Vitamin A Metabolism: An Update

Abstract
:Abbreviations:
1. Introduction
2. Metabolism within the Gastrointestinal (GI) Tract
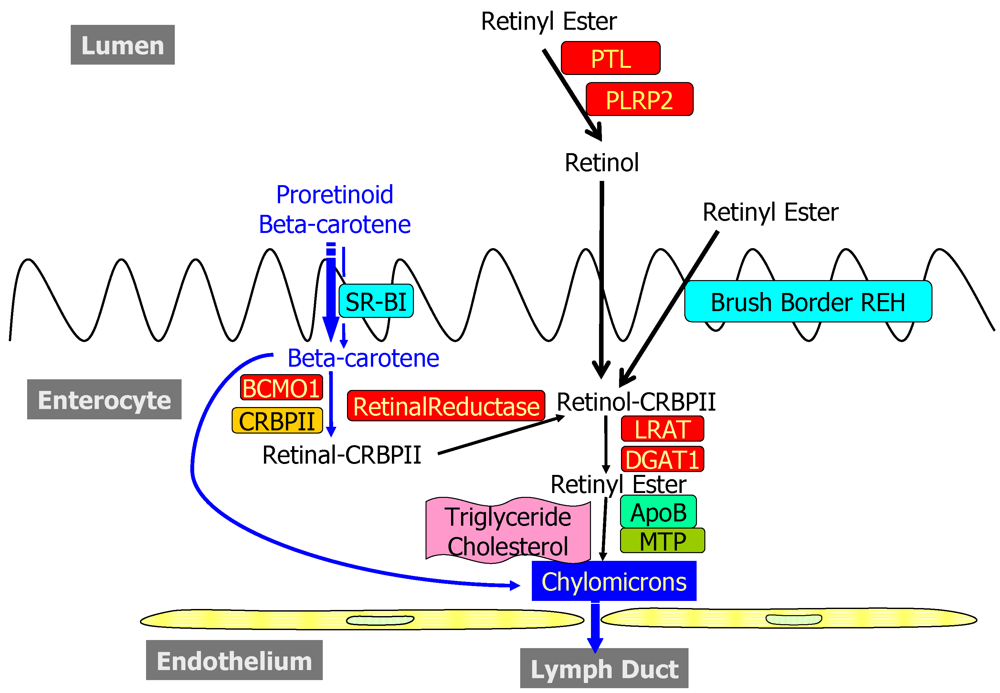
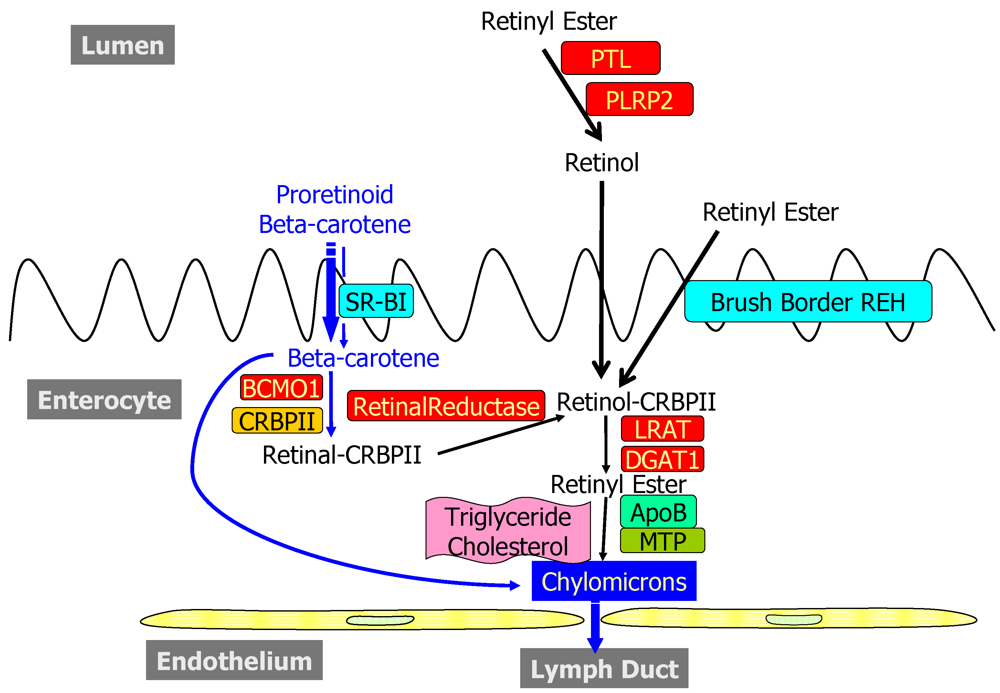
2.1. Dietary Forms and Metabolism in the Lumen of the Intestine
2.2. Metabolism and Processing within the Intestinal Mucosa
2.2.1. Uptake into and Efflux from the Enterocyte
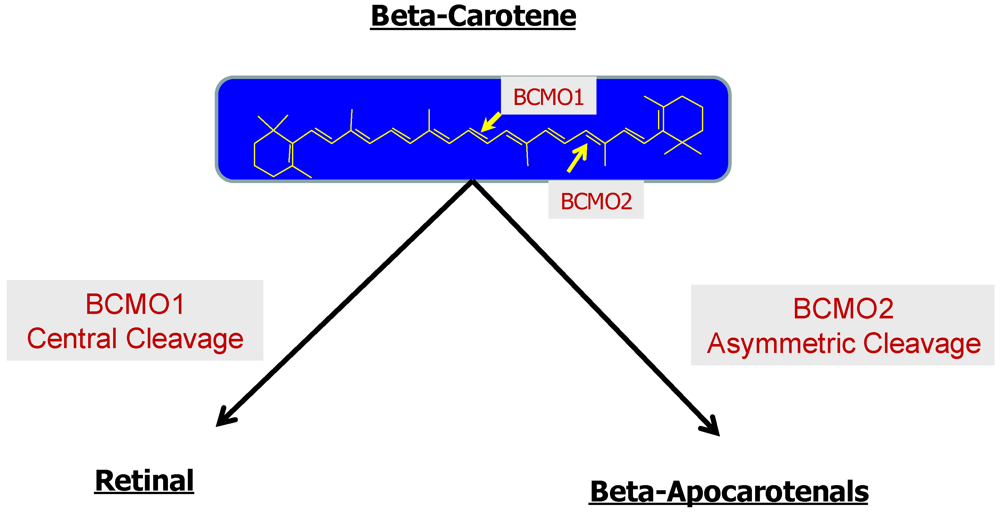
2.2.2. Enzymatic Conversion of Proretinoid Carotenoid to Retinoid
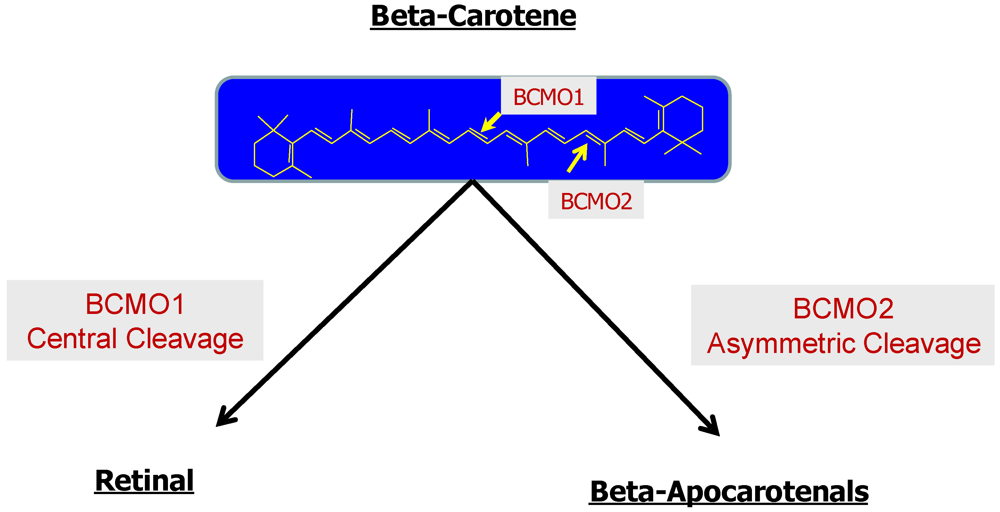
2.2.3. The Bcmo1 and Bcmo2 Genes and Their Expression
2.2.4. Enterocyte Esterification of Retinol
2.2.5. Cellular Retinol-Binding Protein, Type II (CRBPII)
3. Chylomicrons and Their Metabolism in the Circulation

{kind=link}
{kind=link}
{kind=link}
| Protein | Other Designations | Protein Family | Major Retinoid Ligands | Tissue Localization |
|---|---|---|---|---|
| RBP | RBP4 | Lipocalin | all-trans-retinol | Many, with high levels in liver and adipose |
| IRBP | RBP3 | − | all-trans-retinol | Retina |
| 11-cis-retinal | ||||
| CRBPI | RBP1 | iLBP | all-trans-retinol | Many, with high levels in liver, kidney, testis, eye, lung |
| all-trans-retinal | ||||
| CRBPII | RBP2 | iLBP | all-trans-retinol | Small intestine |
| all-trans-retinal | ||||
| CRBPIII | RBP7 | iLBP | all-trans-retinol | Heart, muscle, adipose, mammary |
| CRABPI | RBP5 | iLBP | all-trans-retinoic acid | Ubiquitous expression, with high levels in brain, skin and testes |
| CRABPII | RBP6 b | iLBP | all-trans-retinoic acid | Primarily skin; also found in mammary, uterus, kidney, prostate and olfactory epithelium |
| CRALBP | RLBP1 | CRAL_Trio | 11-cis-retinal | RPE, retina, ciliary body, cornea, pineal gland, optic nerve, brain |
| 11-cis-retinol | ||||
| 9-cis-retinal |
4. Hepatic Retinoid Metabolism
4.1. Uptake and Processing of Chylomicron Retinyl Ester by the Hepatocyte
4.1.1. Hepatic Chylomicron Remnant Receptors
4.1.2. Retinyl Ester Hydrolysis in the Hepatocyte
| Protein | Hepatic Cell Type |
|---|---|
| Bile salt-dependent REH (identified to be CEL) | Hepatocyte |
| Neutral, Bile salt-independent REH | Hepatocyte |
| Acidic, Bile salt-independent REH | Hepatocyte |
| ES-2 | Hepatocyte |
| ES-4 | Hepatocyte > HSC |
| ES-10 | Hepatocyte > HSC |
| ES-22 | Hepatocyte |
| Hepatic Lipase | Hepatocyte |
| LpL | HSC (activated) |
| ATGL | HSC |
4.2. Transfer of Retinol from Hepatocytes to HSCs
4.2.1. RBP-Mediated Transfer
4.2.2. CRBPI-Mediated Transfer
4.3. Storage of Retinoid in the HSC as Retinyl Ester in Lipid Droplets
4.3.1. The role of HSC Lipid Droplets in Retinoid Storage
4.3.2. Esterification of Retinol in the HSC
4.3.3. HSC Lipid Droplet Content and Effects of Dietary Retinoid Status
4.3.4. HSC Lipid Droplets in Hepatic Disease
4.4. Mobilization of Retinol from Hepatic Stores to Peripheral Tissues
4.4.1. Hydrolysis of HSC Lipid Droplet Retinyl Ester
4.4.2. Role of RBP in Hepatic Mobilization of Retinol
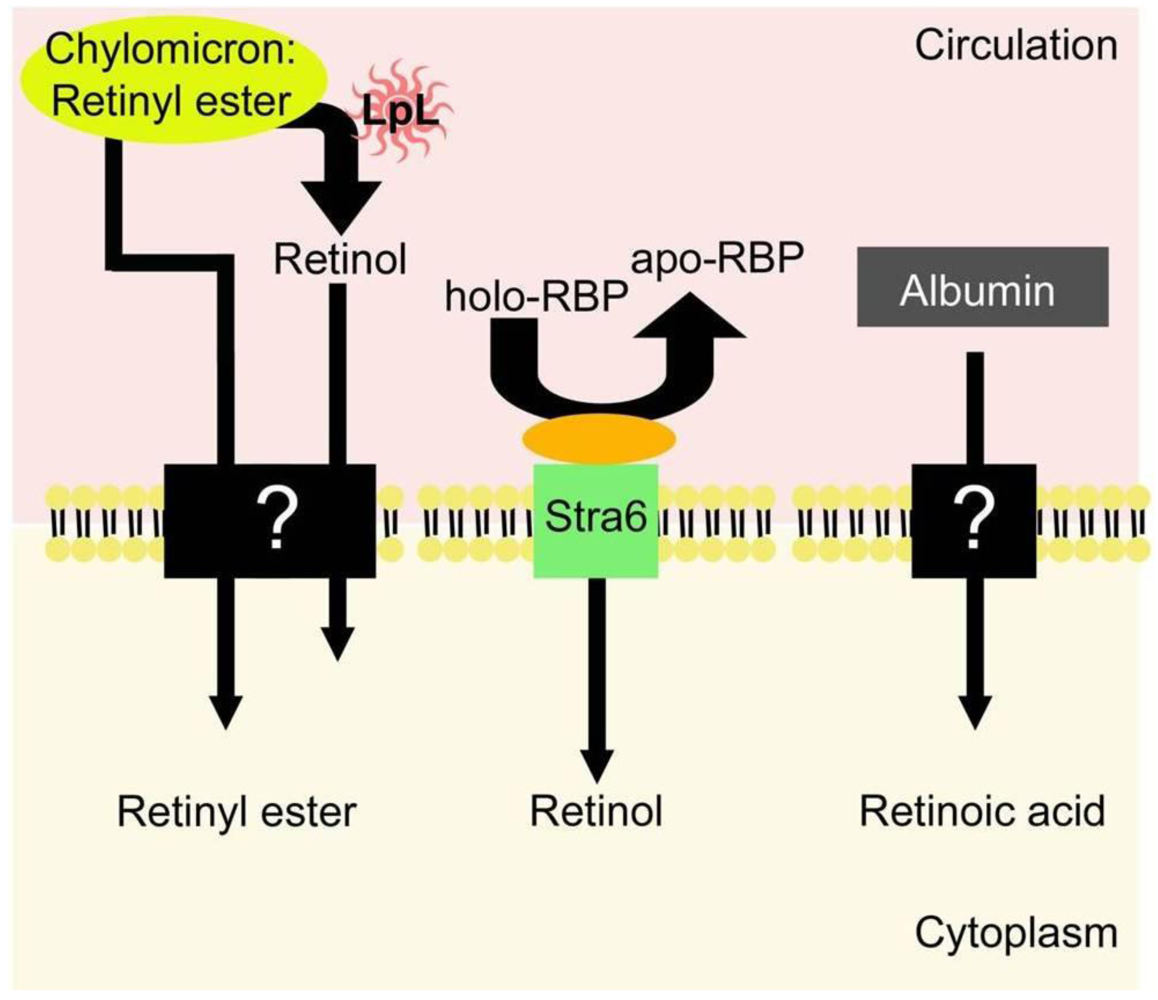
5. Uptake of Retinoids by Extrahepatic Tissues
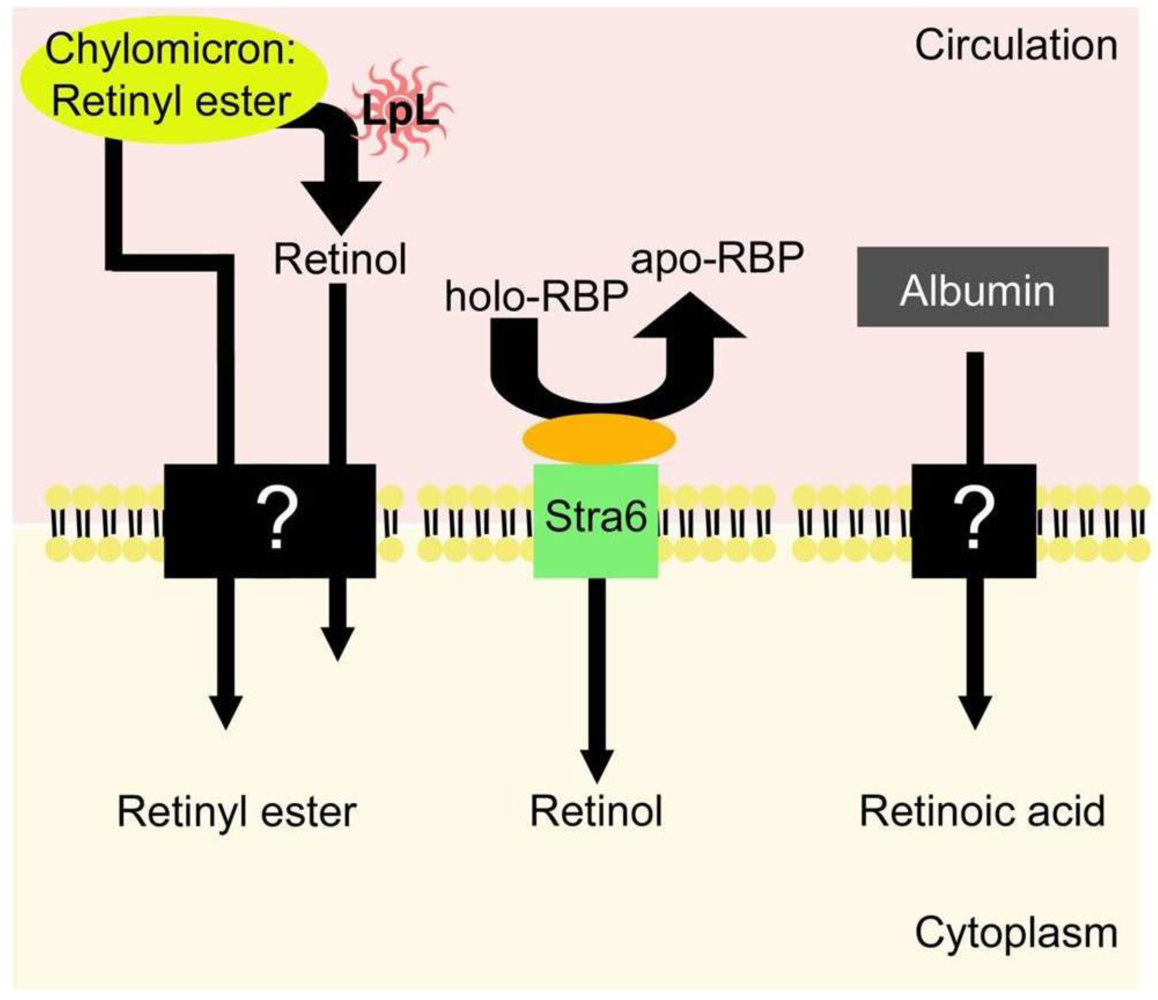
5.1. Uptake of Retinol-RBP from the Blood by Extrahepatic Tissues
5.2. Extrahepatic Uptake of Chylomicron Retinyl Ester
5.3. Retinoid Homeostasis in Extrahepatic Tissues
5.3.1. White Adipose Tissue Retinoid Homeostasis
5.3.2. Adipose-Derived RBP and Metabolic Disease
5.3.3. Retinoid Homeostasis in the Heart
5.3.4. Retinoid Homeostasis in the Eye
Acknowledgements
References and Notes
- To be consistent with our experiences in using and understanding modern nomenclature usage, we will use the term retinoid in place of vitamin A throughout this review. The word retinoid is a generic term that includes both naturally occurring compounds with vitamin A activity and synthetic analogs of retinol, with or without the biological activity (Goodman, D.S. Vitamin A Metabolism. Federation Proc. 1980, 39, 2716–2722; and Goodman, D.S. Vitamin A and Retinoids in Health and Disease. N. Engl. J. Med. 1984, 310, 1023–1031). The focus of this review will be solely on the uptake, storage and metabolism of natural retinoids or their precursor proretinoid carotenoids. Consequently, for the purposes of understanding this review, the terms retinoid and vitamin A should be considered to be synonymous.
- McCollum, E.V.; Davis, M. The necessity of certain lipids during growth. J. Biol. Chem. 1913, 15, 167–175. [Google Scholar]
- Goodman, D.S.; Blaner, W.S. Biosynthesis, absorption, and hepatic metabolism of retinol. In The Retinoids; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Academic Press: New York, NY, USA, 1984; Volume 2, pp. 1–39. [Google Scholar]
- Blomhoff, R.; Green, M.H.; Green, J.B.; Berg, T.; Norum, K.R. Vitamin A metabolism: new perspectives on absorption, transport, and storage. Physiol. Rev. 1991, 71, 951–990. [Google Scholar]
- Blaner, W.S.; Olson, J.A. Retinol and retinoic acid metabolism. In The Retinoids: Biology, Chemistry, and Medicine, 2nd; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Raven Press: New York, NY, USA, 1994; pp. 229–256. [Google Scholar]
- Vogel, S.; Gamble, M.V.; Blaner, W.S. Retinoid uptake, metabolism and transport. In The Handbook of Experimental Pharmacology, The Retinoids; Nau, H., Blaner, W.S., Eds.; Springer Verlag: Heidelberg, Germany, 1999; pp. 31–96. [Google Scholar]
- Bendich, A.; Olson, J.A. Biological action of carotenoids. FASEB J. 1989, 3, 1927–1932. [Google Scholar]
- Weng, W.; Li, L.; van Bennekum, A.M.; Potter, S.H.; Harrison, E.H.; Blaner, W.S.; Breslow, J.L.; Fisher, E.A. Intestinal absorption of dietary cholesteryl ester is decreased but retinyl ester absorption is normal in carboxyl ester lipase knockout mice. Biochemistry 1999, 38, 4143–4149. [Google Scholar]
- Van Bennekum, A.M.; Fisher, E.A.; Blaner, W.S.; Harrison, E.H. Hydrolysis of retinyl esters by pancreatic triglyceride lipase. Biochemistry 2000, 39, 4900–4906. [Google Scholar]
- Reboul, E.; Berton, A.; Moussa, M.; Kreuzer, C.; Crenon, I.; Porel, P. Pancreatic lipase and pancreatic lipase-related protein 2, but not pancreatic lipase-related protein 1, can hydrolyze retinyl palmitate in physiological conditions. Biochim. Biophys. Acta 2006, 1761, 4–10. [Google Scholar]
- Van Bennekum, A.; Werder, M.; Thuahnai, S.T.; Han, C.-H.; Duong, P.; Williams, D.L.; Wettstein, P.; Schulthess, G.; Phillips, M.C.; Hauser, H. Class B scavenger receptor-mediated intestinal absorption of dietary β-carotene and cholesterol. Biochemistry 2005, 44, 4517–4525. [Google Scholar]
- During, A.; Harrison, E.H. Mechanisms of provitamin A (carotenoid) and vitamin A (retinol) transport into and out of intestinal Caco-2 cells. J. Lipid Res. 2007, 48, 2283–2294. [Google Scholar]
- Lobo, G.P.; Hessel, S.; Eichinger, A.; Noy, N.; Moise, A.R.; Wyss, A.; Palczewski, K.; von Lintig, J. ISX is a retinoic acid-sensitive gatekeeper that controls intestinal β,β-carotene absorption and vitamin A production. FASEB J. 2010, 24, 1656–1666. [Google Scholar]
- Reboul, E.; Abou, L.; Mikail, C.; Ghiringhelli, O.; Andre, M.; Portugal, H.; Jourdheuil-Rahmani, D.; Amiot, M.-J.; Lairon, D.; Borel, P. Lutein transport by Caco-2 TC-7 cells occurs partly by a facilitated process involving the scavenger receptor class B type I (SR-BI). Biochem. J. 2005, 387, 455–461. [Google Scholar]
- Moussa, M.; Landrier, J.-F.; Reboul, E.; Ghiringhelli, O.; Coméra, C.; Collet, X.; Fröhlich, K.; Böhm, V.; Borel, P. Lycopene absorption in human intestinal cells and in mice involves scavenger receptor class B type I but not Niemann-Pick C1-like 1. J. Nutr. 2008, 138, 1432–1436. [Google Scholar]
- Reboul, E.; Trompier, D.; Moussa, M.; Klein, A.; Landrier, J.-F.; Chimini, G.; Borel, P. ATP-binding cassette transporter A1 is significantly involved in the intestinal absorption of α- and γ-tocopherol but not in that of retinyl palmitate in mice. Am. J. Clin. Nutr. 2009, 89, 177–184. [Google Scholar]
- Wyss, A.; Wirtz, G.; Woggon, W.; Brugger, R.; Wyss, M.; Frielein, A.; Bachmann, H.; Hunziker, W. Cloning and expression of beta, beta-carotene 15,15′-dioxygenase. Biochem. Biophys. Res. Commun. 2000, 271, 334–336. [Google Scholar]
- Redmond, T.M.; Gentleman, S.; Duncan, T.; Yu, S.; Wiggert, B.; Grantt, E.; Cunningham, F.X., Jr. Identification, expression and substrate specificity of a mammalian beta-carotene 15,15′-dioxygenase. J. Biol. Chem. 2000, 276, 6560–6565. [Google Scholar]
- Paik, J.; During, A.; Harrison, E.H.; Mendelsohn, C.L.; Lai, K.; Blaner, W.S. Expression and characterization of a murine enzyme able to cleave beta-carotene. The formation of retinoids. J. Biol. Chem. 2001, 276, 32160–32168. [Google Scholar]
- Wyss, A.; Wirtz, G.M.; Woggon, W.-D.; Brugger, R.; Wyss, M.; Friedlein, A.; Riss, G.; Bachmann, H.; Hunziker, W. Expression pattern and localization of β,β-carotene 15,15′-doxoygenase in different tissues. Biochem. J. 2001, 354, 521–529. [Google Scholar]
- Takitani, K.; Zhu, C.-L.; Inoue, A.; Tamai, H. Molecular cloning of the rat β-carotene 15,15′-monooxygenase gene and its regulation by retinoic acid. Eur. J. Nutr. 2006, 45, 320–326. [Google Scholar]
- Yan, W.; Jang, G.F.; Haeseleer, F.; Esumi, N.; Chang, J.; Kerrigan, M.; Campochiaro, M.; Campochiaro, P.; Palczewski, K.; Zack, D.J. Cloning and characterization of a human beta, beta-carotene-15,15′-dioxygenase that is highly expressed in the retinal pigment epithelium. Genomics 2001, 72, 193–202. [Google Scholar]
- Lindqvist, A.; Andersson, A. Biochemical properties of purified recombinant human β-carotene 15,15′-monooxygenase. J. Biol. Chem. 2002, 277, 23942–23948. [Google Scholar]
- Von Lintig, J.; Vogt, K. Filling the gap in vitamin A research: Molecular identification of an enzyme cleaving β-carotene to retinal. J. Biol. Chem. 2000, 275, 11915–11920. [Google Scholar]
- Lampert, J.M.; Holzschuh, J.; Hessel, S.; Driever, W.; Vogt, K.; von Lintig, J. Provitamin A conversion to retinal via the beta, beta-carotene-15,15′-oxygenase (bcox) is essential for pattern formation and differentiation during zebrafish embryogenesis. Development 2003, 130, 2173–2180. [Google Scholar]
- Kiefer, C.; Hessel, S.; Lampert, J.M.; Vogt, K.; Lederer, M.O.; Breithaupt, D.E.; von Lintig, J. Identification and characterization of a mammalian enzyme catalyzing the asymmetric oxidative cleavage of provitamin A. J. Biol. Chem. 2001, 276, 14110–14116. [Google Scholar]
- Hu, K.Q.; Liu, C.; Ernst, H.; Krinsky, N.I.; Russell, R.M.; Wang, X.D. The biochemical characterization of ferret carotene-9′,10′-monooxygenase catalyzing cleavage of carotenoids in vitro and in vivo. J. Biol. Chem. 2006, 282, 19327–19338. [Google Scholar]
- Hessel, S.; Eichinger, A.; Isken, A.; Amengual, J.; Hunzelmann, S.; Hoeller, U.; Elste, V.; Hunziker, W.; Goralczyk, R.; Oberhauser, V.; von Lintig, J.; Wyss, A. CMO1 deficiency abolishes vitamin a production from beta-carotene and alters lipid metabolism in mice. J. Biol. Chem. 2007, 282, 33553–33561. [Google Scholar]
- Fierce, Y.; de Moriais Vieira, M.; Piantedosi, R.; Wyss, A.; Blaner, W.S.; Paik, J. In vitro and in vivo characterization of retinoid synthesis from beta-carotene. Arch. Biochem. Biophys. 2008, 472, 126–138. [Google Scholar]
- Leuenberger, M.G.; Engeloch-Jarret, C.; Woggon, W.D. The reaction mechanism of the enzyme‑catalyzed central cleavage of β-carotene to retinal. Angew. Chem. Int. Ed. Engl. 2001, 40, 2613–2617. [Google Scholar]
- Boulanger, A.; McLemore, P.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Yu, S.S.; Gentleman, S.; Redmond, T.M. Identification of beta-carotene 15,15′-monooxygenase as a peroxisome proliferator-activated receptor target gene. FASEB J. 2003, 17, 1304–1306. [Google Scholar]
- Gong, X.; Tsai, S.-W.; Yan, B.; Rubin, L.P. Cooperation between MEF2 and PPARγ in human intestinal β,β-carotene 15,15′-monooxygenase gene expression. BMC Mol. Biol. 2006, 7, 7. [Google Scholar]
- Seino, Y.; Miki, T.; Kiyonari, H.; Abe, T.; Fujimoto, W.; Kimura, K.; Takeuchi, A.; Takahashi, Y.; Oiso, Y.; Iwanaga, T.; Seino, S. ISX participates in the maintenance of vitamin A metabolism by regulation of β-carotene 15,15′-monooxygenase (Bcmo1) expression. J. Biol. Chem. 2008, 283, 4905–4911. [Google Scholar]
- Lindqvist, A.; Sharvill, J.; Sharvill, D.E.; Andersson, S. Loss-of-function mutation in carotenoid 15,15′ monooxygenase identified in a patient with hypercarotenemia and hypovitaminosis A. J. Nutr. 2007, 137, 2346–2350. [Google Scholar]
- Leung, W.C.; Hessel, S.; Meplan, C.; Flint, J.; Oberhauser, V.; Tourniaire, F.; Hesketh, J.E.; von Lintig, J.; Lietz, G. Two common single nucleotide polymorphisms in the gene encoding beta-carotene 15,15′-monoxygenase alter beta-carotene metabolism in female volunteers. FASEB J. 2009, 23, 1041–1053. [Google Scholar]
- Eriksson, J.; Larson, G.; Gunnarsson, U.; Bed’horn, B.; Tixier-Boichard, M.; Stromstedt, L.; Wright, D.; Jungerius, A.; Vereijken, A.; Randi, E.; Jensen, P.; Andersson, L. Identification of the yellow skin gene reveals a hybrid origin of the domestic chicken. PLoS Genet. 2008, 4, e1000010. [Google Scholar] [PubMed]
- Berry, S.D.; Davis, S.R.; Beattie, E.M.; Thomas, N.L.; Burrett, A.K.; Ward, H.E.; Stanfield, A.M.; Biswas, M.; Ankersmit-Udy, A.E.; Oxley, P.E.; Barnett, J.L.; Pearson, J.F.; van der Does, Y.; Macgibbon, A.H.; Spelman, R.J.; Lehnert, K.; Snell, R.G. Mutations in bovine beta-carotene oxygenase affects milk color. Genetics 2009, 182, 923–926. [Google Scholar]
- Våge, D.I.; Boman, I.A. A nonsense mutation in the beta-carotene oxygenase 2 (BCO2) gene is tightly associated with accumulation of carotenoids in adipose tissue in sheep (Ovis aries). BMC Genet. 2010, 11, 10. [Google Scholar] [PubMed]
- O’Byrne, S.M.; Wongsiriroj, N.; Libien, J.; Vogel, S.; Goldberg, I.; Baehr, W.; Palczewski, K.; Blaner, W.S. Retinoid absorption and storage is impaired in mice lacking lecithin: retinol acyltransferase (LRAT). J. Biol. Chem. 2005, 280, 35647–35657. [Google Scholar]
- Wongsiriroj, N.; Piantedosi, R.; Palczewski, K.; Goldberg, I.J.; Johnston, T.P.; Li, E.; Blaner, W.S. The molecular basis of retinoid absorption: a genetic dissection. J. Biol. Chem. 2008, 283, 13510–13519. [Google Scholar]
- Ong, D.E.; Newcomer, M.A.; Chytil, F. Cellular retinol-binding proteins. In The Retinoids: Biology, Chemistry, and Medicine, 2nd; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Raven Press: New York, NY, USA, 1994. [Google Scholar]
- Crow, J.A.; Ong, D.E. Cell-specific immunohistochemical localization of a cellular retinol‑binding protein (type two) in the small intestine of the rat. Proc. Natl. Acad. Sci. USA 1985, 82, 4707–4711. [Google Scholar]
- Zhang, E.X.; Lu, J.; Tso, P.; Blaner, W.S.; Levin, M.S.; Li, E. Increased neonatal mortality in mice lacking cellular retinol-binding protein II. J. Biol. Chem. 2002, 277, 36617–36623. [Google Scholar]
- Goodman, D.S.; Huang, H.S.; Shiratori, T. Tissue distribution and metabolism of newly absorbed vitamin A in the rat. J. Lipid Res. 1965, 6, 390–396. [Google Scholar]
- RBP and RBP4 refer to the identical protein/gene product. From its first description in the late 1960s, the abbreviation RBP had been used for serum retinol-binding protein. In the mid-2000s, the abbreviation RBP4 can into usage largely because the nomenclature for the gene encoding RBP was established as Rbp4. For the purposes of this review, we will use the abbreviation RBP but the reader should be aware that RBP4 is now commonly used to designate this protein.
- Biesalski, H.K.; Frank, J.; Beck, S.C.; Heinrich, F.; Illek, B.; Reifen, R.; Gollnick, H.; Seeliger, M.W.; Wissinger, B.; Zrenner, E. Biochemical but not clinical vitamin A deficiency results from mutations in the gene for retinol binding protein. Am. J. Clin. Nutr. 1999, 69, 931–936. [Google Scholar]
- Quadro, L.; Blaner, W.S.; Salchow, D.J.; Vogel, S.; Piantedosi, R.; Gouras, P.; Freeman, S.; Cosma, M.P.; Colantuoni, V.; Gottesman, M.E. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 1999, 18, 4633–4644. [Google Scholar]
- Kurlandsky, S.B.; Gamble, M.V.; Ramakrishnan, R.; Blaner, W.S. Plasma deliver of retinoic acid to tissues in the rat. J. Biol. Chem. 1995, 270, 17850–17857. [Google Scholar]
- Blaner, W.S.; Obunike, J.C.; Kurlandsky, S.B.; al-Haideri, M.; Piantedosi, R.; Deckelbaum, R.J.; Goldberg, I.J. Lipoprotein lipase hydrolysis of retinyl ester. Possible implications for retinoid uptake by cells. J. Biol. Chem. 1994, 269, 16559–16565. [Google Scholar] [PubMed]
- Van Bennekum, A.M.; Kako, Y.; Weinstock, P.H.; Harrison, E.H.; Deckelbaum, R.J.; Goldberg, I.J.; Blaner, W.S. Lipoprotein lipase expression level influences tissue clearance of chylomicron retinyl ester. J. Lipid Res. 1999, 40, 565–574. [Google Scholar]
- Ross, A.C.; Pasatiempo, A.M.; Green, M.H. Chylomicron margination, lipolysis and vitamin A uptake in the lactating rat mammary gland: implications for milk retinoid content. Exp. Biol. Med. 2004, 229, 46–55. [Google Scholar]
- O’Byrne, S.M.; Kako, Y.; Deckelbaum, R.J.; Hansen, I.H.; Palczewski, K.; Goldberg, I.J.; Blaner, W.S. Multiple pathways ensure retinoid delivery to milk: studies in genetically modified mice. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E862–E870. [Google Scholar]
- Ross, A.C.; Li, N.-Q. Lung retinyl ester is low in young adult rats fed a vitamin A-deficient diet after weaning, despite neonatal vitamin A supplementation and maintenance of normal plasma retinol. J. Nutr. 2007, 137, 2213–2218. [Google Scholar]
- Geerts, A.; Bleser, P.D.; Hautekeete, M.L.; Niki, T.; Wisse, E. Fat-storing (Ito) cell biology. In The Liver: Biology and Pathobiology, 3rd; Arias, I.M., Boyer, J.L., Fausto, N., Jakoby, W.B., Schachter, D., Shafritz, D.A., Eds.; Raven Press: New York, NY, USA, 1994; pp. 819–837. [Google Scholar]
- Geerts, A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis. 2001, 21, 311–335. [Google Scholar]
- Friedman, S.L. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar]
- Wisse, E.; de Zanger, R.B.; van der Smissen, P.; McCuskey, R.S. The liver sieve: considerations concerning the structure and function of endothelial fenestrae, the sinusoidal wall and the space of disse. Hepatolgy 1985, 5, 683–692. [Google Scholar]
- Cooper, A.D. Hepatic uptake of chylomicron remnants. J. Lipid Res. 1997, 38, 2173–2192. [Google Scholar]
- Zeng, B.; Mortimer, B.; Martins, I.J.; Seydel, U.; Redgrave, T.G. Chylomicron remnant uptake is regulated by the expression and function of heparan sulfate proteoglycan in hepatocytes. J. Lipid Res. 1998, 39, 845–860. [Google Scholar]
- Stanford, K.I.; Bishop, J.R.; Foley, E.M.; Gonzales, J.C.; Niesman, I.R.; Witztum, J.L.; Esko, J.D. Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J. Clin. Invest. 2009, 119, 3236–3245. [Google Scholar]
- Crawford, S.E.; Borensztajn, J. Plasma clearance and liver uptake of chylomicron remnants generated by hepatic lipase lipolysis: evidence for a lactoferrin-sensitive and apolipoprotein E-independent pathway. J. Lipid Res. 1999, 40, 797–805. [Google Scholar]
- Out, R.; Kruijt, J.K.; Rensen, P.C.; Hildebrand, R.B.; de Vos, P.; Van Eck, M.; Van Berkel, T.J. Scavenger receptor BI plays a role in facilitating chylomicron metabolism. J. Biol. Chem. 2004, 279, 18401–18406. [Google Scholar]
- Out, R.; Hoekstra, M.; de Jager, S.C.; de Vos, P.; van der Westhuyzen, D.R.; Webb, N.R.; van Eck, M.; Biessen, E.A.; van Berkel, T.J. Adenovirus-mediated hepatic overexpression of scavenger receptor class B type I accelerates chylomicron metabolism in C57BL/6J mice. J. Lipid Res. 2005, 46, 1172–1181. [Google Scholar]
- Harrison, E.H.; Gad, M.Z.; Ross, A.C. Hepatic uptake and metabolism of chylomicron retinyl esters: probable role of plasma membrane/endosomal retinyl ester hydrolases. J. Lipid Res. 1995, 36, 1498–1506. [Google Scholar]
- Blaner, W.S.; Prystowsky, J.H.; Smith, J.E.; Goodman, D.S. Rat liver retinyl palmitate hydrolase activity. Relationship to cholesteryl oleate and triolein hydrolase activities. Biochim. Biophys. Acta 1984, 794, 419–427. [Google Scholar] [PubMed]
- Harrison, E.H.; Gad, M.Z. Hydrolysis of retinyl palmitate by enzymes of rat pancreas and liver. Differentiation of bile salt-dependent and bile salt-independent, neutral retinyl ester hydrolases in rat liver. J. Biol. Chem. 1989, 264, 17142–17147. [Google Scholar] [PubMed]
- Chen, X.; Harrison, E.H.; Fisher, E.A. Molecular cloning of the cDNA for rat hepatic, bile salt-dependent cholesteryl ester/retinyl ester hydrolase demonstrates identity with pancreatic carboxylester lipase. Proc. Soc. Exp. Biol. Med. 1997, 215, 186–191. [Google Scholar]
- Winkler, K.E.; Harrison, E.H.; Marsh, J.B.; Glick, J.M.; Ross, A.C. Characterization of a bile salt-dependent cholesteryl ester hydrolase activity secreted from HepG2 cells. Biochim. Biophys. Acta 1992, 1126, 151–158. [Google Scholar]
- Van Bennekum, A.M.; Li, L.; Piantedosi, R.; Shamir, R.; Vogel, S.; Fisher, E.A.; Blaner, W.S.; Harrison, E.H. Carboxyl ester lipase overexpression in rat hepatoma cells and CEL deficiency in mice have no impact on hepatic uptake or metabolism of chylomicron-retinyl ester. Biochemistry 1999, 38, 4150–4156. [Google Scholar]
- Harrison, E.H.; Gad, M.Z. Hydrolysis of retinyl palmitate by enzymes of rat pancreas and liver. Differentiation of bile salt-dependent and bile salt-independent, neutral retinyl ester hydrolases in rat liver. J. Biol. Chem. 1989, 264, 17142–17147. [Google Scholar] [PubMed]
- Boerman, M.H.; Napoli, J.L. Cholate-independent retinyl ester hydrolysis. Stimulation by apo-cellular retinol-binding protein. J. Biol. Chem. 1991, 266, 22273–22278. [Google Scholar] [PubMed]
- Gad, M.Z.; Harrison, E.H. Neutral and acid retinyl ester hydrolases associated with rat liver microsomes: relationships to microsomal cholesteryl ester hydrolases. J. Lipid Res. 1991, 32, 685–693. [Google Scholar]
- Harrison, E.H. Lipases and Carboxylesterases: Possible roles in the hepatic utilization of vitamin A. J. Nutr. 2000, 130, 340S–344S. [Google Scholar]
- Mentlein, R.; Ronai, A.; Robbi, M.; Heymann, E.; von Deimling, O. Genetic identification of rat liver carboxylesterases isolated in different laboratories. Biochim. Biophys. Acta 1987, 913, 27–38. [Google Scholar]
- Alexson, S.E.; Mentlein, R.; Wernstedt, C.; Hellman, U. Isolation and characterization of microsomal acyl-CoA thioesterase. A member of the rat liver microsomal carboxylesterase multi-gene family. Eur. J. Biochem. 1993, 214, 719–727. [Google Scholar]
- Sun, G.; Alexson, S.E.; Harrison, E.H. Purification and characterization of a neutral, bile salt-independent retinyl ester hydrolase from rat liver microsomes. Relationship to rat carboxylesterase ES-2. J. Biol. Chem. 1997, 272, 24488–24493. [Google Scholar] [PubMed]
- Mentlein, R.; Heymann, E. Hydrolysis of retinyl esters by non-specific carboxylesterases from rat liver endoplasmic reticulum. Biochem. J. 1987, 245, 863–867. [Google Scholar]
- Schreiber, R.; Taschler, U.; Wolinski, H.; Seper, A.; Tamegger, S.N.; Graf, M.; Kohlwein, S.D.; Haemmerle, G.; Zimmermann, R.; Zechner, R.; Lass, A. Esterase 22 and beta-glucuronidase hydrolyze retinoids in mouse liver. J. Lipid Res. 2009, 50, 2514–2523. [Google Scholar]
- Krapp, A.; Ahle, S.; Kersting, S.; Hua, Y.; Kneser, K.; Nielsen, M.; Gliemann, J.; Beisiegel, U. Hepatic lipase mediates the uptake of chylomicrons and β-VLDL into cells via the LDL receptor-related protein (LRP). J. Lipid Res. 1996, 37, 926–936. [Google Scholar]
- Wei, S.; Lai, K.; Patel, S.; Piantedosi, R.; Shen, H.; Colantuoni, V.; Kraemer, F.B.; Blaner, W.S. Retinyl ester hydrolysis and retinol efflux from BFC-1β adipocytes. J. Biol. Chem. 1997, 272, 14159–14165. [Google Scholar]
- Mello, T.; Nakatsuka, A.; Fears, S.; Davis, W.; Tsukamoto, H.; Bosron, W.; Sanghani, S. Expression of carboxylesterase and lipase genes in rat liver cell-types. Biochem. Biophys. Res. Commun. 2008, 374, 460–464. [Google Scholar]
- Blomhoff, R.; Berg, T.; Norum, K.R. Transfer of retinol from parenchymal to stellate cells in liver is mediated by retinol-binding protein. Proc. Natl. Acad. Sci. USA 1988, 85, 3455–3458. [Google Scholar]
- Senoo, H.; Smeland, S.; Malaba, L.; Bjerknes, T.; Stang, E.; Roos, N.; Berg, T.; Norum, K.R.; Blomhoff, R. Transfer of retinol-binding protein from HepG2 human hepatoma cells to cocultured rat stellate cells. Proc. Natl. Acad. Sci. USA 1993, 90, 3616–3620. [Google Scholar]
- Quadro, L.; Blaner, W.S.; Hamberger, L.; Novikoff, P.M.; Vogel, S.; Piantedosi, R.; Gottesman, M.E.; Colantuoni, V. The role of extrahepatic retinol binding protein in the mobilization of retinoid stores. J. Lipid Res. 2004, 45, 1975–1982. [Google Scholar]
- Ghyselinck, N.B.; Bavik, C.; Sapin, V.; Mark, M.; Bonnier, D.; Hindelang, C.; Dierich, A.; Nilsson, C.B.; Hakansson, H.; Sauvant, P.; Azais-Braesco, V.; Frasson, M.; Picaud, S.; Chambon, P. Cellular retinol-binding protein I is essential for vitamin A homeostasis. EMBO J. 1999, 18, 4903–4914. [Google Scholar]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.N.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.C.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta 2009, 1791, 467–473. [Google Scholar]
- O’Byrne, S.M.; Blaner, W.S. Introduction to retinoids. In Carotenoids and Retinoids: Molecular Aspects and Health Issues; Packer, L., Obermuller-Jevic, U., Kraemer, K., Sies, H., Eds.; AOCS Press: Champaign, IL, USA, 2005; pp. 1–22. [Google Scholar]
- Ross, A.C. Retinol esterfication by rat liver microsomes. Evidence for a fatty acyl coenzyme A: retinol acyltransferase. J. Biol. Chem. 1982, 257, 2453–2459. [Google Scholar] [PubMed]
- Yen, C.L.; Monetti, M.; Burri, B.J.; Farese, R.V., Jr. The triacylglycerol synthesis enzyme DGAT1 also catalyzes the synthesis of diacylglycerols, waxes, and retinyl esters. J. Lipid Res. 2005, 46, 1502–1511. [Google Scholar]
- Orland, M.D.; Anwar, K.; Cromley, D.; Chu, C.H.; Chen, L.; Billheimer, J.T.; Hussain, M.M.; Cheng, D. Acyl coenzyme A dependent retinol esterification by acyl coenzyme A:diacylglycerol acyltransferase 1. Biochim. Biophys. Acta 2005, 1737, 76–82. [Google Scholar]
- Shih, M.Y.; Kane, M.A.; Zhou, P.; Yen, C.L.; Streeper, R.S.; Napoli, J.L.; Farese, R.V., Jr. Retinol esterification by DGAT1 is essential for retinoid homeostasis in murine skin. J. Biol. Chem. 2009, 284, 4292–4299. [Google Scholar]
- Liu, L.; Gudas, L.J. Disruption of the lecithin: retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency. J. Biol. Chem. 2005, 280, 40226–40234. [Google Scholar]
- Moriwaki, H.; Blaner, W.S.; Piantedosi, R.; Goodman, D.S. Effects of dietary retinoid and triglyceride on the lipid composition of rat liver stellate cells and stellate cell lipid droplets. J. Lipid Res. 1988, 29, 1523–1534. [Google Scholar]
- Yamada, M.; Blaner, W.S.; Soprano, D.R.; Dixon, J.L.; Kjeldbye, H.M.; Goodman, D.S. Biochemical characteristics of isolated rat liver stellate cells. Hepatology 1987, 7, 1224–1229. [Google Scholar]
- Leo, M.A.; Lieber, C.S. Hepatic vitamin A depletion in alcoholic liver injury. N. Engl. J. Med. 1982, 307, 597–601. [Google Scholar]
- Friedman, S.L. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 2000, 275, 2247–2250. [Google Scholar]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Invest. 2005, 115, 209–218. [Google Scholar]
- De Minicis, S.; Seki, E.; Uchinami, H.; Kluwe, J.; Zhang, Y.; Brenner, D.A.; Schwabe, R.F. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology 2007, 132, 1937–1946. [Google Scholar]
- Liu, C.; Chung, J.; Seitz, H.K.; Russell, R.M.; Wang, X.-D. Chlormethiazole treatment prevents reduced hepatic vitamin A levels in ethanol-fed rats. Alcohol. Clin. Exp. Res. 2002, 26, 1703–1709. [Google Scholar]
- Dan, Z.; Popov, Y.; Patsenker, E.; Preimel, D.; Liu, C.; Wang, X.-D.; Seitz, H.K.; Schuppan, D.; Stickel, F. Hepatotoxicity of alcohol-induced polar retinol metabolites involves apoptosis via loss of mitochondrial membrane potential. FASEB J. 2005, 19, 845–847. [Google Scholar]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.-X.; Diehl, A.M. Diacylglycerol acyltransferase 1 anti-sense oligonucleotides reduce hepatic fibrosis in mice with nonalcoholic steatohepatitis. Hepatology 2008, 47, 625–635. [Google Scholar]
- Zimmerman, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; Zechner, R. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004, 306, 1383–1386. [Google Scholar]
- Lass, A.; Zimmerman, R.; Haemmerle, G.; Riederer, M.; Schoiswohl, G.; Schweiger, M.; Kienesberger, P.; Strauss, J.G.; Gorliewicz, G.; Zechner, R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chararin-Dorfman Syndrome. Cell Metab. 2006, 3, 305–307. [Google Scholar]
- Episkopou, V.; Maeda, S.; Nishiguchi, S.; Shimada, K.; Gaitanaris, G.; Gottesman, M.E.; Robertson, E.J. Disruption of the transthyretin gene results in mice with depressed levels of plasma retinol and thyroid hormone. Proc. Natl. Acad. Sci. USA 1993, 90, 2375–2379. [Google Scholar]
- Soprano, D.R.; Blaner, W.S. Plasma Retinol-Binding Protein. In The Retinoids, Biology, Chemistry and Medicine, 2nd; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Raven Press: New York, NY, USA, 1994; pp. 257–282. [Google Scholar]
- Van Bennekum, A.; Wei, S.; Gamble, M.V.; Vogel, S.; Piantesdosi, R.; Gottesman, M.; Episkopou, V.; Blaner, W.S. Biochemical basis for depressed serum retinol levels in transthyretin-deficient mice. J. Biol. Chem. 2001, 276, 1107–1113. [Google Scholar]
- Wei, S.; Episkopou, V.; Piantedosi, R.; Maeda, S.; Kazunori, S.; Gottesman, M.E.; Blaner, W.S. Studies on the metabolism of retinol and retinol-binding protein in transthyretin-deficient mice produce by homologous recombination. J. Biol. Chem. 1995, 270, 866–870. [Google Scholar]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar]
- Isken, A.; Golczak, M.; Oberhauser, V.; Hunzelmann, S.; Driever, W.; Imanishi, Y.; Palczewski, K.; von Lintig, J. RBP4 disrupts vitamin A uptake homeostasis in a STRA6-deficient animal model for Matthew-Wood syndrome. Cell Metab. 2008, 7, 258–268. [Google Scholar]
- Kim, Y.K.; Wassef, L.; Hamberger, L.; Piantedosi, R.; Palczewski, K.; Blaner, W.S.; Quadro, L. Retinyl ester formation by lecithin: retinol acyltransferase is a key regulator of retinoid homeostasis in mouse embryogenesis. J. Biol. Chem. 2008, 283, 5611–5621. [Google Scholar]
- Blaner, W.S. STRA6, a cell-surface receptor for retinol-binding protein: the plot thickens. Cell Metab. 2007, 5, 164–166. [Google Scholar]
- Taneja, R.; Bouillet, P.; Boylan, J.F.; Gaub, M.P.; Roy, B.; Gudas, L.J.; Chambon, P. Reexpression of retinoic acid receptor (RAR) gamma or overexpression of RAR alpha or RAR beta in RAR gamma-null F9 cells reveals a partial functional redundancy between the three RAR types. Proc. Natl. Acad. Sci. USA 1995, 92, 7854–7858. [Google Scholar]
- Bouillet, P.; Sapin, V.; Chazaud, C.; Messaddeq, N.; Décimo, D.; Dollé, P.; Chambon, P. Developmental expression pattern of Stra6, a retinoic acid-responsive gene encoding a new type of membrane protein. Mech. Dev. 1997, 63, 173–186. [Google Scholar]
- Tice, D.A.; Szeto, W.; Soloviev, I.; Rubinfeld, B.; Fong, S.E.; Dugger, D.L.; Winer, J.; Williams, P.M.; Wieand, D.; Smith, V.; Schwall, R.H.; Pennica, D.; Polakis, P. Synergistic induction of tumor antigens by Wnt-1 signaling and retinoic acid revealed by gene expression profiling. J. Biol. Chem. 2002, 277, 14329–14335. [Google Scholar]
- Cai, K.; Gudas, L.J. Retinoic acid receptors and GATA transcription factors activate the transcription of the human lecithin:retinol acyltransferase gene. Int. J. Biochem. Cell Biol. 2009, 41, 546–553. [Google Scholar]
- Wu, L.; Ross, A.C. Acidic retinoids synergize with vitamin A to enhance retinol uptake and STRA6, LRAT, and CYP26B1 expression in neonatal lung. J. Lipid Res. 2010, 51, 378–387. [Google Scholar]
- Reijntjes, S.; Zile, M.H.; Maden, M. The expression of Stra6 and Rdh10 in the avian embryo and their contribution to the generation of retinoid signatures. Int. J. Dev. Biol. 2010, 54, 1267–1275. [Google Scholar]
- Pasutto, F.; Sticht, H.; Hammersen, G.; Gillessen-Kaesbach, G.; Fitzpatrick, D.R.; Nürnberg, G.; Brasch, F.; Schirmer-Zimmermann, H.; Tolmie, J.L.; Chitayat, D.; et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am. J. Hum. Genet. 2007, 80, 550–560. [Google Scholar]
- Golzio, C.; Martinovic-Bouriel, J.; Thomas, S.; Mougou-Zrelli, S.; Grattagliano-Bessieres, B.; Bonniere, M.; Delahaye, S.; Munnich, A.; Encha-Razavi, F.; Lyonnet, S.; Vekemans, M.; Attie-Bitach, T.; Etchevers, H.C. Matthew-Wood syndrome is caused by truncating mutations in the retinol-binding protein receptor gene STRA6. Am. J. Hum. Genet. 2007, 80, 1179–1187. [Google Scholar]
- Kawaguchi, R.; Yu, J.; Wiita, P.; Honda, J.; Sun, H. An essential ligand-binding domain in the membrane receptor for retinol-binding protein revealed by large-scale mutagenesis and a human polymorphism. J. Biol. Chem. 2008, 283, 15160–15168. [Google Scholar]
- White, T.; Lu, T.; Metlapally, R.; Katowitz, J.; Kherani, F.; Wang, T.Y.; Tran-Viet, K.N.; Young, T.L. Identification of STRA6 and SKI sequence variants in patients with anophthalmia/microphthalmia. Mol. Vis. 2008, 14, 2458–2465. [Google Scholar]
- West, B.; Bove, K.E.; Slavotinek, A.M. Two novel STRA6 mutations in a patient with anophthalmia and diaphragmatic eventration. Am. J. Med. Genet. A 2009, 149A, 539–542. [Google Scholar]
- Chassaing, N.; Golzio, C.; Odent, S.; Lequeux, L.; Vigouroux, A.; Martinovic-Bouriel, J.; Tiziano, F.D.; Masini, L.; Piro, F.; Maragliano, G.; Delezoide, A.L.; Attié-Bitach, T.; Manouvrier-Hanu, S.; Etchevers, H.C.; Calvas, P. Phenotypic spectrum of STRA6 mutations: from Matthew-Wood syndrome to non-lethal anophthalmia. Hum. Mutat. 2009, 30, E673–E681. [Google Scholar]
- Segel, R.; Levy-Lahad, E.; Pasutto, F.; Picard, E.; Rauch, A.; Alterescu, G.; Schimmel, M.S. Pulmonary hypoplasia-diaphragmatic hernia-anophthalmia-cardiac defect (PDAC) syndrome due to STRA6 mutations—what are the minimal criteria? Am. J. Med. Genet. A 2009, 149A, 2457–2463. [Google Scholar]
- Kawaguchi, R.; Sun, H. Techniques to study specific cell-surface receptor-mediated cellular vitamin A uptake. Methods Mol. Biol. 2010, 652, 341–361. [Google Scholar]
- Verma, A.S.; Fitzpatrick, D.R. Anophthalmia and microphthalmia. Orphanet J. Rare Dis. 2007, 2, 47. [Google Scholar]
- Cvekl, A.; Wang, W.L. Retinoic acid signaling in mammalian eye development. Exp. Eye Res. 2009, 89, 280–291. [Google Scholar]
- Biesalski, H.K.; Nohr, D. Importance of vitamin A for lung function and development. Mol. Aspects Med. 2003, 24, 431–440. [Google Scholar]
- Kimura, J.; Deutsch, G.H. Key mechanisms of early lung development. Pediatr. Dev. Pathol. 2007, 10, 335–347. [Google Scholar]
- Pan, J.; Baker, K.M. Retinoic acid and the heart. Vitam. Horm. 2007, 75, 257–283. [Google Scholar]
- Hoover, L.L.; Burton, E.G.; Brooks, B.A.; Kubalak, S.W. The expanding role for retinoid signaling in heart development. ScientificWorldJournal 2008, 8, 194–211. [Google Scholar]
- Greer, J.J.; Babiuk, R.P.; Thebaud, B. Etiology of congenital diaphragmatic hernia: the retinoid hypothesis. Pediatr. Res. 2003, 53, 726–730. [Google Scholar]
- Gallot, D.; Marceau, G.; Coste, K.; Hadden, H.; Robert-Gnansia, E.; Laurichesse, H.; Déchelotte, P.J.; Labbé, A.; Dastugue, B.; Lémery, D.; Sapin, V. Congenital diaphragmatic hernia: a retinoid-signaling pathway disruption during lung development? Birth Defects Res. A Clin. Mol. Teratol. 2005, 73, 523–531. [Google Scholar] [PubMed]
- Clagett-Dame, M.; De Luca, H.F. The role of vitamin A in mammalian reproduction and embryonic development. Annu. Rev. Nutr. 2002, 22, 347–381. [Google Scholar]
- Quadro, L.; Hamberger, L.; Gottesman, M.E.; Colantuoni, V.; Ramakrishnan, R.; Blaner, W.S. Transplacental delivery of retinoid: the role of retinol-binding protein and lipoprotein retinyl ester. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E844–E851. [Google Scholar]
- Vogel, S.; Piantedosi, R.; O’Byrne, S.M.; Kako, Y.; Quadro, L.; Gottesman, M.E.; Goldberg, I.J.; Blaner, W.S. Retinol-binding protein-deficient mice: biochemical basis for impaired vision. Biochemistry 2002, 41, 15360–15368. [Google Scholar]
- Quadro, L.; Hamberger, L.; Gottesman, M.E.; Wang, F.; Colantuoni, V.; Blaner, W.S.; Mendelsohn, C.L. Pathways of vitamin A delivery to the embryo: insights from a new tunable model of embryonic vitamin A deficiency. Endocrinology 2005, 146, 4479–4490. [Google Scholar]
- Vahlquist, A.; Nilsson, S. Mechanisms for vitamin A transfer from blood to milk in rhesus monkeys. J. Nutr. 1979, 109, 1456–1463. [Google Scholar]
- Green, M.H.; Snyder, R.W.; Akohoue, S.A.; Green, J.B. Increased rat mammary tissue vitamin A associated with increased vitamin A intake during lactation is maintained after lactation. J. Nutr. 2001, 131, 1544–1547. [Google Scholar]
- Green, M.H.; Green, J.B.; Akohoue, S.A.; Kelley, S.K. Vitamin A intake affects the contribution of chylomicrons vs. retinol-binding protein to milk vitamin A in lactating rats. J. Nutr. 2001, 131, 1279–1282. [Google Scholar] [PubMed]
- La Rosa, J.C.; Levy, R.I.; Herbert, P.; Lux, S.E.; Fredrickson, D.S. A specific apoprotein activator for lipoprotein lipase. Biochem. Biophys. Res. Commun. 1970, 41, 57–62. [Google Scholar]
- Bharadwaj, K.G.; Hiyama, Y.; Hu, Y.; Huggins, L.A.; Ramakrishnan, R.; Abumrad, N.A.; Shulman, G.I.; Blaner, W.S.; Goldberg, I.J. Chylomicron- and VLDL-derived lipids enter the heart through different pathways: In vivo evidence for receptor and non-receptor mediated fatty acid uptake. J. Biol. Chem. 2010, 285, 37976–37986. [Google Scholar]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar]
- Lamb, T.D.; Pugh, E.N., Jr. Dark adaptation and the retinoid cycle of vision. Prog. Retin. Eye Res. 2004, 23, 308–380. [Google Scholar]
- Travis, G.H.; Golczak, M.; Moise, A.R.; Palczewski, K. Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 469–512. [Google Scholar]
- Tsutsumi, C.; Okuno, M.; Tannous, L.; Piantedosi, R.; Allan, M.; Goodman, D.S.; Blaner, W.S. Retinoids and retinoid-binding protein expression in rat adipocytes. J. Biol. Chem. 1992, 267, 1805–1810. [Google Scholar] [PubMed]
- Bonet, M.L.; Ribot, J.; Felipe, F.; Palou, A. Vitamin A and the regulation of fat reserves. Cell. Mol. Life Sci. 2003, 60, 1311–1321. [Google Scholar]
- Okuno, M.; Caraveo, V.E.; Goodman, D.S.; Blaner, W.S. Regulation of adipocyte gene expression by retinoic acid and hormones: effects on the gene encoding cellular retinol-binding protein. J. Lipid Res. 1995, 36, 137–147. [Google Scholar]
- Vogel, S.; Mendelsohn, C.L.; Mertz, J.R.; Piantedosi, R.; Waldburger, C.; Gottesman, M.E.; Blaner, W.S. Characterization of a new member of the fatty acid-binding protein family that binds all-trans-retinol. J. Biol. Chem. 2001, 276, 1353–1360. [Google Scholar] [PubMed]
- Zizola, C.F.; Schwartz, G.J.; Vogel, S. Cellular retinol-binding protein type III is a PPARgamma target gene and plays a role in lipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1358–E1368. [Google Scholar]
- Caprioli, A.; Zhu, H.; Sato, T.N. CRBP-III:lacZ expression pattern reveals a novel heterogeneity of vascular endothelial cells. Genesis 2004, 40, 139–145. [Google Scholar]
- Haq, R.; Chytil, F. Expression of nuclear retinoic acid receptors in rat adipose tissue. Biochem. Biophys. Res. Commun. 1991, 176, 1539–1544. [Google Scholar]
- Kamei, Y.; Kawada, T.; Kazuki, R.; Sugimoto, E. Retinoic acid receptor gamma 2 gene expression is up-regulated by retinoic acid in 3T3-L1 preadipocytes. Biochem. J. 1993, 193, 807–812. [Google Scholar]
- Berry, D.C.; Noy, N. All-trans-retinoic acid represses obesity and insulin resistance by activating both peroxisome proliferation-activated receptor beta/delta and retinoic acid receptor. Mol. Cell Biol. 2009, 29, 3286–3296. [Google Scholar]
- Kuri-Harcuch, W. Differentiation of 3T3-F442A cells into adipocytes is inhibited by retinoic acid. Differentiation 1982, 23, 164–169. [Google Scholar]
- Zizola, C.F.; Frey, S.K.; Jitngarmkusol, S.; Kadereit, B.; Yan, N.; Vogel, S. Cellular retinol-binding protein type I (CRBP-I) regulates adipogenesis. Mol. Cell. Biol. 2010, 30, 3412–3420. [Google Scholar]
- Abel, E.D.; Peroni, O.; Kim, J.K.; Kim, Y.B.; Boss, O.; Hadro, E.; Minnemann, T.; Shulman, G.I.; Kahn, B.B. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 2001, 409, 729–733. [Google Scholar]
- Muoio, D.M.; Newgard, C.B. Metabolism: A is for adipokine. Nature 2005, 436, 337–338. [Google Scholar]
- Tamori, Y.; Sakaue, H.; Kasuga, M. RBP4, an unexpected adipokine. Nat. Med. 2006, 12, 30–31. [Google Scholar]
- Wolf, G. Serum retinol-binding protein: a link between obesity, insulin resistance, and type 2 diabetes. Nutr. Rev. 2007, 65, 251–256. [Google Scholar]
- Graham, T.E.; Yang, Q.; Blüher, M.; Hammarstedt, A.; Ciaraldi, T.P.; Henry, R.R.; Wason, C.J.; Oberbach, A.; Jansson, P.A.; Smith, U.; Kahn, B.B. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N. Engl. J. Med. 2006, 354, 2552–2563. [Google Scholar]
- Li, W.P.; Cheng, Q.F.; Ma, S.K.; Liu, X.R.; Qin, H.; Bai, X.S.; Zhang, S.H.; Li, Q.F. Elevated serum RBP4 is associated with insulin resistance in women with polycystic ovary syndrome. Endocrine 2006, 30, 283–287. [Google Scholar]
- Klöting, N.; Graham, T.E.; Berndt, J.; Kralisch, S.; Kovacs, P.; Wason, C.J.; Fasshauer, M.; Schön, M.R.; Stumvoll, M.; Blüher, M.; Kahn, B.B. Serum retinol-binding protein is more highly expressed in visceral than in subcutaneous adipose tissue and is a marker of intra-abdominal fat mass. Cell Metab. 2007, 6, 79–87. [Google Scholar]
- Aeberli, I.; Biebinger, R.; Lehmann, R.; L’allemand, D.; Spinas, G.A.; Zimmermann, M.B. Serum retinol-binding protein 4 concentration and its ratio to serum retinol are associated with obesity and metabolic syndrome components in children. J. Clin. Endocrinol. Metab. 2007, 92, 4359–4365. [Google Scholar]
- Mills, J.P.; Furr, H.C.; Tanumihardjo, S.A. Retinol to retinol-binding protein (RBP) is low in obese adults due to elevated apo-RBP. Exp. Biol. Med. (Maywood) 2008, 233, 1255–1261. [Google Scholar]
- Erikstrup, C.; Mortensen, O.H.; Nielsen, A.R.; Fischer, C.P.; Plomgaard, P.; Petersen, A.M.; Krogh-Madsen, R.; Lindegaard, B.; Erhardt, J.G.; Ullum, H.; Benn, C.S.; Pedersen, B.K. RBP-to-retinol ratio, but not total RBP, is elevated in patients with type 2 diabetes. Diabetes Obes. Metab. 2009, 11, 204–212. [Google Scholar]
- Janke, J.; Engeli, S.; Boschmann, M.; Adams, F.; Böhnke, J.; Luft, F.C.; Sharma, A.M.; Jordan, J. Retinol-binding protein 4 in human obesity. Diabetes 2006, 55, 2805–2810. [Google Scholar]
- Vitkova, M.; Klimcakova, E.; Kovacikova, M.; Valle, C.; Moro, C.; Polak, J.; Hanacek, J.; Capel, F.; Viguerie, N.; Richterova, B.; Bajzova, M.; Hejnova, J.; Stich, V.; Langin, D. Plasma levels and adipose tissue messenger ribonucleic acid expression of retinol-binding protein 4 are reduced during calorie restriction in obese subjects but are not related to diet-induced changes in insulin sensitivity. J. Clin. Endocrinol. Metab. 2007, 92, 2330–2335. [Google Scholar]
- Broch, M.; Vendrell, J.; Ricart, W.; Richart, C.; Fernández-Real, J.M. Circulating retinol-binding protein-4, insulin sensitivity, insulin secretion, and insulin disposition index in obese and nonobese subjects. Diabetes Care 2007, 30, 1802–1806. [Google Scholar]
- Promintzer, M.; Krebs, M.; Todoric, J.; Luger, A.; Bischof, M.G.; Nowotny, P.; Wagner, O.; Esterbauer, H.; Anderwald, C. Insulin resistance is unrelated to circulating retinol binding protein and protein C inhibitor. J. Clin. Endocrinol. Metab. 2007, 92, 4306–4312. [Google Scholar]
- Bajzová, M.; Kováciková, M.; Vítková, M.; Klimcáková, E.; Polák, J.; Kovácová, Z.; Viguerie, N.; Vedral, T.; Mikulásek, L.; Srámková, P.; Srp, A.; Hejnová, J.; Langin, D.; Stich, V. Retinol-binding protein 4 expression in visceral and subcutaneous fat in human obesity. Physiol. Res. 2008, 57, 927–934. [Google Scholar]
- Von Eynatten, M.; Humpert, P.M. Retinol-binding protein-4 in experimental and clinical metabolic disease. Expert Rev. Mol. Diagn. 2008, 8, 289–299. [Google Scholar]
- Graham, T.E.; Kahn, B.B. Tissue-specific alterations of glucose transport and molecular mechanisms of intertissue communication in obesity and type 2 diabetes. Horm. Metab. Res. 2007, 39, 717–721. [Google Scholar]
- Esteve, E.; Ricart, W.; Fernández-Real, J.M. Adipocytokines and insulin resistance: the possible role of lipocalin-2, retinol binding protein-4, and adiponectin. Diabetes Care 2009, 32, S362–S367. [Google Scholar]
- Zhou, M.D.; Sucov, H.M.; Evans, R.M.; Chien, K.R. Retinoid-dependent pathways suppress myocardial cell hypertrophy. Proc. Natl. Acad. Sci. USA 1995, 92, 7391–7395. [Google Scholar]
- Wu, J.; Garami, M.; Cheng, T.; Gardner, D.G. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J. Clin. Invest. 1996, 97, 1577–1588. [Google Scholar]
- Wang, H.J.; Zhu, Y.C.; Yao, T. Effects of all-trans retinoic acid on angiotensin II-induced myocyte hypertrophy. J. Appl. Physiol. 2002, 92, 2162–2168. [Google Scholar]
- He, Y.; Huang, Y.; Zhou, L.; Lu, L.M.; Zhu, Y.C.; Yao, T. All-trans retinoic acid inhibited angiotensin II-induced increase in cell growth and collagen secretion of neonatal cardiac fibroblasts. Acta Pharmacol. Sin. 2006, 27, 423–429. [Google Scholar]
- Paiva, S.A.; Matsubara, L.S.; Matsubara, B.B.; Minicucci, M.F.; Azevedo, P.S.; Campana, A.O.; Zornoff, L.A. Retinoic acid supplementation attenuates ventricular remodeling after myocardial infarction in rats. J. Nutr. 2005, 135, 2326–2328. [Google Scholar]
- Choudhary, R.; Palm-Leis, A.; Scott, R.C., III; Guleria, R.S.; Rachut, E.; Baker, K.M.; Pan, J. All-trans retinoic acid prevents development of cardiac remodeling in aortic banded rats by inhibiting the renin-angiotensin system. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H633–H644. [Google Scholar] [PubMed]
- Palace, V.P.; Hill, M.F.; Khaper, N.; Singal, P.K. Metabolism of vitamin A in the heart increases after a myocardial infarction. Free Radic. Biol. Med. 1999, 26, 1501–1507. [Google Scholar]
- Azevedo, P.S.; Minicucci, M.F.; Chiuso-Minicucci, F.; Justulin, L.A., Jr.; Matsubara, L.S.; Matsubara, B.B.; Novelli, E.; Seiva, F.; Ebaid, G.; Campana, A.O.; Zornoff, L.A.; Paiva, S.A. Ventricular remodeling induced by tissue vitamin A deficiency in rats. Cell. Physiol. Biochem. 2010, 26, 395–402. [Google Scholar]
- Manna, A.; Cadenotti, L.; Motto, A.; Ballo, P. Reversible cardiac dysfunction without myocytolysis related to all-trans retinoic acid administration during induction therapy of acute promyelocytic leukemia. Ann. Hematol. 2009, 88, 91–92. [Google Scholar]
- Wald, G. Molecular basis of visual excitation. Science 1968, 162, 230–239. [Google Scholar]
- Saari, J.C. Retinoids in mammalian vision. In The Handbook of Experimental Pharmacology, The Retinoids; Nau, H., Blaner, W.S., Eds.; Springer Verlag: Heidelberg, Germany, 1999; pp. 563–588. [Google Scholar]
- Gollapalli, D.R.; Rando, R.R. All-trans-retinyl esters are the substrates for isomerization in the vertebrate visual cycle. Biochemistry 2003, 42, 5809–5818. [Google Scholar]
- Moiseyev, G.; Crouch, R.K.; Goletz, P.; Oatis, J.D.; Redmond, T.M.; Ma, J.X. Retinyl esters are the substrate for isomerohydrolase. Biochemistry 2003, 42, 2229–2238. [Google Scholar]
- Mata, N.L.; Moghrabi, W.N.; Lee, J.S.; Bui, T.V.; Radu, R.A.; Tsin, A.C. Rpe65 is a retinyl ester binding protein that presents insoluble substrate to the isomerase in retinal pigment epithelial cells. J. Biol. Chem. 2004, 279, 635–643. [Google Scholar]
- Jin, M.; Li, S.; Moghrabi, W.N.; Sun, H.; Travis, G.H. Rpe65 is the retinoid isomerase in bovine retinal pigment epithelium. Cell 2005, 122, 449–459. [Google Scholar]
- Moiseyev, G.; Chen, Y.; Takahashi, Y.; Wu, B.X.; Ma, J.X. RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc. Natl. Acad. Sci. USA 2005, 102, 12413–12418. [Google Scholar]
- Redmond, T.M.; Poliakov, E.; Yu, S.; Tsai, J.Y.; Lu, Z.; Gentleman, S. Mutation of key residues of Rpe65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc. Natl. Acad. Sci. USA 2005, 102, 13658–13663. [Google Scholar]
- Redmond, T.M.; Yu, S.; Lee, E.; Bok, D.; Hamasaki, D.; Chen, N.; Goletz, P.; Ma, J.X.; Crouch, R.K.; Pfeifer, K. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat. Genet. 1998, 20, 344–351. [Google Scholar]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar]
- Illing, M.; Molday, L.L.; Molday, R.S. The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J. Biol. Chem. 1997, 272, 10303–10310. [Google Scholar]
- Azarian, S.M.; Travis, G.H. The photoreceptor rim protein is an ABC transporter encoded by the gene for recessive Stargardt’s disease. FEBS Lett. 1997, 409, 247–252. [Google Scholar]
- Weng, J.; Mata, N.L.; Azarian, S.M.; Tzekov, R.T.; Birch, D.G.; Travis, G.H. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell 1999, 98, 13–23. [Google Scholar]
- Mata, N.L.; Weng, J.; Travis, G.H. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc. Natl. Acad. Sci. USA 2000, 97, 7154–7159. [Google Scholar]
- Sun, H.; Molday, R.S.; Nathans, J. Retinal stimulates ATP hydrolysis by purified adn reconstituted ABCR, the photoreceptor-specific ATP-binding cassette transporter responsible for Stargradt disease. J. Biol. Chem. 2004, 275, 53972–53979. [Google Scholar]
- Beharry, S.; Zhong, M.; Molday, R.S. N-retinylidene-phosphatidylethanolamine is the preferred retinoid substrate for the photoreceptor-specific ABC transporter ABCA4 (ABCR). J. Biol. Chem. 2004, 273, 21790–21799. [Google Scholar]
- Rattner, A.; Smallwood, P.M.; Nathans, J. Identification and characterization of all-trans-retinol dehydrogenase from photoreceptor outer segments, the visual cycle enzyme that reduces all-trans-retinal to all-trans-retinol. J. Biol. Chem. 2000, 275, 11034–11043. [Google Scholar]
- Maeda, A.; Maeda, T.; Imanishi, Y.; Kuksa, V.; Alekseev, A.; Bronson, J.D.; Zhang, H.; Sun, W.; Saperstein, D.A.; Rieke, F.; Baehr, W.; Palczewski, K. Role of photoreceptor-specific retinol dehydrogenase in the retinoid cycle in vivo. J. Biol. Chem. 2005, 280, 45537–45546. [Google Scholar]
- Parker, R.O.; Crouch, R.K. Retinol dehydrogenases (RDHs) in the visual cycle. Exp. Eye Res. 2010, 91, 788–792. [Google Scholar]
- Maeda, A.; Maeda, T.; Sun, W.; Zhang, H.; Baehr, W.; Palczewski, K. Redundant and unique roles of retinol dehydrogenases in the mouse retina. Proc. Natl. Acad. Sci. USA 2007, 104, 19565–19570. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A Metabolism: An Update. Nutrients 2011, 3, 63-103. https://doi.org/10.3390/nu3010063
D’Ambrosio DN, Clugston RD, Blaner WS. Vitamin A Metabolism: An Update. Nutrients. 2011; 3(1):63-103. https://doi.org/10.3390/nu3010063
Chicago/Turabian StyleD’Ambrosio, Diana N., Robin D. Clugston, and William S. Blaner. 2011. "Vitamin A Metabolism: An Update" Nutrients 3, no. 1: 63-103. https://doi.org/10.3390/nu3010063
APA StyleD’Ambrosio, D. N., Clugston, R. D., & Blaner, W. S. (2011). Vitamin A Metabolism: An Update. Nutrients, 3(1), 63-103. https://doi.org/10.3390/nu3010063