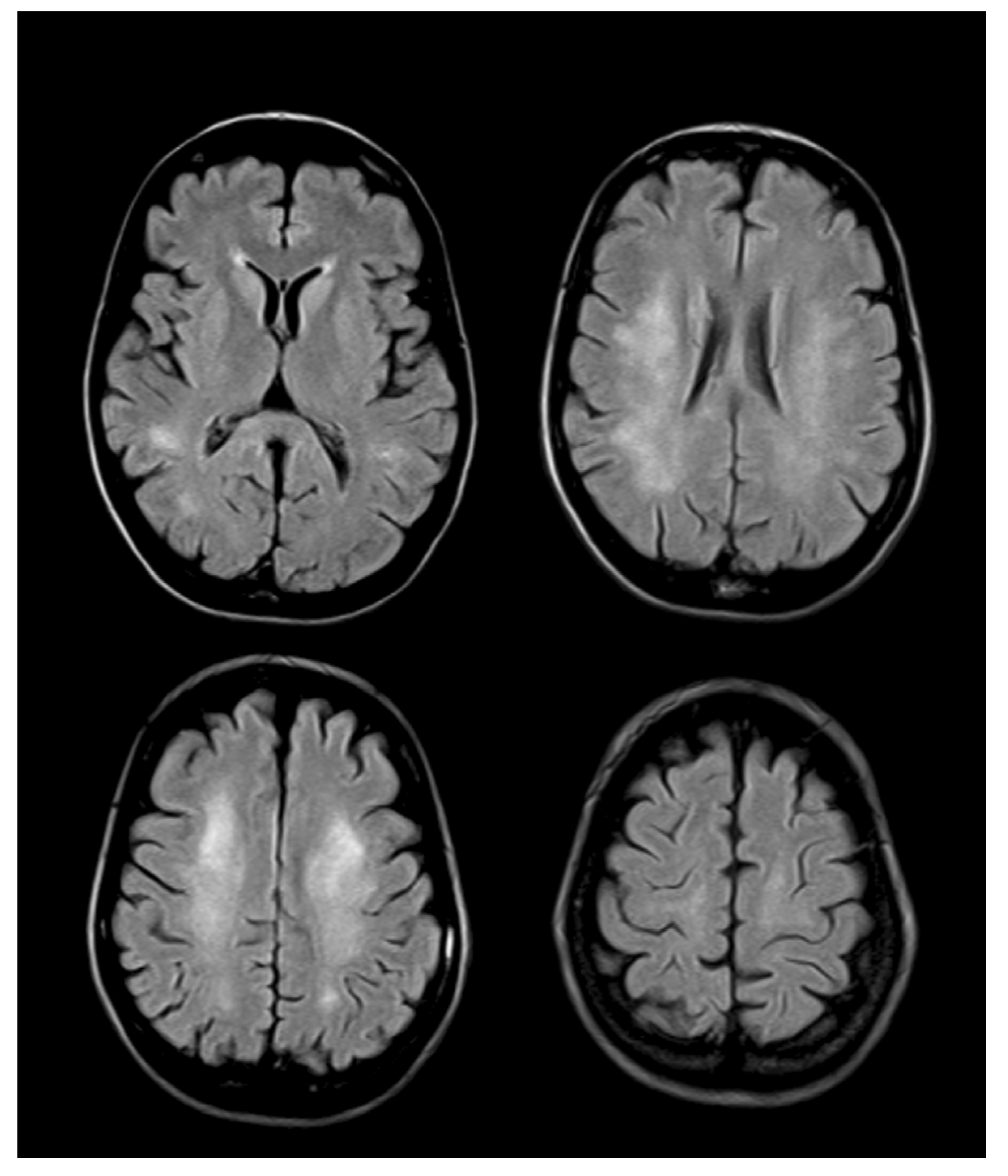
5. Therapy
Neurologic Changes after Effective Therapy
6. Conclusions
Conflicts of Interest
References
- Antony, A.C. Megaloblastic Anemias. In Hematology: Basic Principles and Practice, 5th ed.; Hoffman, R., Benz, E.J., Jr., Shattil, S.J., Eds.; Elsevier Churchill Livingstone: Philadelphia, PA, USA, 2009; Volume 3, p. 491. [Google Scholar]
- Watanabe, F. Vitamin B12 sources and bioavailability. Exp. Biol. Med. 2007, 232, 1266–1274. [Google Scholar] [CrossRef]
- Antony, A.C. Vegetarianism and vitamin B-12 (cobalamin) deficiency. Am. J. Clin. Nutr. 2003, 78, 3–6. [Google Scholar]
- Herbert, V.; Jacob, E.; Wong, K.T.; Scott, J.; Pfeffer, R.D. Low serum vitamin B12 levels in patients receiving ascorbic acid in megadoses: Studies concerning the effect of ascorbate on radioisotope vitamin B12 assay. Am. J. Clin. Nutr. 1978, 31, 253–258. [Google Scholar]
- Koop, H.; Bachem, M.G. Serum iron, ferritin, and vitamin B12 during prolonged omeprazole therapy. J. Clin. Gastroenterol. 1992, 14, 288–292. [Google Scholar] [CrossRef]
- Kristiansen, M.; Aminoff, M.; Jacobsen, C.; de la Chapelle, A.; Krahe, R.; Verroust, P.J.; Moestrup, S.K. Cubilin p1297l mutation associated with hereditary megaloblastic anemia 1 causes impaired recognition of intrinsic factor-vitamin B(12) by cubilin. Blood 2000, 96, 405–409. [Google Scholar]
- Nielsen, M.J.; Rasmussen, M.R.; Andersen, C.B.; Nexo, E.; Moestrup, S.K. Vitamin B12 transportfrom food to the body’s cells—A sophisticated, multistep pathway. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 345–354. [Google Scholar] [CrossRef]
- Herrmann, W.; Schorr, H.; Obeid, R.; Geisel, J. Vitamin B-12 status, particularly holotranscobalamin II and methylmalonic acid concentrations, and hyperhomocysteinemia in vegetarians. Am. J. Clin. Nutr. 2003, 78, 131–136. [Google Scholar]
- Honzik, T.; Adamovicova, M.; Smolka, V.; Magner, M.; Hruba, E.; Zeman, J. Clinical presentation and metabolic consequences in 40 breastfed infants with nutritional vitamin B12 deficiency—What have we learned? Eur. J. Paediatr. Neurol. 2010, 14, 488–495. [Google Scholar]
- Dror, D.K.; Allen, L.H. Effect of vitamin B12 deficiency on neurodevelopment in infants: Current knowledge and possible mechanisms. Nutr. Rev. 2008, 66, 250–255. [Google Scholar] [CrossRef]
- Pennypacker, L.C.; Allen, R.H.; Kelly, J.P.; Matthews, L.M.; Grigsby, J.; Kaye, K.; Lindenbaum, J.; Stabler, S.P. High prevalence of cobalamin deficiency in elderly outpatients. J. Am. Geriatr. Soc. 1992, 40, 1197–1204. [Google Scholar]
- Carmel, R. How I treat cobalamin (vitamin B12) deficiency. Blood 2008, 112, 2214–2221. [Google Scholar] [CrossRef]
- Lewerin, C.; Jacobsson, S.; Lindstedt, G.; Nilsson-Ehle, H. Serum biomarkers for atrophic gastritis and antibodies against helicobacter pylori in the elderly: Implications for vitamin B12, folic acid and iron status and response to oral vitamin therapy. Scand. J. Gastroenterol. 2008, 43, 1050–1056. [Google Scholar] [CrossRef]
- Stabler, S.P. Clinical practice. Vitamin B12 deficiency. N. Engl. J. Med. 2013, 368, 149–160. [Google Scholar] [CrossRef]
- Saperstein, D.S.; Wolfe, G.I.; Gronseth, G.S.; Nations, S.P.; Herbelin, L.L.; Bryan, W.W.; Barohn, R.J. Challenges in the identification of cobalamin-deficiency polyneuropathy. Arch. Neurol. 2003, 60, 1296–1301. [Google Scholar] [CrossRef]
- Langan, R.C.; Zawistoski, K.J. Update on vitamin B12 deficiency. Am. Fam. Physician 2011, 83, 1425–1430. [Google Scholar]
- Lindenbaum, J.; Rosenberg, I.H.; Wilson, P.W.; Stabler, S.P.; Allen, R.H. Prevalence of cobalamin deficiency in the framingham elderly population. Am. J. Clin. Nutr. 1994, 60, 2–11. [Google Scholar]
- Carmel, R.; Green, R.; Rosenblatt, D.S.; Watkins, D. Update on cobalamin, folate, and homocysteine. Hematol. Am. Soc. Hematol. Educ. Program 2003, 2003, 62–81. [Google Scholar] [CrossRef]
- Scalabrino, G. The multi-faceted basis of vitamin B-12 (cobalamin) neurotrophism in adult central nervous system: Lessons learned from its deficiency. Prog. Neurobiol. 2009, 88, 203–220. [Google Scholar] [CrossRef]
- Senol, M.G.; Sonmez, G.; Ozdag, F.; Saracoglu, M. Reversible myelopathy with vitamin B12 deficiency. Singap. Med. J. 2008, 49, E330–E332. [Google Scholar]
- Hemmer, B.; Glocker, F.X.; Schumacher, M.; Deuschl, G.; Lucking, C.H. Subacute combined degeneration: Clinical, electrophysiological, and magnetic resonance imaging findings. J. Neurol. Neurosurg. Psychiatry 1998, 65, 822–827. [Google Scholar] [CrossRef]
- Kumar, N. Nutritional neuropathies. Neurol. Clin. 2007, 25, 209–255. [Google Scholar] [CrossRef]
- Healton, E.B.; Savage, D.G.; Brust, J.C.M.; Garrett, T.J.; Lindenbaum, J. Neurologic aspects of cobalamin deficiency. Medicine 1991, 70, 229–245. [Google Scholar]
- Puri, V.; Chaudhry, N.; Goel, S.; Gulati, P.; Nehru, R.; Chowdhury, D. Vitamin B12 deficiency: A clinical and electrophysiological profile. Electromyogr. Clin. Neurophysiol. 2005, 45, 273–284. [Google Scholar]
- Huang, C.R.; Chang, W.N.; Tsai, N.W.; Lu, C.H. Serial nerve conduction studies in vitamin B12 deficiency-associated polyneuropathy. Neurol. Sci. 2011, 32, 183–186. [Google Scholar]
- Dommisse, J. Subtle vitamin-B12 deficiency and psychiatry: A largely unnoticed but devastating relationship? Med. Hypotheses 1991, 34, 131–140. [Google Scholar] [CrossRef]
- Hutto, B.R. Folate and cobalamin in psychiatric illness. Compr. Psychiatry 1997, 38, 305–314. [Google Scholar] [CrossRef]
- Obeid, R.; McCaddon, A.; Herrmann, W. The role of hyperhomocysteinemia and B-vitamin deficiency in neurological and psychiatric diseases. Clin. Chem. Lab. Med. 2007, 45, 1590–1606. [Google Scholar]
- Bjelland, I.; Tell, G.S.; Vollset, S.E.; Refsum, H.; Ueland, P.M. Folate, vitamin B12, homocysteine, and the MTHFR 677C->T polymorphism in anxiety and depression: The hordaland homocysteine study. Arch. Gen. Psychiatry 2003, 60, 618–626. [Google Scholar] [CrossRef]
- Bottiglieri, T.; Laundy, M.; Crellin, R.; Toone, B.K.; Carney, M.W.; Reynolds, E.H. Homocysteine, folate, methylation, and monoamine metabolism in depression. J. Neurol. Neurosurg. Psychiatry 2000, 69, 228–232. [Google Scholar] [CrossRef]
- Papakostas, G.I. Evidence for S-adenosyl-l-methionine (SAM-e) for the treatment of major depressive disorder. J. Clin. Psychiatry 2009, 70, 18–22. [Google Scholar] [CrossRef]
- Tiemeier, H.; van Tuijl, H.R.; Hofman, A.; Meijer, J.; Kiliaan, A.J.; Breteler, M.M. Vitamin B12, folate, and homocysteine in depression: The rotterdam study. Am. J. Psychiatry 2002, 159, 2099–2101. [Google Scholar] [CrossRef]
- Penninx, B.W.; Guralnik, J.M.; Ferrucci, L.; Fried, L.P.; Allen, R.H.; Stabler, S.P. Vitamin B(12) deficiency and depression in physically disabled older women: Epidemiologic evidence from the women’s health and aging study. Am. J. Psychiatry 2000, 157, 715–721. [Google Scholar] [CrossRef]
- Hintikka, J.; Tolmunen, T.; Tanskanen, A.; Viinamaki, H. High vitamin B12 level and good treatment outcome may be associated in major depressive disorder. BMC Psychiatry 2003, 3, 17. [Google Scholar]
- Hector, M.; Burton, J.R. What are the psychiatric manifestations of vitamin B12 deficiency? J. Am. Geriatr. Soc. 1988, 36, 1105–1112. [Google Scholar]
- Buchman, N.; Mendelsson, E.; Lerner, V.; Kotler, M. Delirium associated with vitamin B12 deficiency after pneumonia. Clin. Neuropharmacol. 1999, 22, 356–358. [Google Scholar]
- Lerner, V.; Kanevsky, M. Acute dementia with delirium due to vitamin B12 deficiency: A case report. Int. J. Psychiatry Med. 2002, 32, 215–220. [Google Scholar] [CrossRef]
- Goebels, N.; Soyka, M. Dementia associated with vitamin B(12) deficiency: Presentation of two cases and review of the literature. J. Neuropsychiatry Clin. Neurosci. 2000, 12, 389–394. [Google Scholar] [CrossRef]
- Van Goor, L.; Woiski, M.D.; Lagaay, A.M.; Meinders, A.E.; Tak, P.P. Review: Cobalamin deficiency and mental impairment in elderly people. Age Ageing 1995, 24, 536–542. [Google Scholar] [CrossRef]
- Morris, M.C.; Schneider, J.A.; Tangney, C.C. Thoughts on B-vitamins and dementia. J. Alzheimer's Dis. 2006, 9, 429–433. [Google Scholar]
- Stabler, S.P.; Allen, R.H.; Savage, D.G.; Lindenbaum, J. Clinical spectrum and diagnosis of cobalamin deficiency. Blood 1990, 76, 871–881. [Google Scholar]
- Whyte, E.M.; Mulsant, B.H.; Butters, M.A.; Qayyum, M.; Towers, A.; Sweet, R.A.; Klunk, W.; Wisniewski, S.; DeKosky, S.T. Cognitive and behavioral correlates of low vitamin B12 levels in elderly patients with progressive dementia. Am. J. Geriatr. Psychiatry 2002, 10, 321–327. [Google Scholar]
- Chiu, H.F.K. Vitamin B12 deficiency and dementia. Int. J. Geriatr. Psychiatry 1996, 11, 851–858. [Google Scholar] [CrossRef]
- Blundo, C.; Marin, D.; Ricci, M. Vitamin B12 deficiency associated with symptoms of frontotemporal dementia. Neurol. Sci. 2011, 32, 101–105. [Google Scholar] [CrossRef]
- Schreinzer, D.; Barnas, C.; Fischer, P. Frontotemporal dementia associated with vitamin B12 deficiency. J. Am. Geriatr. Soc. 2003, 51, 280–281. [Google Scholar] [CrossRef]
- Akdal, G.; Yener, G.G.; Kurt, P. Treatment responsive executive and behavioral dysfunction associated with vitamin B12 deficiency. Neurocase 2008, 14, 147–150. [Google Scholar] [CrossRef]
- Osimani, A.; Berger, A.; Friedman, J.; Porat-Katz, B.S.; Abarbanel, J.M. Neuropsychology of vitamin B12 deficiency in elderly dementia patients and control subjects. J. Geriatr. Psychiatry Neurol. 2005, 18, 33–38. [Google Scholar] [CrossRef]
- Eastley, R.; Wilcock, G.K.; Bucks, R.S. Vitamin B12 deficiency in dementia and cognitive impairment: The effects of treatment on neuropsychological function. Int. J. Geriatr. Psychiatry 2000, 15, 226–233. [Google Scholar] [CrossRef]
- Malouf, R.; Areosa Sastre, A. Vitamin B12 for cognition. Cochrane Database Syst. Rev. 2003. [Google Scholar] [CrossRef]
- Douaud, G.; Refsum, H.; de Jager, C.A.; Jacoby, R.; Nichols, T.E.; Smith, S.M.; Smith, A.D. Preventing alzheimer’s disease-related gray matter atrophy by B-vitamin treatment. Proc. Natl. Acad. Sci. USA 2013, 110, 9523–9528. [Google Scholar]
- Yamada, K.; Shrier, D.A.; Tanaka, H.; Numaguchi, Y. A case of subacute combined degeneration: MRI findings. Neuroradiology 1998, 40, 398–400. [Google Scholar] [CrossRef]
- Okada, S.; Kuwako, T.; Nakajo, H.; Ishihara, M.; Uchiyama, F.; Obo, R.; Yokose, N.; Hamamoto, M. Two cases of subacute combined degeneration: Magnetic resonance findings. J. Nippon Med. Sch. 2006, 73, 328–331. [Google Scholar] [CrossRef]
- Timms, S.R.; Cure, J.K.; Kurent, J.E. Subacute combined degeneration of the spinal cord: MR findings. AJNR 1993, 14, 1224–1227. [Google Scholar]
- Tajima, Y.; Mito, Y.; Owada, Y.; Moriwaka, F.; Tashiro, K. MR appearance of subacute combined degeneration of the spinal cord. Jpn. J. Psychiatry Neurol. 1994, 48, 611–614. [Google Scholar]
- Katsaros, V.K.; Glocker, F.X.; Hemmer, B.; Schumacher, M. MRI of spinal cord and brain lesions in subacute combined degeneration. Neuroradiology 1998, 40, 716–719. [Google Scholar] [CrossRef]
- Berlit, P.; Ringelstein, A.; Liebig, T. Spinal MRI precedes clinical improvement in subacute combined degeneration with B12 deficiency. Neurology 2004, 63, 592. [Google Scholar]
- Ravina, B.; Loevner, L.A.; Bank, W. MR findings in subacute combined degeneration of the spinal cord: A case of reversible cervical myelopathy. Am. J. Roentgenol. 2000, 174, 863–865. [Google Scholar] [CrossRef]
- Nakamura, K.; Mukherjee, P.; Swanson, R.A. Vitamin B12 deficiency. Arch. Neurol. 2004, 61, 960. [Google Scholar] [CrossRef]
- Karantanas, A.H.; Markonis, A.; Bisbiyiannis, G. Subacute combined degeneration of the spinal cord with involvement of the anterior columns: A new MRI finding. Neuroradiology 2000, 42, 115–117. [Google Scholar]
- Tian, C. Hyperintense signal on spinal cord diffusion-weighted imaging in a patient with subacute combined degeneration. Neurol. India 2011, 59, 429–431. [Google Scholar] [CrossRef]
- Scalabrino, G. Cobalamin (vitamin B(12)) in subacute combined degeneration and beyond: Traditional interpretations and novel theories. Exp. Neurol. 2005, 192, 463–479. [Google Scholar] [CrossRef]
- Spinazzi, M.; de Lazzari, F.; Tavolato, B.; Angelini, C.; Manara, R.; Armani, M. Myelo-optico-neuropathy in copper deficiency occurring after partial gastrectomy. Do small bowel bacterial overgrowth syndrome and occult zinc ingestion tip the balance? J. Neurol. 2007, 254, 1012–1017. [Google Scholar]
- Lovblad, K.; Ramelli, G.; Remonda, L.; Nirkko, A.C.; Ozdoba, C.; Schroth, G. Retardation of myelination due to dietary vitamin B12 deficiency: Cranial MRI findings. Pediatr. Radiol. 1997, 27, 155–158. [Google Scholar] [CrossRef]
- Kori, S. Hyperintense splenium in vitamin B12 deficiency. Neurol. India 2005, 53, 377–378. [Google Scholar] [CrossRef]
- Cech, I.; Burau, K.D. Serological differences in folate/vitamin B12 in pregnancies affected by neural tube defects. South. Med. J. 2010, 103, 419–424. [Google Scholar] [CrossRef]
- Wenz, H.; Eisele, P.; Artemis, D.; Forster, A.; Brockmann, M.A. Acute marchiafava-bignami disease with extensive diffusion restriction and early recovery: Case report and review of the literature. J. Neuroimaging 2012. [Google Scholar] [CrossRef]
- Misra, U.K.; Kalita, J.; Das, A. Vitamin B12 deficiency neurological syndromes: A clinical, MRI and electrodiagnostic study. Electromyogr. Clin. Neurophysiol. 2003, 43, 57–64. [Google Scholar]
- De Lau, L.M.; Smith, A.D.; Refsum, H.; Johnston, C.; Breteler, M.M. Plasma vitamin B12 status and cerebral white-matter lesions. J. Neurol. Neurosurg. Psychiatry 2009, 80, 149–157. [Google Scholar]
- Scott, T.M.; Tucker, K.L.; Bhadelia, A.; Benjamin, B.; Patz, S.; Bhadelia, R.; Liebson, E.; Price, L.L.; Griffith, J.; Rosenberg, I.; et al. Homocysteine and B vitamins relate to brain volume and white-matter changes in geriatric patients with psychiatric disorders. Am. J. Geriatr. Psychiatry 2004, 12, 631–638. [Google Scholar]
- Graber, J.J.; Sherman, F.T.; Kaufmann, H.; Kolodny, E.H.; Sathe, S. Vitamin B12-responsive severe leukoencephalopathy and autonomic dysfunction in a patient with “normal” serum B12 levels. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1369–1371. [Google Scholar] [CrossRef]
- Lindenbaum, J.; Healton, E.B.; Savage, D.G.; Brust, J.C.; Garrett, T.J.; Podell, E.R.; Marcell, P.D.; Stabler, S.P.; Allen, R.H. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N. Engl. J. Med. 1988, 318, 1720–1728. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Briani, C.; Dalla Torre, C.; Citton, V.; Manara, R.; Pompanin, S.; Binotto, G.; Adami, F. Cobalamin Deficiency: Clinical Picture and Radiological Findings. Nutrients 2013, 5, 4521-4539. https://doi.org/10.3390/nu5114521
Briani C, Dalla Torre C, Citton V, Manara R, Pompanin S, Binotto G, Adami F. Cobalamin Deficiency: Clinical Picture and Radiological Findings. Nutrients. 2013; 5(11):4521-4539. https://doi.org/10.3390/nu5114521
Chicago/Turabian StyleBriani, Chiara, Chiara Dalla Torre, Valentina Citton, Renzo Manara, Sara Pompanin, Gianni Binotto, and Fausto Adami. 2013. "Cobalamin Deficiency: Clinical Picture and Radiological Findings" Nutrients 5, no. 11: 4521-4539. https://doi.org/10.3390/nu5114521
APA StyleBriani, C., Dalla Torre, C., Citton, V., Manara, R., Pompanin, S., Binotto, G., & Adami, F. (2013). Cobalamin Deficiency: Clinical Picture and Radiological Findings. Nutrients, 5(11), 4521-4539. https://doi.org/10.3390/nu5114521