Abstract
The requirement for vitamin A in reproduction was first recognized in the early 1900’s, and its importance in the eyes of developing embryos was realized shortly after. A greater understanding of the large number of developmental processes that require vitamin A emerged first from nutritional deficiency studies in rat embryos, and later from genetic studies in mice. It is now generally believed that all-trans retinoic acid (RA) is the form of vitamin A that supports both male and female reproduction as well as embryonic development. This conclusion is based on the ability to reverse most reproductive and developmental blocks found in vitamin A deficiency induced either by nutritional or genetic means with RA, and the ability to recapitulate the majority of embryonic defects in retinoic acid receptor compound null mutants. The activity of the catabolic CYP26 enzymes in determining what tissues have access to RA has emerged as a key regulatory mechanism, and helps to explain why exogenous RA can rescue many vitamin A deficiency defects. In severely vitamin A-deficient (VAD) female rats, reproduction fails prior to implantation, whereas in VAD pregnant rats given small amounts of carotene or supported on limiting quantities of RA early in organogenesis, embryos form but show a collection of defects called the vitamin A deficiency syndrome or late vitamin A deficiency. Vitamin A is also essential for the maintenance of the male genital tract and spermatogenesis. Recent studies show that vitamin A participates in a signaling mechanism to initiate meiosis in the female gonad during embryogenesis, and in the male gonad postnatally. Both nutritional and genetic approaches are being used to elucidate the vitamin A-dependent pathways upon which these processes depend.
1. Background
It has been nearly 100 years since the essential micronutrient, vitamin A, was first described. In 1913 McCollum and Davis reported that the addition of an ether extract from egg yolk or butter, but not lard or olive oil, could reinstate growth in rats maintained for several months on a purified ration of casein, carbohydrates and salt mixtures [1]. We now know that this essential accessory article in foodstuffs is vitamin A. Using dietary manipulation to induce deficiency in rats, the importance of vitamin A in both male and female reproduction was soon discovered [2,3]. For a time, there was confusion over how both yellow-colored foods and those that were colorless, for example extract from pork liver or cod liver oil, could both yield vitamin A activity [4]. This problem was solved when Moore fed carotenoids to vitamin A-deficient (VAD) rats in amounts that enabled the animals to resume normal growth, and showed that only the “colorless” form of vitamin A was found in the livers collected from these animals [5]. Thus, it was realized that carotenoids (at least a subset) could be converted to vitamin A, a fact that was fully appreciated when Karrer et al. published the chemical structures of both carotene and vitamin A [6,7]. In 1946, Arens and van Dorp synthesized vitamin A acid (retinoic acid), and reported it was as potent as vitamin A in supporting the growth of VAD rats but could not be converted back to vitamin A [8,9,10]. The metabolic scheme in which vitamin A (retinol) generates the vitamin A aldehyde (retinaldehyde) to support synthesis of the visual pigments, and its further irreversible oxidation to the vitamin A acid (all-trans retinoic acid, RA) that supports growth and tissue maintenance was first reported in the landmark paper by Dowling and Wald and this metabolic scheme stands essentially unchanged today (Figure 1A) [11].
All-trans retinol (retinol, vitamin A) is obtained in the diet from plant sources (carotenoids with vitamin A activity) or as retinyl esters from animal sources. Retinol has two major fates: (1) esterification and tissue storage, and (2) oxidative metabolism to all-trans retinaldehyde and further oxidation to RA. The enzyme lecithin:retinol acyltransferase (LRAT) is responsible for esterifying the majority of retinol into retinyl esters [12]. The first and rate-limiting step in the production of RA from retinol results from the action of cytosolic alcohol dehydrogenases (ADH) and microsomal retinol dehydrogenases (RDH) yielding all-trans retinaldehyde [13]. The irreversible oxidation of all-trans retinaldehyde to all-trans retinoic acid is catalyzed by several aldehyde dehydrogenases (RALDH), of the ALDH1A class (ALDH 1A1, 1A2, and 1A3 also known as RALDH 1, 2 and 3) [14,15]. RALDH-independent generation of RA from retinol by CYP1B1 has also been reported [16]. The metabolism of RA at the C4 and C18 positions to oxidative metabolites including 4-hydroxy-RA, 18-hydroxy-RA, and 4-oxo-RA occurs by the action of cytochrome P450 enzymes of the CYP26 family (A1, B1 and C1) [17,18,19,20,21,22,23]. Vitamin A and metabolites are lipophilic compounds that are generally found in association with serum and cellular binding proteins [24]. Retinol-binding protein (RBP or RBP4) carries the majority of retinol in the circulation [25], and a membrane receptor, STRA6, binds to RBP to enable efficient retinol uptake by a number of cells [26]. In addition, cellular proteins that bind to retinol (CRBP I, II, and III), and RA (CRABP I and II) have been studied in null mutant mice; some have been found to be dispensable, while roles for others have been revealed when animals are fed a vitamin A-restricted diet [12,27,28].
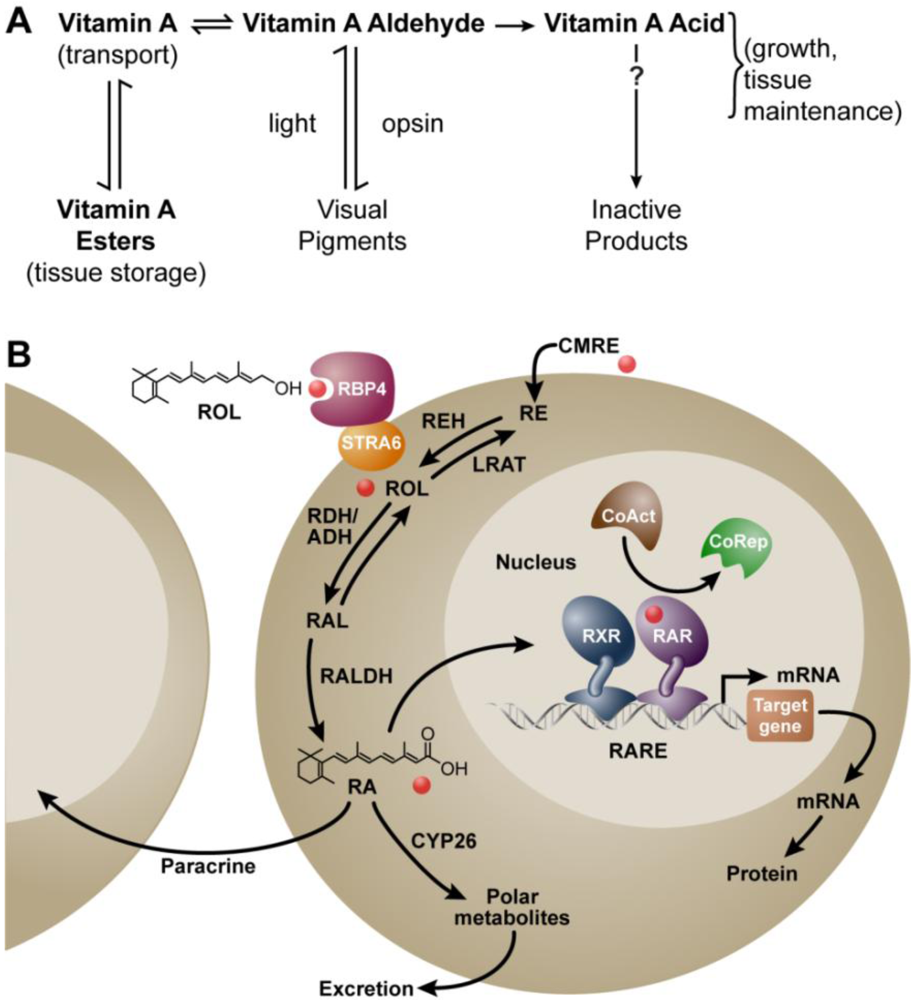
Figure 1.
Metabolism of vitamin A (retinol) to all-trans retinoic acid (RA), and the mechanism of RA action. (A) Metabolic scheme proposed by Dowling and Wald in 1960 [11]; (B) Mechanism ca. 2011. Vitamin A (retinol, ROL) circulates bound to the plasma retinol-binding protein (RBP4) and transthyretin (not shown). RBP4 binds to the membrane receptor STRA6 to facilitate the cellular uptake of retinol in some cells. Vitamin A circulating as part of a chylomicron remnant (CMRE) can also serve as a source of vitamin A for the cell. Note that cellular retinol and RA binding proteins have been omitted for simplicity. Retinol is either esterified by lecithin:retinol acyltransferase (LRAT) and stored, or is oxidized reversibly to retinaldehyde (RAL) by retinol dehydrogenases (RDH/ADH), and further oxidized in irreversible fashion to RA by retinaldehyde dehydrogenase (RALDH 1, 2, or 3). In the nucleus, the RAR/RXR complex is bound to a specific sequence of DNA called the retinoic acid response element (RARE). Binding of RA to the RAR leads to release of the corepressor complex (CoRep) and association with coactivator proteins (CoAct), followed by altered transcription of downstream target genes and ultimately changes in cellular function. RA also undergoes further oxidation by the cytochrome P450 (CYP) 26 family to more polar metabolites. The lipophilic molecule, RA, can act within the same cell in which it is synthesized (autocrine) or can diffuse through the cell membrane to act in nearby cells (paracrine). Abbreviations: ADH, alcohol dehydrogenase; RDH, retinol dehydrogenase; REH, retinyl ester hydrolase; RE, retinyl ester.
Figure 1.
Metabolism of vitamin A (retinol) to all-trans retinoic acid (RA), and the mechanism of RA action. (A) Metabolic scheme proposed by Dowling and Wald in 1960 [11]; (B) Mechanism ca. 2011. Vitamin A (retinol, ROL) circulates bound to the plasma retinol-binding protein (RBP4) and transthyretin (not shown). RBP4 binds to the membrane receptor STRA6 to facilitate the cellular uptake of retinol in some cells. Vitamin A circulating as part of a chylomicron remnant (CMRE) can also serve as a source of vitamin A for the cell. Note that cellular retinol and RA binding proteins have been omitted for simplicity. Retinol is either esterified by lecithin:retinol acyltransferase (LRAT) and stored, or is oxidized reversibly to retinaldehyde (RAL) by retinol dehydrogenases (RDH/ADH), and further oxidized in irreversible fashion to RA by retinaldehyde dehydrogenase (RALDH 1, 2, or 3). In the nucleus, the RAR/RXR complex is bound to a specific sequence of DNA called the retinoic acid response element (RARE). Binding of RA to the RAR leads to release of the corepressor complex (CoRep) and association with coactivator proteins (CoAct), followed by altered transcription of downstream target genes and ultimately changes in cellular function. RA also undergoes further oxidation by the cytochrome P450 (CYP) 26 family to more polar metabolites. The lipophilic molecule, RA, can act within the same cell in which it is synthesized (autocrine) or can diffuse through the cell membrane to act in nearby cells (paracrine). Abbreviations: ADH, alcohol dehydrogenase; RDH, retinol dehydrogenase; REH, retinyl ester hydrolase; RE, retinyl ester.
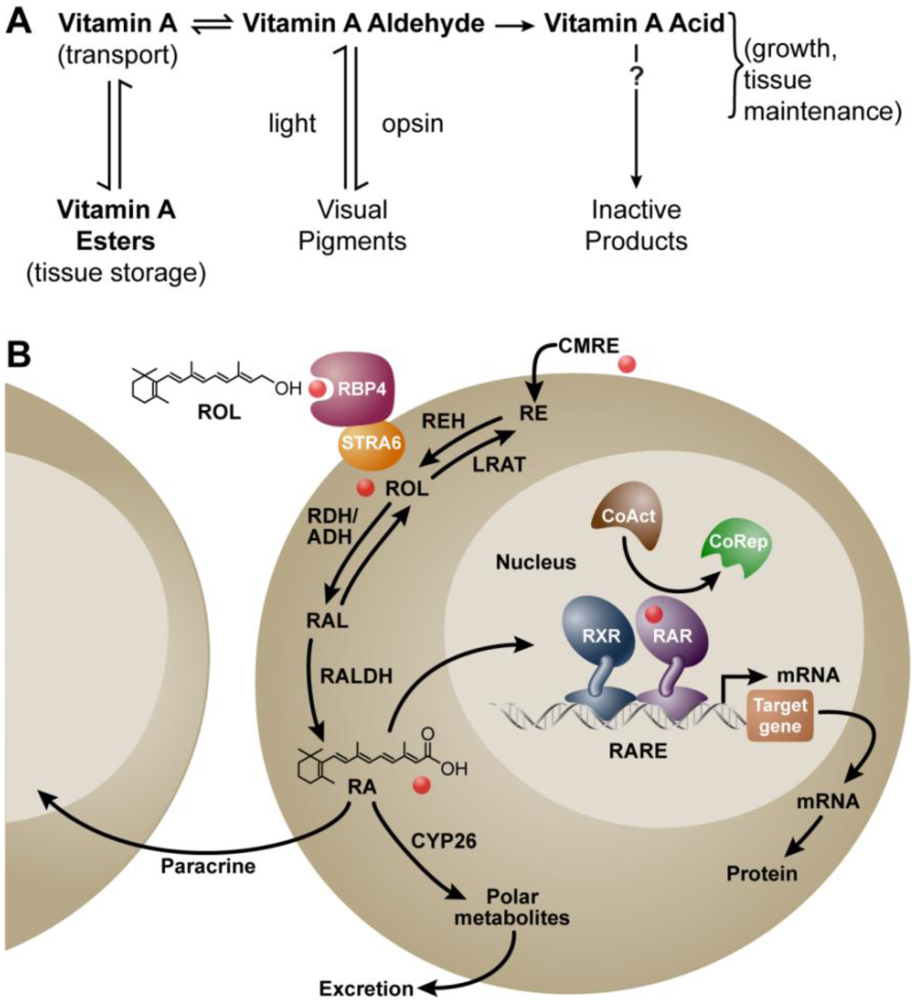
RA is a ligand for the nuclear retinoic acid receptor (RAR) proteins. There are three major subtypes of RAR protein (α, β and γ) with additional isoforms resulting from alternate promoter usage and splicing [29]. The nuclear RARs act as ligand-activated transcription factors to regulate gene transcription in a cell-type and tissue-specific manner [30]. The all-trans isomer of RA is the highest affinity endogenous ligand for the RAR [31]. Members of a second protein family, the retinoid (rexinoid)-X receptors (RXR) heterodimerize with RAR to confer high-affinity binding to DNA. The DNA to which the RAR/RXR heterodimer binds is called a retinoic acid response element (RARE). The consensus RARE is composed of two direct repeats of PuG(G/T)TCA separated most often by 5 bases [32]. However, more complex elements that do not adhere to this rule have been described [33]. RAREs serve as enhancer elements and when occupied by the RA/RAR/RXR complex, facilitate chromatin opening and changes in RA target gene transcriptional activity [34,35]. A large number of genes are altered when cells or tissues are exposed to RA, however, only a small subset are primary (direct) targets via RARE-mediated transcription, with the remainder representing downstream targets [36,37]. A schematic summarizing the metabolism of vitamin A and the cellular mechanism of RA action is shown in Figure 1B. Note in this review retinoid is a term that refers to compounds structurally-related to retinol, and in this review, is used to refer to vitamin A and its metabolites.
2. Vitamin A and Reproduction
2.1. Male Reproduction
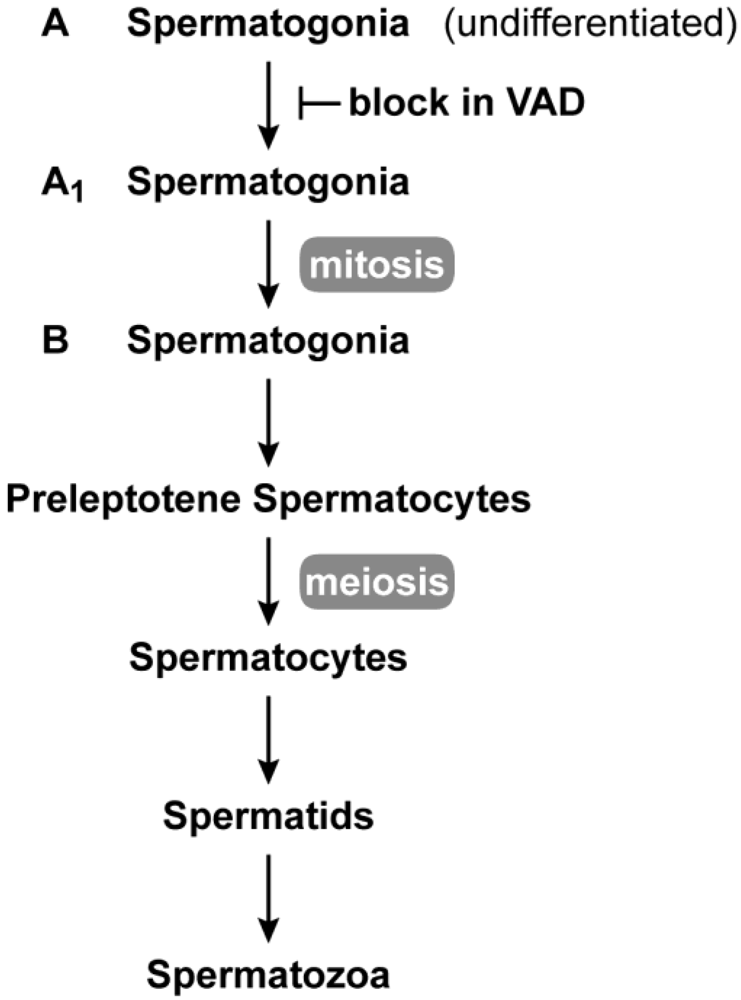
Vitamin A is required for male reproduction. Early work in the laboratories of Wolbach and Howe as well as Mason showed that in vitamin A deficiency, the epithelia of the epididymis, prostate, and seminal vesicle is replaced with stratified squamous keratinizing epithelium, and spermatogenesis ceases [2,38]. Later work showed that in the VAD rat testes, undifferentiated spermatogonia, Sertoli cells and a small number of preleptotene spermatocytes remain [39,40,41], whereas in the mouse, spermatogenesis is arrested at the spermatogonia stage [42]. Upon addition of vitamin A, spermatogenesis can be reinstituted by stimulating A to A1 spermatogonial differentiation in a synchronized manner [42,43,44]. The block in adult spermatogenesis resulting from vitamin A deficiency is shown in Figure 2. Recent work supports the conclusion that the vitamin A metabolite, RA, is needed both for adult male spermatogonial differentiation (transition to A1) and the entrance into meiosis [45,46,47].
In 1991, Van Pelt and de Rooij determined that a large dose of RA (5 mg) given by injection twice a week, when combined with a RA-containing diet, supported the development of spermatocytes, and their subsequent development into spermatids in VAD rats supporting that RA is the active form of vitamin A in male reproduction [48]. A CYP26-mediated catabolic barrier comprised of peritubular myoid cells surrounds the seminiferous tubule, and may prevent RA in the general circulation from reaching cells in the interior of the tubule, thus explaining why such high doses of exogenous RA were required [49,50]. Within the normal tubule, the Sertoli cell is believed to generate RA by the action of Raldh1 [49,51,52], and possibly Raldh2[50]. Raldh2 is also found in late pachytene and diplotene spermatocytes, and early stage spermatids.
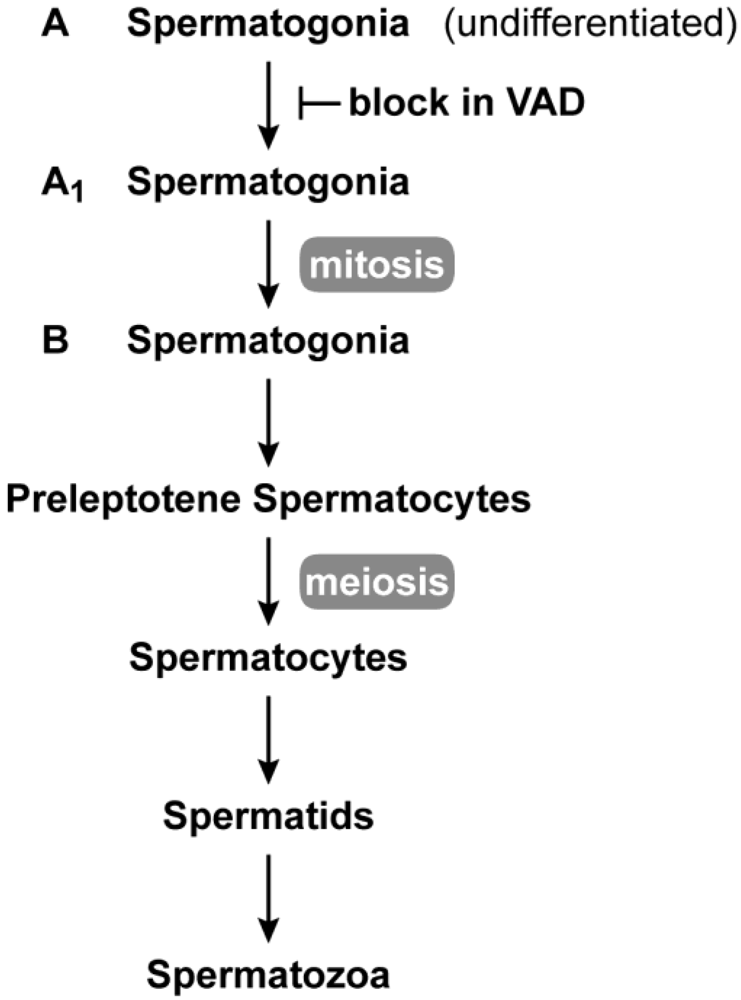
Figure 2.
Spermatogenesis in the adult. Spermatogenesis takes place in the seminiferous epithelium of testis tubules from puberty through adulthood. Undifferentiated (A-type) spermatogonia at the base of the seminiferous epithelium divide mitotically until they enter the differentiation pathway to become A1 spermatogonia. A1 spermatogonia undergo division to A1-A4 and finally B spermatogonia. B spermatogonia divide to produce preleptotene (primary) spermatocytes that migrate away from the base of the seminiferous tubule to undergo meiosis. The first meiotic division produces secondary spermatocytes, and after the second meiotic division, spermatids (haploid cells) begin the differentiation process (spermiogenesis) to spermatozoa. In vitamin A deficiency, the transition from A to A1 spermatogonia is blocked [42,43,44].
Figure 2.
Spermatogenesis in the adult. Spermatogenesis takes place in the seminiferous epithelium of testis tubules from puberty through adulthood. Undifferentiated (A-type) spermatogonia at the base of the seminiferous epithelium divide mitotically until they enter the differentiation pathway to become A1 spermatogonia. A1 spermatogonia undergo division to A1-A4 and finally B spermatogonia. B spermatogonia divide to produce preleptotene (primary) spermatocytes that migrate away from the base of the seminiferous tubule to undergo meiosis. The first meiotic division produces secondary spermatocytes, and after the second meiotic division, spermatids (haploid cells) begin the differentiation process (spermiogenesis) to spermatozoa. In vitamin A deficiency, the transition from A to A1 spermatogonia is blocked [42,43,44].
RARγ null males are sterile and exhibit squamous metaplasia of the seminal vesicles and prostate glands [53]. RARα null mutants are sterile and show a reduction in spermatozoa, indicating that nuclear RAR is needed for spermiation [54]. RARα is expressed primarily in the Sertoli cell, Rarβ in spermatids and RARγ in A spermatogonia [49]. Expression of RARα is needed for differentiation of the spermatogonia during prepuberty [55]. However, RAR signaling in the Sertoli cell cannot account for the VAD-induced arrest in spermatogonia differentiation, as deletion of all RARs in the Sertoli cell does not cause arrest of spermatogonia differentiation in the adult mouse. The cell types in which RA and its receptors act to support spermatogenesis continues to be a subject of active investigation [45,49,55,56,57].
2.2. Female Reproduction
In the female, the effect of vitamin A deficiency on reproductive outcome is dependent upon the time when deficiency is imposed, as well as its severity [58]. When severe vitamin A deficiency is imposed prior to mating, cornified cells are continuously present in vaginal smears [59,60] and reproduction fails prior to implantation [3]. VAD female rats continue to ovulate and form corpora lutea irregularly or at normal intervals, however, degenerated eggs are found in the last portion of the tube, and there is no evidence that blastogenesis has occurred.
Warkany and Schraffenberger showed that when limited amounts of provitamin A carotenoid are provided to VAD female rats prior to mating, a less severe maternal vitamin A deficiency is produced which enables fertilization and implantation to occur, but embryonic death at midgestation often results [61]. If provided in adequate amounts, retinol will support reproduction and embryonic development in full [62]. RA in amounts ranging from 2 to 12 mcg/g diet or 40 to 230 mcg/rat/day given to a VAD female rat is sufficient to maintain normal fertilization, implantation and early embryogenesis. However, pregnant animals maintained on this level of RA will invariably resorb all the fetuses. Higher amounts of RA (250 mcg/g diet or approximately 4.5 mg/rat/day) or retinol is needed by embryonic day 8.5 (E8.5) (late gastrula/early neurula) to support normal embryonic development and overcome midgestational resorption [63,64,65,66,67].
Maternal vitamin A also plays a role in placental development and/or maintenance, as the chorioallantoic placenta undergoes widespread necrosis by E15.5 in VAD rats supported on insufficient amounts of RA [68]. Microscopic analysis of the placenta from E14 to E17 reveals changes in the central region of the junctional zone and the labyrinthine zone of VAD rats supported on insufficient vitamin A acid, whereas addition of retinyl acetate to the diet prevents these changes [69]. Microscopic findings suggest the differentiation of the parenchymal cells of the junctional zone to glycogen cells, and of the chorionic trophoblast cells to the inner trophoblastic lamina of the trichorial placenta is affected. In summary, the relative vitamin A status of the female, both at the time of conception and throughout pregnancy, is a critical determinant in reproductive outcome, and deficiency can lead to either a complete failure of reproduction prior to implantation or fetal resorption or malformation.
2.3. Germ Cell Development
The generation of sperm and oocytes requires germ cells to undergo meiosis, the process in which diploid cells give rise to haploid cells. In the female, germ cells enter meiosis I during embryogenesis, whereas in the male, this process occurs postnatally (Figure 3A). It has been proposed that access of primordial germ cells (gonocytes) to RA plays an important role in determining when they will enter meiosis, with female germ cells entering into meiosis after exposure to embryonic RA, whereas, in the male embryo, this pathway is blocked by the action of CYP26B1, but is enabled after birth [70,71,72,73]. However, a report appearing when this manuscript was under review raises questions concerning a role for RA in this model [74].
The culture of embryonic rat ovaries with RA promotes meiotic entry [75], whereas murine embryonic ovaries (E11.5) cultured in the presence of the RAR panantagonist BMS-204493 for 2 days do not express the RA-responsive gene Stra8 [70], a gene that is required for meiotic initiation in female gonocytes [76]. However, a recent report shows that female RALDH2 and RALDH2/3 knockout gonads express Stra8 and undergo meiosis in the absence of detectable RA activity as assessed using a β2-RARE-lacZ (RARE-lacZ) reporter [74]. If the reporter accurately detects all RA activity, this new work indicates that meiotic initiation can occur independently of RA signaling.
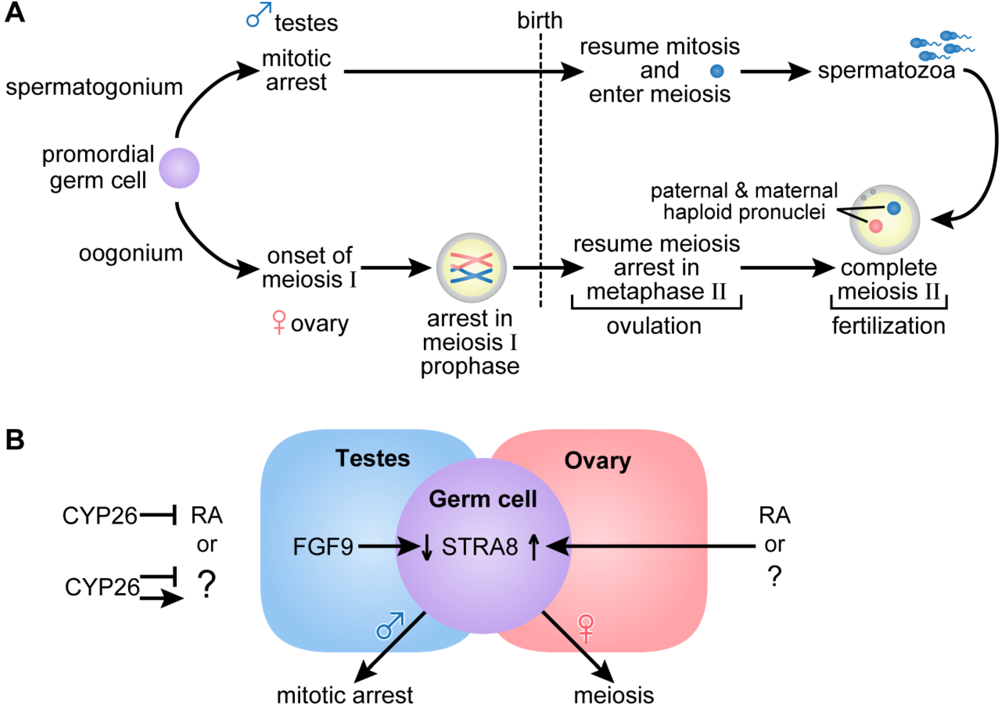
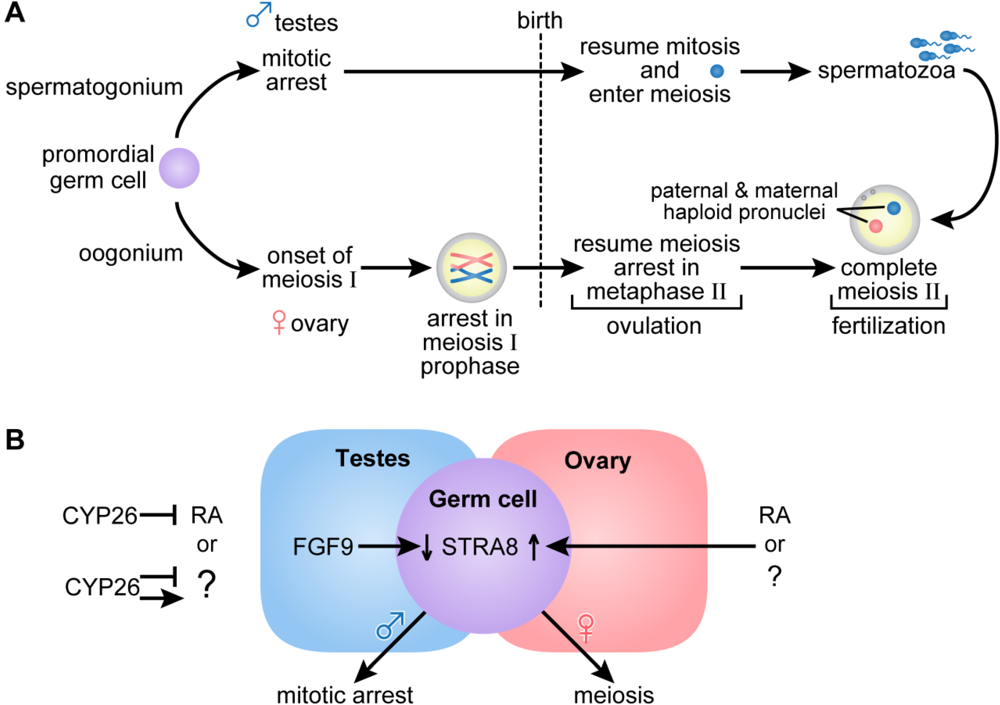
Figure 3.
(A) Germ cell development and gametogenesis. Primordial germ cells colonize the gonad in both male and female embryos. The first morphological marker of sex-specific germ cell development is seen in the female embryo when the oogonia enter meiosis. Primary oocytes proceed through the leptotene, zygotene and pachytene stages of meiotic prophase before birth, when they arrest in diplotene of meiosis I. At ovulation, meiosis I is completed, and the secondary oocytes enter meiosis II and arrest again in metaphase. Meiosis II is completed after fertilization. In the male embryo, germ cells are committed to the spermatogenic program but arrest in G0/G1, and do not complete mitosis and enter meiosis until after they are born. Primary spermatocytes entering meiosis I are seen during the first week of life; secondary spermatocytes complete meiosis II forming spermatids and functional gametes called spermatozoa or sperm. In the male, waves of meiosis continue throughout life. (B) In the female embryo access to RA or alternatively, another factor indicated as (?), promotes entry into meiosis whereas embryonic male germ cells are maintained in a pluripotent state. Either RA or another factor acts in the embryonic female germ cell to increase Stra8, essential for entry into meiosis. In the ovary, Fgf9 levels are low. In the male embryo, entry into meiosis is prevented by the action of CYP26B1; high levels of Fgf9 in the testes antagonize Stra8 expression and maintain germ cells in a pluripotent state. (Adapted from [77,78]).
Figure 3.
(A) Germ cell development and gametogenesis. Primordial germ cells colonize the gonad in both male and female embryos. The first morphological marker of sex-specific germ cell development is seen in the female embryo when the oogonia enter meiosis. Primary oocytes proceed through the leptotene, zygotene and pachytene stages of meiotic prophase before birth, when they arrest in diplotene of meiosis I. At ovulation, meiosis I is completed, and the secondary oocytes enter meiosis II and arrest again in metaphase. Meiosis II is completed after fertilization. In the male embryo, germ cells are committed to the spermatogenic program but arrest in G0/G1, and do not complete mitosis and enter meiosis until after they are born. Primary spermatocytes entering meiosis I are seen during the first week of life; secondary spermatocytes complete meiosis II forming spermatids and functional gametes called spermatozoa or sperm. In the male, waves of meiosis continue throughout life. (B) In the female embryo access to RA or alternatively, another factor indicated as (?), promotes entry into meiosis whereas embryonic male germ cells are maintained in a pluripotent state. Either RA or another factor acts in the embryonic female germ cell to increase Stra8, essential for entry into meiosis. In the ovary, Fgf9 levels are low. In the male embryo, entry into meiosis is prevented by the action of CYP26B1; high levels of Fgf9 in the testes antagonize Stra8 expression and maintain germ cells in a pluripotent state. (Adapted from [77,78]).
There is clear in vivo evidence that vitamin A is required for the normal onset of meiotic prophase in ovarian germ cells [79]. While fetal ovarian germ cell number is unaffected by vitamin A deficiency, the germ cells in embryos with the most severe vitamin A deficiency fail to enter meiosis as evidenced by a lack of immunostaining for SYCP3 (a gene that encodes a component of the synaptonemal complex), and the critical RA-responsive gene, Stra8 is nearly undetectable. When RA is included in the maternal diet at a slightly higher level, but one insufficient to support most other vitamin A-dependent embryonic processes, a small number of cells undergo meiosis (30%) compared to 75% of cells in the vitamin A-sufficient group. Thus, it is possible that only a very low level of RA is needed to initiate meiosis, or alternatively, vitamin A may support meiotic entry by an alternative mechanism.
The mesonephros has been proposed as the source of RA that initiates the entry of primordial gonocytes into meiosis [71]. Raldh2 is largely responsible for RA synthesis in the mesonephros, with some contribution from Raldh3 in the region of the mesonephric duct [71,80]. Expression of Raldh1 mRNA is reported in the adjacent male gonad from E11.5 to 13.5, and to a lesser extent in the female gonad by E13.5 [81]. The gene encoding the RA-metabolizing cytochrome P450 enzyme, Cyp26b1, is expressed early in the genital ridge, but is decreased after E12.5 in the female mouse embryo, thus enabling access of the gonocyte to RA [71]. In contrast, the expression of Cyp26b1 persists in the male embryonic gonad, and has been proposed to prevent RA from stimulating the gonocytes to undergo meiosis [70,71]. Embryonic male germ cells undergo G0/G1 mitotic cell cycle arrest and do not enter meiosis until a week after birth. In CYP26B1 null mutant male embryos, RA levels in the testes are increased and germ cells prematurely enter meiosis at embryonic day 13.5, and this is followed by apoptosis [73]. CYP26B1 is also required later in embryogenesis to maintain cells in an undifferentiated state [82]. However, a new report shows that inhibition of CYP26 activity with ketoconazole results in a similar increase in Stra8 mRNA in cultured wild-type and RALDH2 null mutant testis/mesonephros complexes, whereas no expression is seen in testes cultured in isolation [74]. This suggests that some factor other than RA from the mesonephros may be affected by CYP26, and thus control entry into meiosis. Thus, either a very low level of RA that is undetectable by the RARE-lacZ reporter and that is generated by an enzyme other than RALDH2 is responsible for meiotic entry, or alternatively, CYP26B1 functions by degrading an unknown inducer of Stra8 or participates in the synthesis of a factor that inhibits Stra8 expression.
Recently, Li et al. showed that vitamin A is required in the male neonate for germ cells to enter the first round of spermatogenesis [83]. Vitamin A deficiency was produced in prepubertal life by maintaining LRAT−/− female mice on a diet devoid of all vitamin A during pregnancy and lactation. It was possible to generate deficiency at this time because LRAT−/− mice cannot store retinol in the form of retinyl esters in most tissues, and thus the mothers and pups can be more rapidly depleted of their vitamin A stores than wild-type mice. In the wild-type mouse, Stra8 begins to increase in the testis by postnatal day 6 [84] and spermatogenesis begins shortly thereafter [85]. However, in the VAD LRAT−/− gonad the expected increase in Stra8 did not occur. Germ cells, although present in normal numbers, did not undergo meiosis as was observed in the vitamin A-sufficient controls and instead remained undifferentiated [83]. The addition of retinol to VAD LRAT−/− pups at postnatal day 5 prevented meiotic failure. Thus, it appears that an environment depleted of vitamin A maintains germ cells in an undifferentiated state, and vitamin A sufficiency enables entry into meiosis I in both the developing female and male gonad.
3. Vitamin A and Embryonic Development
3.1. Embryonic Vitamin A Deficiency Studies
Very early evidence that vitamin A is required for embryonic development came from reports of abnormalities in pigs born to gilts on a VAD diet [86,87]. A range of reproductive outcomes were reported in gilts fed a vitamin A-free ration from 160 days before breeding and for 30 days thereafter. Some gilts on the deficient diet failed to show the symptoms of estrous, whereas others became pregnant, but resorbed all the fetuses. The remaining offspring from litters that survived to term showed a lack of eye development or no eyes. Cleft palate, accessory ears and arrested ascension of the kidneys were also observed, but at a lower frequency. In the 1940’s, Warkany and colleagues published their landmark studies describing a syndrome of defects in rat embryos from VAD mothers given limited amounts of carotene [61,88,89,90,91,92,93,94,95]. Approximately 70% of the VAD female rats supplemented in this fashion conceived, however, the majority of pregnancies did not continue beyond midgestation [61]. Ocular defects were most frequently observed in offspring from the few pregnancies that progressed to term. In order to understand how maternal vitamin A deficiency affected development, fetuses ranging from embryonic days 12.5 to 20.5 (near term) were taken by cesarean section whenever evidence of maternal bleeding was observed, and histology was performed. Of the fetuses recovered in this fashion, defects in eye development were most frequent (49% with one or more abnormality) and included coloboma, retinal eversion, penetration of the retina by mesodermal tissue, low insertion of the optic stalk and the cup, and defects in the iris. Defects at lower penetrance were noted in other systems including the genitourinary tract (42%), kidney (38%), diaphragm (31%), lung (4%), aortic arch (9%) and heart (4%) [95]. The low penetrance of many of these defects likely resulted from variation both in the timing and severity of maternal vitamin A deficiency.
Taking advantage of the fact that RA will support early fetal development in VAD rats [65], See et al. was able to generate the previously described vitamin A deficiency syndrome in 100% of fetuses (with the exception of cardiovascular defects) from mothers supported on a sufficient amount of RA up to E10.5, and fed a suboptimal amount of RA thereafter (late VAD rat model, Table 1) [67]. Rapid initiation of VAD is possible using this approach because RA, unlike retinol, is not stored and has a very short biological half-life. The low penetrance of cardiovascular defects was due to institution of deficiency after aortic arch and septal development had largely been completed. The addition of retinol after E10.5 prevented all fetal anomalies from appearing, and the addition of a higher level of RA led to either a complete or partial rescue of fetal anomalies, supporting the idea that RA is the functional form of vitamin A in embryonic development. Using nutritional models in which RA deficiency is imposed either early, or after the 12-15 somite stage, a number of defects have been added to the original fetal vitamin A deficiency syndrome. Nervous system, cardiovascular, and axial patterning defects result from early deficiency [63,64,65,66,96], whereas a less well-developed nasal region, salivary gland hypoplasia, agenesis of the Harderian glands, hypoplasia of the intestinal villi and a number of skeletal abnormalities arise if RA is limiting at later times [67].

Table 1.
Summary of abnormalities in VAD rat embryos given insufficient RA (late VAD) * [67], insufficient carotene (VAD syndrome) [95], or observed in RAR null mutants [97,98,99].
| Late VAD | VAD syndrome | Observed in RAR null mutants | |
|---|---|---|---|
| Ocular | 100% | 49% | Yes |
| Types | |||
| Eversion of retina | 100% | 27% | Yes |
| Fibrous retrolenticular membrane | 100% | 49% | Yes |
| Coloboma | 100% | 18% | Yes |
| Heart-interventricular septal defect | 17% | 4% | Yes |
| Lung-hypoplasia | 100% | 4% | Yes |
| Diaphragmatic hernia | 100% | 31% | Yes |
| Intestinal villi hypoplastic | 83% | Not reported | Not reported |
| Kidney | 100% | 38% | Yes |
| Types | |||
| Kidneys too close or fused | 100% | 20% | Not reported |
| Ectopia | 100% | 4% | Yes |
| Ureter-ectopic termination | 100% | 36% | Yes |
| Undescended testes | 100% | 54% | Not reported |
| Skeletal defects | 100% | Not reported | Yes |
| Glandular defects | 100% | Not reported | Yes |
| Nasal region less developed | 100% | Not reported | Yes |
* Dietary RA restricted after E10.5 (12-15 somite stage) of development.
3.2. Role of the Retinoic Acid Receptors
The Rars are widely expressed in development [100]. A series of genetic experiments revealed the importance of the RAR in mediating the actions of RA in developing embryos [99,101,102]. These key experiments showed that compound RAR mutants die either in utero or shortly after birth, and recapitulate most of the defects described as part of the vitamin A deficiency syndrome [61,88,89,90,91,92,93,94,95,103] and the late VAD rat embryo model [67,104]. These genetic studies provide clear evidence that the vitamin A metabolite, RA, acts through a RAR signaling mechanism in support of normal embryonic development.
RAR/RXR heterodimers form the functional unit that transduces the RA signal, with one genomic copy of the Rxrα sufficient to support RXR function in embryogenesis [105]. Ocular defects are present in some RXRα mutants lacking only the AF-2 domain (activation function) in the ligand-binding domain [99,106,107]. However, work from the Duester group indicates that ligand binding to the RAR is sufficient to transduce the signal, as a RAR- but not a RXR- selective ligand is able to rescue the developmental defects in RALDH2 null mutant embryos [108]. Thus, the need for a RXR ligand in RAR-driven events remains to be established.
3.3. Transport of Retinoid from Maternal to Fetal Compartment
There are at least two ways in which retinoid is transferred from the maternal blood to the fetus. The major form of transport is the binding of retinol to a specific binding protein (RBP or RBP4) [109,110]. Retinyl esters can also be transported in the form of chylomicron remnants or as a part of very low-density and low-density lipoproteins. During early placentation, RBP is localized in the endoderm of the yolk sac visceral wall surrounding the embryo, and after definitive placentation, in the yolk sac membranes as well as in the uterus (decidua basalis) [111,112]. RBP does not cross the placenta into the fetal circulation [113]. Maternal retinol must be transferred to fetal RBP [114], or alternatively, retinol may be esterified in the placenta, and delivered to tissues in lipoproteins [115,116]. Although RBP null mutant embryos from null mutant mothers develop normally on a vitamin A-sufficient diet, embryogenesis is perturbed when maternal vitamin A intake is restricted [110]. Thus, when RBP is not present, circulating retinyl ester appears to represent the main pathway for the provision of retinoid from mother to embryo, provided maternal vitamin A intake is adequate [113].
3.4. Embryonic RA Synthesis and Catabolism
It is clear that RA is essential for embryonic development. However, too much RA at critical stages can result in embryo lethality or malformation [117,118,119,120]. Thus, regulation of the amount of RA that is available to the embryo at specific times and to a given tissue site is of critical importance. Both the distribution and function of enzymes involved in RA synthesis and degradation have been studied intensively in developing embryos. Interestingly, both the levels and domains of Cyp26 gene expression in the developing embryo are affected by retinoid status, whereas the Raldhs are unaffected [121].
Of the retinol dehydrogenase enzymes, RDH10 plays a key role, as loss results in embryonic lethality by approximately E13.0 [122]. These embryos (trex mutants) show a spectrum of defects consistent with vitamin A deficiency, although they do not die as early in gestation as severely RA-depleted embryos [65,123], indicating that one or more additional enzymes must also be active in generating retinaldehyde for RA synthesis. Supplementation with RA rescues the abnormalities, supporting a role for RDH10 in RA biosynthesis. A recent detailed examination of Rdh10 in the avian embryo shows that the expression domain is often smaller than that of the corresponding Raldh, leading the authors to suggest that retinaldehyde may be transferred between cells [124]. Recent work in Xenopus shows that RA down regulates XRDH10 transcripts, and this may serve as an additional mechanism to regulate endogenous retinoid levels [125]. Adh 1, 3 and 4 enzyme family members are also expressed in developing embryos [126,127], however, mutation does not result in embryo lethality [128,129]. It should be noted, that ADH3 and ADH4 null mutant mice do undergo early postnatal lethality if maintained for two generations on a VAD diet [129]. Thus, to date, RDH10 is the only retinol dehydrogenase shown to be indispensable for embryonic development under normal dietary conditions.
Numerous retinaldehyde dehydrogenase single and compound deletion mutants have been generated and studied [123,130,131,132,133,134,135]. Deletion of RALDH2 is lethal early in development, whereas RALDH3 null mutants die at birth [131], and RALDH1 mutants are viable [133]. Thus, RALDH2 is the earliest expressed family member to produce RA in the embryo, and is believed responsible for all RA signaling activity from E7.5-E8.5 in the mouse [136]. Shortly thereafter, RALDH1 and RALDH3 contribute to RA synthesis in the eye and olfactory pit. Cyp1B1 can also generate RA and is expressed at known sites of RALDH-independent retinoid signaling [16], however, null mutant mice for this gene are viable [137]. Recent work in zebrafish indicates that the availability of retinaldehyde for RA synthesis may represent another point of control, with a short-chain dehydrogenase/reductase family member, DHRS3a, which catalyzes reduction of retinaldehyde to retinol serving as a RA-induced feedback inhibitor of RA biosynthesis [138].
The importance of CYP26 enzymes in restricting RA distribution and availability is illustrated by studies in which one or more is genetically ablated. Mutation of Cyp26A1 is embryonic lethal and mutants die during mid- to late gestation with symptoms mirroring those observed in RA toxicity including caudal defects and occasionally exencephaly [139,140,141]. CYP26B1 null mutants die immediately after birth and exhibit limb, male germ cell, and craniofacial abnormalities [73,142,143]; whereas loss of CYP26C1 alone does not produce an overt phenotype [144]. Deletion of both mouse CYP26 A1 and C1 is lethal at E9.5 to E10.5 [144], and deletion of all three CYP26 family members in the mouse is also lethal early in embryonic life, and produces a duplication of the body axis [145]. Thus, CYP26 enzymes play a very early and important role in regulating access of the embryo to RA.
3.5. RA in the Early Embryo
Using direct identification by HPLC, all-trans retinaldehyde is detected in embryos as early as the egg cylinder or pre-primitive streak stage, whereas RA is detected between the mid-primitive streak stage and the late allantoic bud stage [146]. Expression of a β2-RARE-lacZ (RARE-lacZ) transgene that is activated in reporter mice when retinoid interacts with the RAR, has been used extensively as a marker of retinoid-signaling activity in vivo [147]. In the post-implantation embryo, definitive staining is noted throughout the length of the primitive streak and the appearance of staining is coincident with formation of the neural plate. The time of initiation of RA synthesis within the embryo proper has also been deduced from studies of retinaldehyde dehydrogenase mRNA (Raldh2). It is first detected at the mid-primitive streak stage adjacent to the node and primitive streak [148,149]. The early activity of Raldh2 in the embryo, along with expression in the visceral endoderm (extraembryonic) is required for vascular development in both the embryo proper, as well as the yolk sac [150,151,152]. Prior to this time RA in the embryo is believed to be of maternal origin [145]. HPLC studies of embryonic tissues at later times confirm that all-trans retinol and RA are the primary retinoids detected, with retinol clearly the most abundant [153,154,155].
The importance of protecting the embryo from RA at times when it is not needed is highlighted by recent work on embryos null for all three CYP26 family members. RA is available from the maternal circulation, and Raldh2 is expressed in the endometrial stroma and decidua [156]. Raldh2 is highly expressed in the endometrial stroma by E2.5, remains high after implantation, and expression is reduced as stromal cells undergo decidualization. Uehara et al. report that the developing embryo is protected from maternal sources of RA by Cyp26 family members expressed in extraembryonic cell types including extraembryonic endoderm and ectoderm, visceral endoderm and ectoplacental cone that surround the murine embryo proper at E5.5 and E6.25 [145]. When all three CYP26 family members are deleted, embryos show a duplication of the body axis resulting from expanded Nodal expression driven by RA throughout the epiblast. LacZ staining that results from RA-driven expression of the RARE-lacZ reporter is not normally detected in wild-type embryos at E6.25, but is abundantly expressed in CYP26A1/B1/C1 triple knockout embryos. Thus, the CYP26 catabolic enzymes play a key role in regulating the exposure of the early embryo to maternal RA.
3.6. Early Nervous System Development
Vitamin A is required for many aspects of nervous system development including patterning and neural differentiation [157,158]. One of the best-studied functions of RA is in the developing hindbrain where it contributes to the anteroposterior patterning of the neural plate [159]. When induced, the neural plate is initially anterior or forebrain-like. Induction of more posterior domains occurs by the actions of RA, Wnts and FGFs, with RA specifying the posterior hindbrain and cervical spinal cord [160]. Patterning in the vertebrate hindbrain involves segmentation, a strategy that is used to direct the diverse range of nerves and craniofacial structures essential to hindbrain function [161]. Hindbrain segments called rhombomeres (r), are transient structures that express a distinct set of cellular and molecular properties, including an ordered pattern of Hox gene expression. Using the VAD quail model, the Maden and Zile laboratories first observed that the posterior region of the hindbrain (rhombomeres 4-7) does not develop in VAD embryos [162]. The effect of depleting vitamin A by a number of means in a variety of organisms was then studied in great detail. Loss of function mutation of RALDH2 in both mice and zebrafish results in anteriorization of the hindbrain [163,164,165]. VAD rat embryos generated by severely restricting the amount of RA fed to deficient mothers at the beginning of pregnancy results in a loss of posterior rhombomere segmentation, with a significant shortening of the hindbrain [65]. In addition, ectopic otic-like vesicles appear posterior to the orthotopic vesicle. By varying the amount of RA added back to the maternal diet, this study provided evidence that an increasing amount of retinoid is needed for the correct specification of more posterior rhombomeres.
RARα/γ compound mutants show a hindbrain phenotype similar to that seen in vitamin A deficiency [166], whereas the RARα/β compound null mutants present a less severe phenotype [167], indicating that RARα and RARγ play an important role at early stages of hindbrain patterning. Elimination of all RARs in zebrafish severely disrupts hindbrain patterning [168]. Using cultured chick embryos treated with RAR antagonist at various stages of development, distinct developmental time windows were identified when RA specifies specific rhombomeres in a rostrocaudal sequence [169].
The RA needed for antero-posterior patterning is produced in the anterior paraxial mesoderm by RALDH2 and diffuses into the adjacent central nervous system (Figure 4A). After this, Raldh2 mRNA is found in somitic as well as presomitic mesoderm, but not in the node [170]. The generation and diffusion of RA has been proposed to form a gradient (higher caudal/lower rostral) that patterns the hindbrain. However, the ability of exogenous RA to rescue development implies that mechanisms other than simple diffusion of RA from a localized posterior source must be involved in generating differential responsiveness along the hindbrain anterior to posterior axis.
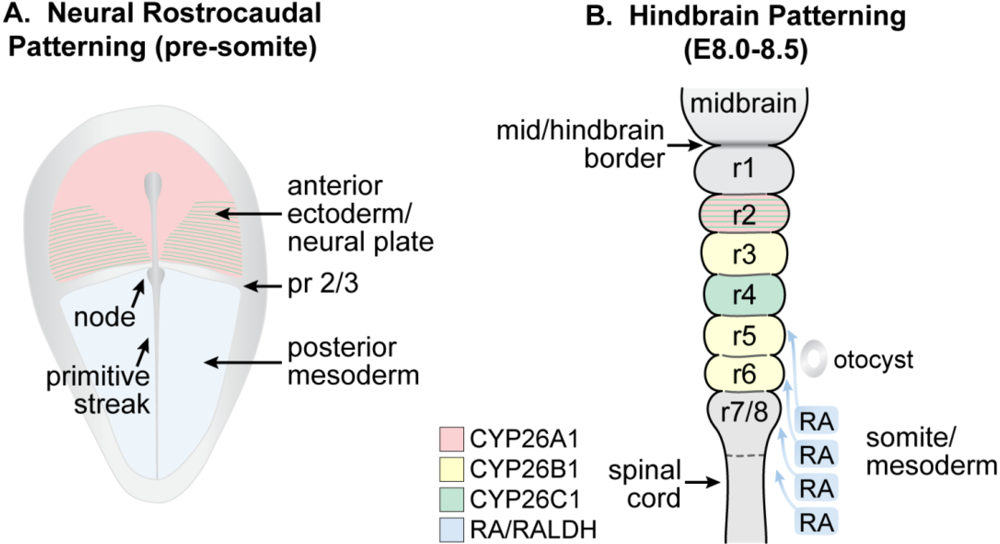
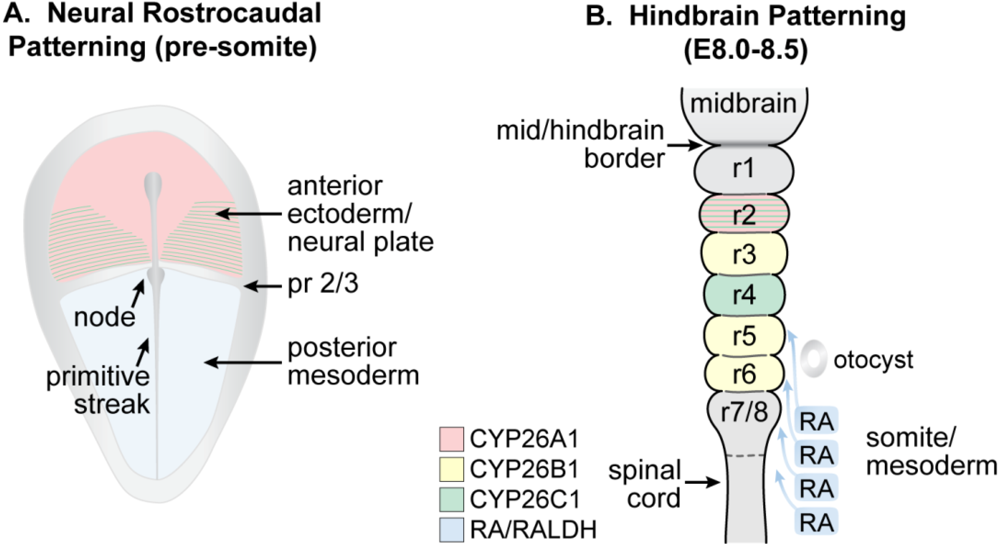
Figure 4.
Schematic showing the location of RA and Cyp26 expression in a presomitic mouse embryo undergoing (A) patterning of the neural plate and (B) later during the course of hindbrain patterning. (A) RA generated by RALDH2 in the posterior mesoderm forms an early anterior boundary of activity in the neural plate at presumptive rhombomere (pr) 2/pr3, whereas Cyp26A1 and Cyp26C1 are expressed rostral to the pr2 border; (B) By E8.0 to E8.5, RA is being expressed by the somites and anterior presomitic mesoderm, and acts on the overlying hindbrain and spinal cord. The activity of the CYP26 enzymes regulate access of the neuroepithelium to RA (Adapted from [139]).
Figure 4.
Schematic showing the location of RA and Cyp26 expression in a presomitic mouse embryo undergoing (A) patterning of the neural plate and (B) later during the course of hindbrain patterning. (A) RA generated by RALDH2 in the posterior mesoderm forms an early anterior boundary of activity in the neural plate at presumptive rhombomere (pr) 2/pr3, whereas Cyp26A1 and Cyp26C1 are expressed rostral to the pr2 border; (B) By E8.0 to E8.5, RA is being expressed by the somites and anterior presomitic mesoderm, and acts on the overlying hindbrain and spinal cord. The activity of the CYP26 enzymes regulate access of the neuroepithelium to RA (Adapted from [139]).
The importance of CYP26 family members in restricting access of the developing hindbrain to RA was first exemplified by genetic ablation of CYP26A1, resulting in hindbrain posteriorization. The rhombomere-specific expression pattern of the Cyp26 mRNAs led to the proposal that boundaries of RA activity are created by their expression [23,141,171] (Figure 4B). For example, Cyp26A1 is initially expressed in the anterior epiblast and neural plate [18] and forms a boundary at presumptive r2/r3 [141]. This is followed by the later expression of Cyp26A1 in r2 and Cyp26C1 in r2 and r4 [18,21,139,144]. Accordingly, CYP26A1 mutants show hindbrain abnormalities just rostral to the region affected by retinoid deficiency, including posterior transformation of r2/3 to a r4-like character [139,140], however, individual CYP26B1 and CYP26C1 mutants develop normally [143,144]. A series of detailed studies using mouse embryos carrying the RARE-lacZ transgene led to a further posit that the initial gradient of RA entering the posterior hindbrain is converted into RA boundaries that shift over time [172]. It was proposed that CYP26C1 could play a role in restricting RA activity to the rhombomere 4/5 border, although CYP26C1 null mutants reportedly do not show abnormalities at this boundary as assessed by HoxB1 and RARE-lacZ staining [144]. However, CYP26A1/C1 double knockouts do exhibit even more severe hindbrain abnormalities than CYP26A1 mutants alone, including a grossly enlarged posterior hindbrain at the expense of anterior structures [144]. Interestingly, in Cyp26 morpholino studies in zebrafish, restriction of the RA-responsive gene, Vhnff1, is determined by the posterior limit of Cyp26C1 at the r4/r5 boundary, a function that can be compensated for by CYP26B1 [173]. Thus, the combined action of these enzymes along with the dynamic changes in where they are expressed during development must prevent RA from reaching specific anterior structures. Although not identical, recent models explaining how RA patterns the developing hindbrain all include an important role for CYP26 family members in regulating the formation of waves or pulses of RA in this dynamic process [23,144,172,173].
When hindbrain patterning is underway, exposure of the anterior brain to RA is prevented by the action of CYP26A1 and CYP26C1, and loss of CYP26A1 renders embryos more sensitive to the teratogenic effects of excess exogenous RA on head truncation [174]. By regulating levels of RA in the anterior brain, the CYPs may enable a ligand independent function of the RARs. Based on studies in Xenopus embryos, unliganded RARs in the head region at this early time are proposed to function as transcriptional repressors, that if activated inappropriately by RA, lead to activation of target genes that should remain off [175]. Thus, teratogenesis may result from over activation of genes that are normally upregulated by RA and/or a loss of gene repression by unliganded RAR.
A role for RA in forebrain patterning was initially proposed based on studies in chick embryos exposed to RAR and RXR antagonists [176] and from the VAD quail model in which the size of the telencephalic vessel is reduced and the diencephalon is abnormally patterned [177]. Additional studies in VAD quail indicated that the vitamin was needed to correctly position anterior and dorsal boundaries in the forebrain via modulation of FGF8 and Wnt signaling [178]. RA was also reported in chick to contribute to regionalization of the telencephalon along the dorsoventral axis, and RALDH3 produced by the head ectoderm was proposed as the retinoid source [179]. However, RALDH2/3 compound null mutant mice reportedly do not show defects in the early molecular determinants of forebrain patterning [178,180]. Instead, Molotkova and colleagues propose that RA functions at later times in forebrain development, and provide evidence that RA generated by RALDH3 in the lateral ganglionic eminence is required for normal expression of the dopamine D2 receptor in the nucleus accumbens and medial striatum [180].
Additional roles for RA in later development of the central nervous system (CNS) are beginning to emerge [181]. McCaffery’s group found that RA is produced by RALDH2 in the meninges overlying the hindbrain at mouse embryonic day 13, and that RA activity as assessed by RARE-lacZ is present in precerebellar neurons migrating around the hindbrain circumference to form the inferior olive and pontine nuclei [182]. This group went on to show that RA is required for the generation of posterior neurons in the inferior olive, as the posterior inferior olive is significantly smaller in late VAD rat embryos compared to vitamin A-sufficient controls [183]. In the meninges covering the cortex, levels of Raldh2 increase in the mouse embryo from E13 to E14 onward, to a maximal level in the newborn brain [184]. Recently, RA from the meninges was reported to function in cortical development by regulating the switch of radial glia cells in the ventricular zone from symmetric to asymmetric division, resulting in the formation of neurons or intermediate progenitor cells [185]. It will be important to determine whether RA regulates stem cell division/differentiation in any other areas of the CNS.
3.7. Spinal Cord and Other Neuronal Development
RA plays a role in the development of caudal structures, including the neural tube that forms the spinal cord and in somite development. These tissues arise from the node-streak border, a region that comprises the caudal end of the node and the rostral end of the primitive streak [186]. Although primary neurulation occurs in both RALDH2 mutant mice and VAD chick and rat embryos [65,123,187], the RALDH2 mutant is reported to have a thinner neuroepithelium [149] and VAD quail embryos have fewer spinal cord neurons and smaller somites [162,188]. Raldh2 is first expressed during gastrulation in the mesenchyme adjacent to the node and the primitive streak [148]. In the mouse and chick embryonic axis, RA from the anterior presomitic mesoderm and somites promotes neural differentiation by inhibiting the expression of Fgf8[189]. In RALDH2 mutants, primitive streak and mesodermal markers are expanded at the expense of those representative of the prospective neuroepithelium [149].
RA from the adjacent somites is needed to generate certain interneurons and ventral spinal cord motor neurons [190,191]. RA generated from within the spinal cord is required for the differentiation of a subset of lateral motor column (LMC) neurons that extend axons into the limb [192,193]. Interactions between HOXD10, which defines rostral boundaries of the lumbosacral LMC, and HOXD11, which opposes the effects of HOXD10, influence the distribution of motor neuron subtypes along the rostrocaudal axis. HOXD11 may also serve to restrict RALDH expression to regions where it is needed to support LMC neuron survival and maturation [194].
There is evidence that RA promotes the survival of sympathetic neurons by inducing responsiveness to neurotrophins [195,196,197]. RA is also linked to the promotion of neurite outgrowth [157,162]. Recently, a number of genes including neuron navigator 2 (Nav2) and Nedd9 were discovered in a screen for RA-responsive genes in a neuroblastoma cell line that elaborates neurites in response to RA [198,199,200]. Nedd9 is a direct downstream target of RA, whereas a functional RARE has not yet been identified in the Nav2 gene [33]. When NAV2 is knocked down, the neuroblastoma cell is no longer able to extend neurites in response to RA [201]. The human Nav2 gene can rescue defects in axonal elongation in the C. elegans mutant, UNC-53 (the homolog of Nav2 in the worm), and a mouse mutant hypomorphic for NAV2 also shows defects in cranial nerve development and general neurite density [202]. NAV2 may function as a link between actin remodeling and microtubule dynamics during neurite outgrowth.
3.8. Eye Development
Recognition of the importance of vitamin A in eye development first came from the experiments of Hale in which piglets from deficient mothers were born blind [86,87]. The morphologic consequences of vitamin A deficiency in rat embryos were examined in great detail by Warkany and Scharffenberger [61,88] and Wilson et al. [95]. They noted dysmorphogenesis of the anterior eye segment, retina and optic disc, although the penetrance of the defects was extremely variable. Using a model in which normal development is supported in VAD rat embryos up to embryonic day 10.5 (12-15 somite stage) with RA and deficiency is induced thereafter, See et al. was able to produce eye defects in 100% of embryos which included; coloboma, absence of the anterior chamber of the eye, rudimentary iris or loss, fusion of the lens and cornea, thickening of the eyelid tissues, reduced size of the conjunctival sac, folding of the retina, absence of the vitreous body and the presence of a fibrous retrolenticular membrane [67,104]. The addition of retinol at E10.5 provided a full rescue of all defects, showing that all the defects were attributable to retinoid deficiency imposed after this time. The addition of a high level of RA after E10.5 dramatically improved eye development, supporting that vitamin A acid is the active moiety.
The RA synthesizing enzymes, RALDHs, show a very dynamic pattern of expression in the developing eye. In the mouse, Raldh2 (also called V2 activity) is transiently expressed in the optic vesicle neuroepithelium at E8.5 [132,148,203], after which Raldh3 (also called V1 activity) is expressed in the ventral neural retina and pigmented neuroepithelium [134,204,205,206] and Raldh1 (also called ADH-2) is expressed in the dorsal retina from E9.5 onward [134,207]. RARα is expressed in all layers of the developing murine neural retina, whereas RARβ is expressed in the inner nuclear layer from embryonic day 14 to postnatal day 7 [208,209]. Genetic ablation of two or more retinoid receptors in mice results in eye defects that are largely similar to those seen in VAD rat embryos [97,209,210,211]. A number of eye defects in RALDH3 and RALDH1/3 compound mutants [134,135,178] are also similar to those observed in late VAD rat embryos.
Using the late VAD rat embryo model, it is possible to identify the times when vitamin A must be present to support eye development by adding back retinol at successively later times ranging from E11.5 to E15.5, and evaluating eye development at E18.5 [104]. A full rescue of all structures is achieved if retinol is added by E11.5, whereas addition on or after E14.5 is ineffective. Retinol added as late as E13.5 completely prevents retinal folding, formation of a fibrous retrolenticular membrane, and loss of the vitreous body but is ineffective in preventing anterior eye segment defects, whereas addition by E12.5 improves or rescues the majority of defects in anterior eye segment development (absence of anterior chamber, rudimentary iris, corneal-lenticular stalk fusion, and small conjunctival sac) and prevents coloboma of the retina and optic disc in the majority of fetuses. This study also revealed that the cells within the retina of late VAD embryos lose their characteristic shape and orientation along the apical-to-basal axis of the retina, with the appearance of gaps or holes in the neural retina that worsen as retinol is added at successively later times. Interestingly, the increase in cell adhesion proteins, N-cadherin and β-catenin, that normally occurs with development is not seen in VAD retinas. Additionally, a reduction in cyclin D1 labeling is observed in the retina of late VAD fetuses, suggesting that cell proliferation is also disrupted. These effects may contribute to retinal collapse seen in VAD rat embryos receiving no supplemental retinol, or retinol on or after E14.5. Using this temporal model, See et al. also clearly showed that a lack of optic fissure closure does not lead to retinal folding/collapse, as a well formed retina is produced by adding retinol at a time that is too late to prevent coloboma [104]. Retinal thinning and disruption of cellular organization and adhesion is a more likely reason for collapse of the retina seen at late stages of severe deficiency.
RARα has been reported to be the sole receptor transducing the RA signal in the retina beyond E10.5 based on the inability to detect RARE-lacZ expression in the RARα null mutant mouse retina [212]. In the absence of this receptor, however, the mouse retina appears to develop normally leading to the conclusion that RARα is unnecessary for the developing mouse retina [213]. If RARE-lacZ expression is an accurate readout of all RAR-mediated signaling in the neural retina of RARα null mutants, then the neural retina would appear not to be a direct target of RA and its receptors. If so, then it is possible that adverse effects of vitamin A deficiency on neural retina development could occur by a loss of paracrine action of RA in a secondary tissue, as is proposed for retinoid support of anterior eye segment development (Figure 5). Thus, it is unknown whether RA generated in the retina acts locally to regulate retina development.
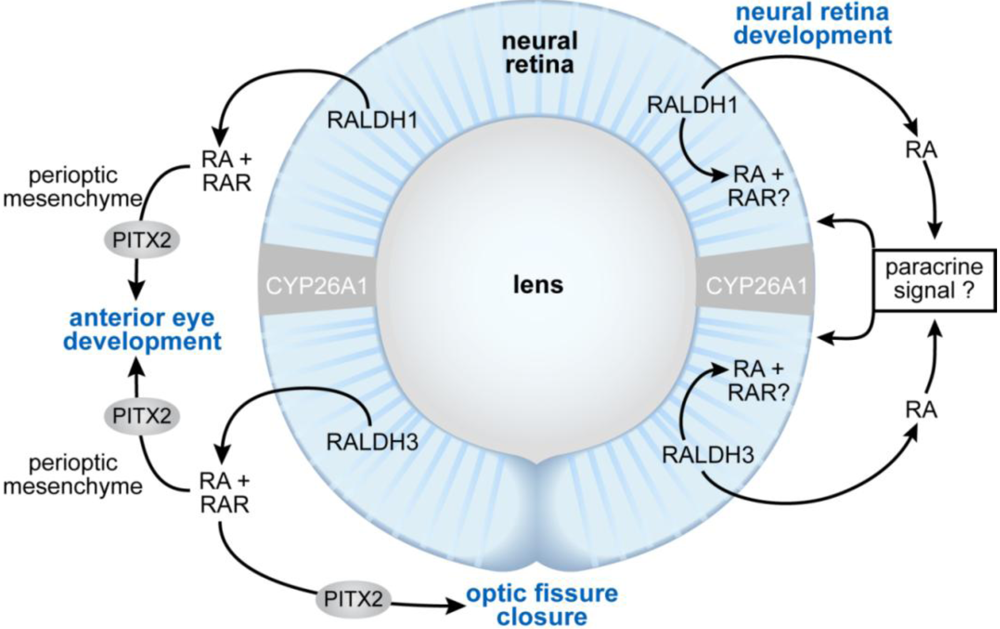
Figure 5.
Schematic showing the proposed sites of RA function during eye morphogenesis (left) and differentiation (right). At early stages of eye development, RA generated by RALDH1 and RALDH3 acts as a paracrine signal binding to RARs located in the perioptic mesenchyme to support anterior eye segment development and closure of the optic fissure. Pitx2 is a RA/RAR-regulated transcription factor that is required both for anterior eye segment morphogenesis, as well as closure of the optic fissure. At later stages of development, RA promotes differentiation of the neural retina. The mechanism is unclear, but could involve either a paracrine effect of RA outside of the neural retina, or a direct effect on the cells within the retina itself.
Figure 5.
Schematic showing the proposed sites of RA function during eye morphogenesis (left) and differentiation (right). At early stages of eye development, RA generated by RALDH1 and RALDH3 acts as a paracrine signal binding to RARs located in the perioptic mesenchyme to support anterior eye segment development and closure of the optic fissure. Pitx2 is a RA/RAR-regulated transcription factor that is required both for anterior eye segment morphogenesis, as well as closure of the optic fissure. At later stages of development, RA promotes differentiation of the neural retina. The mechanism is unclear, but could involve either a paracrine effect of RA outside of the neural retina, or a direct effect on the cells within the retina itself.
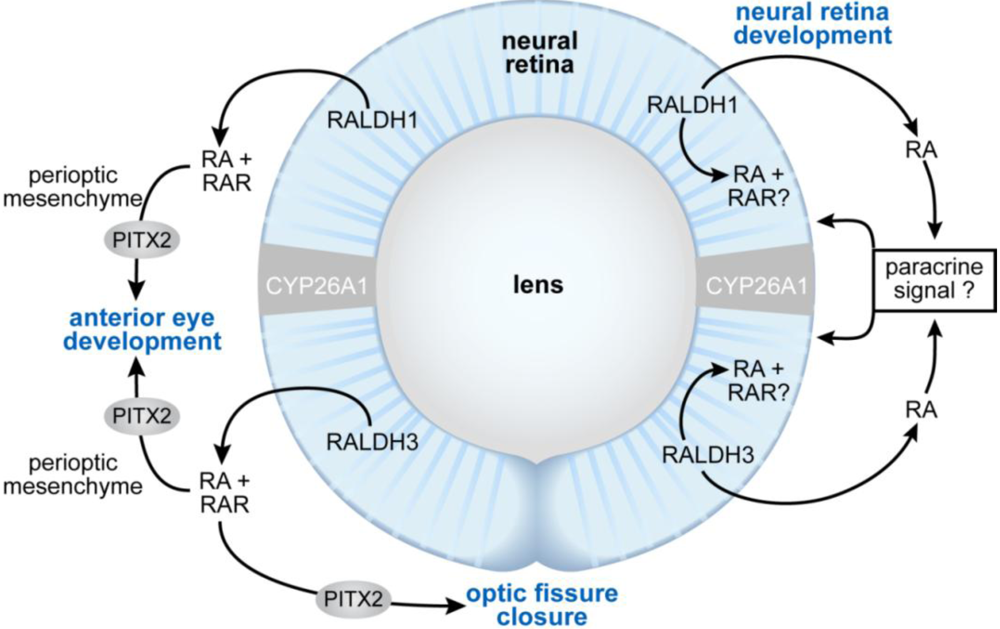
The molecular basis for the vitamin A deficiency-induced defects in anterior eye segment development and in the appearance of coloboma has been the subject of several recent studies. It does not involve abnormalities in dorsoventral patterning genes, but rather, disruption of paracrine signaling from the retina to the perioptic mesenchyme. Compound deletion of both RARβ and RARγ in the neural crest cell-derived periocular mesenchyme using Wnt1-cre to direct excision results in eye defects including abnormalities of the cornea and anterior chamber [134,212]. Similarly, RALDH1/3 compound null mutants show malformations of the anterior segment of the eye, including agenesis of the corneal and iris stroma, agenesis of the anterior chamber and less frequently, coloboma of the retina [134,135]. Thus, RA from retina is believed to signal in paracrine fashion to the perioptic mesenchyme to support the development of anterior eye structures. Defects result from the loss of control of programmed cell death in this region, and by loss of RA-mediated Pitx2, Foxc1, Eya2 and Dkk2 expression, and subsequent loss of repression of Wnt/β-catenin signaling [134,135,212,214]. Pitx2 encodes a homeodomain transcription factor that is essential for anterior eye segment development, and is also required for optic fissure closure [215]. Pitx2 is down regulated in the periocular mesenchyme of late VAD embryos that not only show defects in anterior eye segment development, but also retinal/optic disc coloboma at 100% penetrance [67,104]. Very recently it was shown that the Pitx2 gene contains a RARE located 4.3 kb upstream of the promoter, and thus is a direct target of RA and its receptors [214]. Duester and colleagues propose that RA signaling activates Pitx2 in the perioptic mesenchyme, which in turn, induces Dkk2 and suppresses local Wnt signaling in the regulation of normal anterior segment development. Zacharias et al. has extended this model to include a role for canonical Wnt signaling in the maintenance of Pitx2 expression after its initiation by RA, and prior to the time that Wnt signaling is suppressed [216].
3.9. Somites and Skeleton
RA plays a role in the initiation of differentiation of the anterior region of the presomitic mesoderm from which the new somites originate [217]. VAD quail embryos have smaller somites than their vitamin A-sufficient counterparts, and also have an expanded FGF8 domain in the presomitic mesoderm [188]. In RALDH2 null mutants, the expression of Fgf8 mRNA in the primitive ectoderm (epiblast) is shifted anteriorly such that it enters the node ectoderm and neuroectoderm, and somite development is abnormal [170]. FGF signaling is important in the process of somitogenesis, and the opposition of FGF and RA signaling is also important in regulating somite size. A reduction in FGF in the presomitic mesoderm below a threshold value is needed to position the future somite boundary [218,219]. Higher levels of FGF8 and Wnt3a signaling maintain cells in an undifferentiated state, whereas exposure to RA along with lower FGF8 and Wnt3a initiates differentiation, and these factors are believed to control the developmental switch at this boundary [217]. Recent work in chick indicates that RA may also be involved in the termination of the process of segmentation [220].
RA signaling is involved both in the specification of the axial identity of future somites as well as in later stages of skeletal development. Somites, although similar by appearance, develop into distinct structures dependent upon their axial position. Determination of initial axial identity is believed to occur in mesodermal cells prior to somite formation [221]. VAD rat embryos show anterior vertebral transformations throughout the axial skeleton that can be rescued only if vitamin A is provided on or before embryonic day 8.75, a time that precedes appearance of the first somite [66]. Genetic deletion of several RARs produces defects in axial development with anterior cervical transformations similar to VAD embryos [53,97,209]. However, in these mutants, either posteriorization at the cervical/thoracic junction is observed, or no vertebral changes caudal to this region are reported [97,209]. Alteration of Hox gene expression appears to be major way in which retinoids affect positional information along the anteroposterior body axis [222,223,224,225].
Vitamin A is also required for skeletal development beyond its role in presomitic embryos. Rat embryos given sufficient RA up to embryonic day 10.5 (approximately the 12-15 somite stage), but made deficient thereafter exhibit hypoplastic cranial bones, defects of the thyroid, cricoid and tracheal cartilages as well as agenesis of the neural arch of cervical vertebrae 1 (C1) and ectopic bone in the dorsal regions of C1 [67]; defects in this region bear many similarities to those observed in RARα/γ compound null mutants [97,98]. Late VAD rat embryos also exhibit gross malformation of the sternal and pelvic regions [67]. Sternal malformations have been reported in RBP null mutant embryos from RBP null mothers fed inadequate vitamin A [110]. Surprisingly, late VAD rat embryos have anterior vertebral transformations in the cervical axial skeleton up to and including the thoracic juncture, along with posteriorization events at the thoracic and sacral levels of the skeleton [67]. As discussed above, a number of RAR mutants show both cervical anteriorizations and posteriorization at the cervical thoracic junction [97,209]. It is also interesting that administration of excess RA to the mouse at E7 (presomitic; equivalent to E8.5-9 rat) produces posteriorizing transformations throughout the skeleton; however, excess RA at E8.5 (equivalent to E10-10.5 rat), produces anterior transformations starting at vertebrae 15 (thoracic vertebra 8, T8) whereas excess RA at even later times, yields both rostral posteriorizing and caudal anteriorizations [223,224]. Thus, vertebral identities, initially specified at the late primitive streak phase, can also be respecified at later times in mouse development when the vertebrae precursors (the somites), differentiate, and the sclerotome cells begin to form vertebrae. In summary, vitamin A plays a normal role in the maintenance of vertebral identity as well as in the development of skeletal elements.
Much of the craniofacial skeleton originates from neural crest cells [226]. Frontonasal agenesis has been reported in compound RAR null mutant mice [97] and RBP null mutant mice on a vitamin A-deficiency diet [110]. Dupe and Pellerin recently showed that selective ablation of RARα and RARγ subtypes using the Wnt1-Cre promoter leads to agenesis of the frontonasal skeletal elements, but does not appear to adversely affect the early survival and migration of neural crest cells [227]. Deletion of all three RAR subtypes using the selective promoter produces a similar phenotype, indicating that RARα and RARγ act cell-autonomously in neural crest cells to direct morphogenesis of these skeletal elements.
3.10. Heart Development
Cardiac and aortic arch defects were observed as a part of the early vitamin A deficiency syndrome in rat embryos [93,94] and in RAR compound null mutant mouse embryos [98,102]. When severe VAD is imposed in quail, the initiation of heart morphogenesis is disrupted [88,228,229]. In VAD quail embryos, vascular networks are absent and the heart appears ballooned and non-compartmentalized, and is randomly-positioned without an inflow tract at the posterior site [228]. Inability of the heart tube to undergo looping is also observed in RALDH2 mouse mutants [230]. The sinuatrial (venous) valve, a transient structure that flanks the orifice between the sinus venosus and right atrium, is not formed properly in rat embryos deprived of RA for one day (E9.5-10.5), and results in improper channeling of venous blood and anterior cardinal vein distension [64]. If RA deficiency is imposed after E10.5 in the rat embryo, a time when the cardiac primordium has completed looping and the primitive vasculature is established, early cardiac and aortic arch defects are not observed [67]. In summary, roles for vitamin A in mammalian heart development include: heart tube patterning and looping, chamber and outflow tract septation, ventricular trabeculation, cardiomyocyte differentiation and coronary vessel development [231,232,233]. A more in depth discussion of the molecular pathways involved in heart morphogenesis can be found in several recent reviews [229,232,234].
3.11. Kidney and Urinary Tract Development
The requirement for RA in the developing kidney and urogenital tract has been illustrated in several animal models. In rodents, maternal vitamin A deficiency results in embryonic renal hypoplasia, the severity of which depends on the extent of vitamin A deprivation [67,91,110,235]. Ectopic kidneys, renal fusion and failure of the renal pelvis and calyx to undergo dilatation, in addition to ectopic ureteric openings and other genitourinary tract defects have been reported in VAD rat fetuses [67,91]. RALDH2 null mutant mice lack nephric ducts [123] and in RARα/β2 double receptor mutants, nephron progenitors, stromal cells and ureteric bud tips are all greatly reduced or completely absent at birth [98,236]. These compound RAR mutants also have incorrectly positioned distal ureters, hydronephrosis and megaureter.
Signaling between the ureteric bud epithelium that forms the collecting duct system, the metanephric mesenchyme that differentiates into nephron, and the stromal mesenchyme that differentiates into the renal interstitium is important during early kidney development. In the embryonic kidney, Raldh2 is localized in stromal mesenchyme of the outer cortex and Raldh3 is expressed in the ureteric bud [130,237,238]. RA and its receptors are needed to maintain Ret expression [237], a gene critically required for formation of the ureteric bud and its branching in the kidney [239,240,241]. The forced expression of Ret in ureteric bud cells in RARα/β2 double receptor mutants rescues renal development, restoring ureteric bud growth and stromal cell patterning [237]. Recent work shows that RA generated by RALDH2 in stromal mesenchyme acts in paracrine fashion to activate RA-receptor signaling and Ret expression in ureteric bud cells [242].
In kidney, the number of embryonic branching events determines the final number of nephrons an individual will have for life, and it has been proposed that suboptimal nephron number at birth increases susceptibility to acquired renal disease and essential hypertension later in life [243,244,245,246]. Studies in rodents suggest that even mild vitamin A deficiency (a 50% decrease in circulating vitamin A concentrations) can lead to impaired branching and a 20% reduction in the number of nephrons [235]. Quite remarkably, a single injection of RA to a control group of pregnant rats at midgestation (E11) led to supernumerary nephron endowment in the kidneys of their offspring. RA given intraperitoneally at E11.5 is also able to increase nephron endowment in offspring exposed to maternal protein restriction without affecting body weight or kidney size, such that the number of nephrons per volume of kidney tissue is increased in these rat pups above that seen in the kidneys of control offspring [247]. Hence mild vitamin A deficiency in pregnancy may correlate to sub-clinical deficiencies in nephron number and slight nephron deficits that are not recognized at birth, but could possibly contribute in the long-term to renal failure and hypertension.
3.12. Diaphragm
The diaphragm functions as the primary muscle of respiration and forms a physical barrier between the thoracic and abdominal cavities. Congenital diaphragmatic hernia (CDH) occurs in approximately one in 3000 births, and is associated with high neonatal mortality [248]. Vitamin A is essential for normal diaphragm development, and it has been hypothesized that disruption of retinoid signaling may contribute to the etiology of the human disorder. A recent report shows that there is a relationship between low cord retinol and RBP levels and CDH in newborn infants [249].
A herniated diaphragm was observed as a part of the early vitamin A deficiency syndrome [89], and appears at a 100% penetrance in RA-supported VAD embryos given insufficient RA after E10.5 of development, whereas a normal diaphragm results if the mother is supplemented with retinol after this time [67]. Diaphragmatic hernia is also observed at low penetrance in RARα/β2 compound null mutant mice [98]. Congenital diaphragmatic hernia in humans is linked to mutations in Stra6, which encodes for a membrane RBP receptor [26,250], as well as to CDH-affected chromosome loci encoding for several other retinoid-related genes [251].
The pleuroperitoneal fold (PPF) is a transient structure formed at the union of the pleuro-pericardial folds and the septum transversum and represents the component of the primordial diaphragm through which muscle precursor cells and pioneer axons of the phrenic nerve migrate to form the mature diaphragm. The PPF is fully formed by E13.5 in vitamin A-sufficient rat embryos, but it is abnormal in late VAD embryos [252]. Similar defects in PPF development are seen in rat embryos treated with nitrofen and several other CDH-inducing teratogens that interfere with the synthesis of RA and RA signaling in vivo [253,254]. Raldh2 mRNA is expressed in the PPF, and RARs α, γ and RXRα are most strongly expressed in the nonmuscular mesenchymal cells of the PPF. Thus, vitamin A signaling in the developing PPF appears to play a key role in the developing diaphragm.
3.13. Lung and Upper Respiratory Tract and Airways
Respiratory defects including left lung agenesis, bilateral lung hypoplasia, and agenesis of the esophagotracheal septum were described in early VAD syndrome embryos but were characterized as rare anomalies [90,95]. Lung hypoplasia is observed with 100% penetrance in rat embryos in which RA deficiency is imposed after E10.5, and the severity is increased as RA in the maternal diet is reduced [67]. Rars are expressed throughout lung development and RAR compound null mutant mice (RARα/β) show left lung agenesis and hypoplasia [209]. Additionally, a wide array of RA synthesizing, metabolizing and binding proteins are found in developing lung [255,256,257]. Mice null for RALDH2 or RDH10, also have lung agenesis or hypoplastic phenotypes [122,258,259]. The lung is second only to the liver as the major retinoid storage organ [260].
The lung arises from foregut endoderm during early development of the embryo. RA from the splanchnic mesoderm surrounding the foregut endoderm has been found to be essential for primordial lung bud formation at E9.5 in the mouse [256,258,259,261]. It was recently shown that in the foregut mesoderm, RA controls the Fgf10 expression required for bud formation by balancing the activation of canonical Wnt signaling through direct transcriptional repression of its antagonist Dkk1, and repression of Tgfβ signaling [262]. This conclusion is reinforced by work showing that simultaneous activation of Wnt and repression of Tgfβ in RA-deficient foregut rescues lung bud formation.
While RA signaling is required for initial budding, by E10.5-E11.5, as secondary buds form, levels are down regulated by the appearance of the RA-degrading enzyme CYP26A1 to enable more distal branching and distal airway formation to proceed to completion [256,263]. A role for RA in alveoli formation is supported by the finding that RARs are required for correct lung alveoli septation, and reports that exogenous RA can stimulate alveoli formation in immature rat and mouse lung [264,265,266,267,268,269]. Figure 6 summarizes the proposed functions of RA in lung development.
A recent report in the New England Journal of Medicine showed that, in a region with endemic vitamin A (retinol) deficiency, children whose mothers had received vitamin A supplementation before, during, and for 6 months after pregnancy had better lung function when they were tested at 9 to 11 years of age than children whose mothers had received beta carotene supplementation or placebo. Additionally, they found that the period during which supplementation with vitamin A was most important was from gestation through a postnatal age of 6 months [270].
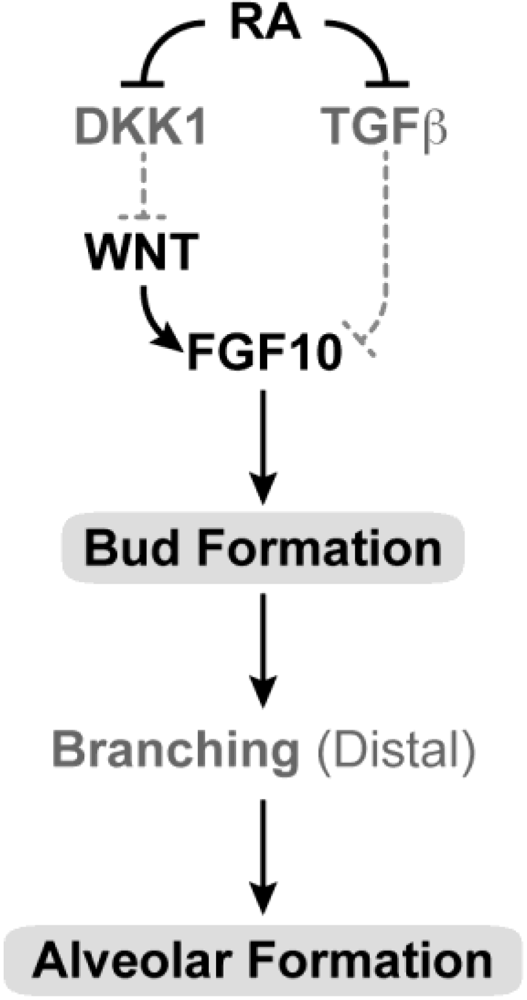
Figure 6.
Schematic showing the proposed sites of RA action in lung development. In the developing embryo, RA is needed for primary bud formation (grey oval), but signaling is down-regulated during the time that lung differentiation occurs. A later role for RA in alveolar formation (grey oval) is also proposed. During induction of the lung buds, RA regulates mesodermal Fgf10 levels by negatively regulating Tgfβ and enabling induction of the Wnt pathway by repression of the Dickkopf homolog 1 (Dkk1) known to antagonize Wnt ligand-receptor binding. RA may also influence the response of the foregut endoderm (origin of lung progenitors) to Fgf10. (Adapted from [262]).
Figure 6.
Schematic showing the proposed sites of RA action in lung development. In the developing embryo, RA is needed for primary bud formation (grey oval), but signaling is down-regulated during the time that lung differentiation occurs. A later role for RA in alveolar formation (grey oval) is also proposed. During induction of the lung buds, RA regulates mesodermal Fgf10 levels by negatively regulating Tgfβ and enabling induction of the Wnt pathway by repression of the Dickkopf homolog 1 (Dkk1) known to antagonize Wnt ligand-receptor binding. RA may also influence the response of the foregut endoderm (origin of lung progenitors) to Fgf10. (Adapted from [262]).
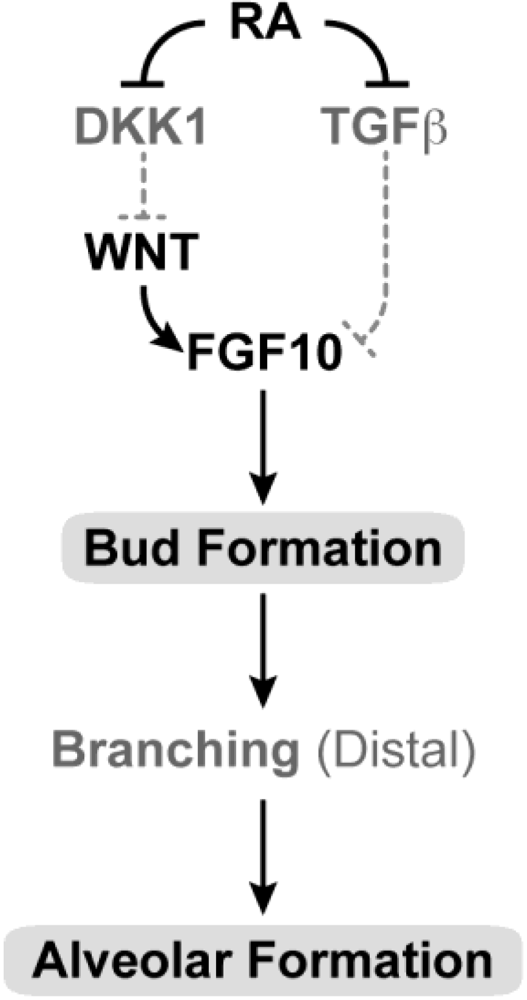
RA acting through the RAR is also necessary for the morphogenesis of other components of the respiratory tract including partitioning of the primitive foregut into oesophagus and trachea, and for the opening between the nasal and oropharyngeal cavities [67,131,209,256,258,261].
3.14. Pancreas
Although not observed either as a part of the vitamin A deficiency syndrome or in compound RAR null mutant mice, a requirement for RA in pancreatic development has been proposed based on studies in RALDH null mutant mice [271,272] and in several other organisms in which retinoid signaling is deficient or inhibited, including Xenopus, zebrafish, chick, and quail [273,274,275,276]. RDH10 knockout mice do not have a pancreas [122]. RA produced by RALDH2 is required for early development of the dorsal pancreas in the mouse [271,272]. Specification of dorsal pancreatic tissue can be rescued in RALDH2-deficient embryos by low-dose maternal administration of RA.
The effects of RA deficiency on pancreatic lineages appear to be due to the loss of pancreatic field specification within the endoderm [274]. Specification of the pancreas occurs between E8.0-8.5 of mouse development, and is followed by the development of the dorsal and ventral pancreatic buds at E9.0-9.5 [271]. RALDH2 null mutant embryos do not develop a dorsal pancreatic bud. It has been suggested that the differential expression of retinoic acid receptors (RARs) in gastrula stage endoderm is at least partially responsible for the distinct responsiveness of dorsal versus ventral pancreas [277]. Both defects in RA signaling and RA treatment have been shown to affect the expression of PDX-1 [271,272,275,278] an essential regulator of early pancreas development required for the pancreatic buds to grow and differentiate [279,280,281]. RALDH2 mutant embryos lack PDX1 expression in dorsal but not ventral endoderm [271,272]. Conversely, exogenous RA has been shown to expand the pancreatic field [273,274,275], and CYP26A1 has recently been shown to play a critical role in setting the anterior limit of the pancreas field endodermal Cyp26 expression [282]. Additional information regarding the role of RA in pancreas development can be found in several recent reviews [283,284].
3.15. Limb Development and Interdigital Cell Death
Functions for RA in limb development have been forwarded and debated for many years. Inhibitors of RA synthesis and vitamin A deficiency inhibit limb outgrowth in the quail [285,286]. In the absence of RALDH2, murine forelimb buds do not develop and embryonic growth ceases prior to the stage when hindlimb buds are initiated [287,288]. Zebrafish RALDH2 mutants lack pectoral fins and fin bud induction does not occur [164,165,276,289]. Limb defects in mice also result from treatment with excess exogenous RA, or inappropriate exposure to endogenous levels of RA due to the absence of CYP26B1. These studies show that controlled exclusion of RA from the limb bud is essential for proper limb morphology [142].
RA has been proposed to play a role in patterning the developing limb by regulating the pattern of expression of genes such as Hand2 (activates SHH), Shh (patterns the anterior-to-posterior axis), and Meis1/2 (transcription factors that mark the proximal limb bud mesenchyme) [287,290,291,292]. However, a new report from Zhao and colleagues using a RALDH2/3 double mouse knockout suggests that RA acts as a permissive signal at an earlier stage to allow limb bud initiation rather than acting in an instructive manner, and that the role of RA is to antagonize early axial FGF signals which otherwise inhibit the limb field [80,141,293]. According to this new model, RA signaling within the forelimb bud proper is not required for normal patterning to occur. A similar idea was proposed by Gilbert et al., who found that axial retinoic acid signals played a permissive role in the induction of zebrafish pectoral fins [289]. RA appears to be dispensable for hindlimb budding and patterning [80,287]. It is notable that trex embryos carrying a mutation in Rdh10, have small, abnormal forelimbs but have hindlimbs that are relatively unaffected [122].
At later stages of development, RA is also essential for interdigital cell death, the mechanism by which digit separation occurs. Webbed digits have been described in compound RAR mutant mice due to a loss of apoptosis [209,294]. Around E12.5 (mouse) RA signaling becomes confined to the interdigital zones by a combination of interdigital Raldh2 expression and Cyp26B1 expression in the developing digits [295]. The mechanism by which RA activates cell death is currently a subject of active investigation [294,295,296,297].
4. Conclusions
In summary, the vitamin A metabolite, RA, is essential for reproduction in both the male and female, as well as for many events in the developing embryo. Nutritional as well as genetic approaches are being used to identify the cell types and pathways that are dependent upon RA signaling in support of these processes. Paracrine signaling appears to play a prominent role in RA action. Regulation of RA synthesis as well as its catabolism is important in determining when and where RA signaling will be activated. Future studies are needed to develop a more detailed understanding of when in development, and in what specific cell types RA and its receptors are acting. Elucidation of the pathways that are involved in support of vitamin A functions in stem/germ cell division/differentiation, patterning and tissue/organ development remain major tasks for future work.
Acknowledgements
The work from the author’s laboratory discussed in this review was supported in part by funds from NIH grant DK-14881, a fellowship to EMM NIH T32 DK07665, and the USDA CSREES WIS04305, WIS04768 and CSRSG 9400543 and 9900802. Support to the author was also provided by NIH CA49837. We also thank Laura Vanderploeg in the Biochemistry Media Lab for the artwork.
References
- McCollum, E.V.; Davis, M. The necessity of certain lipins in the diet during growth. J. Biol. Chem. 1913, 15, 167–175. [Google Scholar]
- Wolbach, S.B.; Howe, P.R. Tissue changes following deprivation of fat-soluble A vitamin. J. Exp. Med. 1925, 42, 753–777. [Google Scholar]
- Evans, H.M. The effects of inadequate vitamin A on the sexual physiology of the female. J. Biol. Chem. 1928, 77, 651–654. [Google Scholar]
- Carpenter, K.J.; Harper, A.E.; Olson, R.E. Experiments That Changed Nutritional Thinking. J. Nutr. 1997, 127, 1017S–1053S. [Google Scholar]
- Moore, T. Vitamin A and carotene: The absence of the liver oil vitamin A from carotene. VI. The conversion of carotene to vitamin A in vivo. Biochem. J. 1930, 24, 692–702. [Google Scholar] [PubMed]
- Karrer, P.; Morf, R.; Schoepp, K. Zur Kenntnis des Vitamins A in Gischtranen. Helv. Chim. Acta 1931, 14, 1431–1436. [Google Scholar]
- Karrer, P. Carotenoids, flavins and vitamin A and B2: Nobel lecture, December 11, 1937. In Nobel Lectures, Chemistry (1922-1941); Elsevier: Amsterdam, The Netherlands, 1966; pp. 433–448. [Google Scholar]
- Arens, J.F.; van Dorp, D.A. Synthesis of some compounds possessing vitamin A activity. Nature 1946, 157, 190–191. [Google Scholar]
- Arens, J.F.; van Dorp, D.A. Activity of vitamin A-acid in the rat. Nature 1946, 158, 622. [Google Scholar]
- van Dorp, D.A.; Arens, J.F. Biological activity of vitamin A acid. Nature 1946, 158, 60. [Google Scholar]
- Dowling, J.E.; Wald, G. The biological function of vitamin A acid. Proc. Natl. Acad. Sci. USA 1960, 46, 587–608. [Google Scholar]
- Moise, A.R.; Noy, N.; Palczewski, K.; Blaner, W.S. Delivery of retinoid-based therapies to target tissues. Biochemistry 2007, 46, 4449–4458. [Google Scholar]
- Pares, X.; Farres, J.; Kedishvili, N.; Duester, G. Medium- and short-chain dehydrogenase/reductase gene and protein families: Medium-chain and short-chain dehydrogenases/reductases in retinoid metabolism. Cell. Mol. Life Sci. 2008, 65, 3936–3949. [Google Scholar]
- Duester, G. Families of retinoid dehydrogenases regulating vitamin A function: Production of visual pigment and retinoic acid. Eur. J. Biochem. 2000, 267, 4315–4324. [Google Scholar]
- Sima, A.; Parisotto, M.; Mader, S.; Bhat, P.V. Kinetic characterization of recombinant mouse retinal dehydrogenase types 3 and 4 for retinal substrates. Biochim. Biophys. Acta 2009, 1790, 1660–1664. [Google Scholar]
- Chambers, D.; Wilson, L.; Maden, M.; Lumsden, A. RALDH-independent generation of retinoic acid during vertebrate embryogenesis by CYP1B1. Development 2007, 134, 1369–1383. [Google Scholar]
- White, J.A.; Beckett-Jones, B.; Guo, Y.D.; Dilworth, F.J.; Bonasoro, J.; Jones, G.; Petkovich, M. cDNA cloning of human retinoic acid-metabolizing enzyme (hP450RAI) identifies a novel family of cytochromes P450. J. Biol. Chem. 1997, 272, 18538–18541. [Google Scholar]
- Fujii, H.; Sato, T.; Kaneko, S.; Gotoh, O.; Fujii-Kuriyama, Y.; Osawa, K.; Kato, S.; Hamada, H. Metabolic inactivation of retinoic acid by a novel P450 differentially expressed in developing mouse embryos. EMBO J. 1997, 16, 4163–4173. [Google Scholar]
- Chithalen, J.V.; Luu, L.; Petkovich, M.; Jones, G. HPLC-MS/MS analysis of the products generated from all-trans-retinoic acid using recombinant human CYP26A. J. Lipid Res. 2002, 43, 1133–1142. [Google Scholar]
- White, J.A.; Ramshaw, H.; Taimi, M.; Stangle, W.; Zhang, A.; Everingham, S.; Creighton, S.; Tam, S.P.; Jones, G.; Petkovich, M. Identification of the human cytochrome P450, P450RAI-2, which is predominantly expressed in the adult cerebellum and is responsible for all-trans-retinoic acid metabolis. Proc. Natl. Acad. Sci. USA 2000, 97, 6403–6408. [Google Scholar]
- Tahayato, A.; Dolle, P.; Petkovich, M. Cyp26C1 encodes a novel retinoic acid-metabolizing enzyme expressed in the hindbrain, inner ear, first branchial arch and tooth buds during murine developmen. Gene Expr. Patterns 2003, 3, 449–454. [Google Scholar]
- Taimi, M.; Helvig, C.; Wisniewski, J.; Ramshaw, H.; White, J.; Amad, M.; Korczak, B.; Petkovich, M. A novel human cytochrome P450, CYP26C1, involved in metabolism of 9-cis and all-trans isomers of retinoic aci. J. Biol. Chem. 2004, 279, 77–85. [Google Scholar]
- White, R.J.; Schilling, T.F. How degrading: Cyp26s in hindbrain development. Dev. Dyn. 2008, 237, 2775–2790. [Google Scholar]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A Metabolism: An Update. Nutrients 2011, 3, 63–103. [Google Scholar]
- Soprano, D.R.; Blaner, W.S. Plasma Retinol-Binding Protein. In The Retinoids: Biology, Chemistry, and Medicine, 2nd; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Raven Press: New York, NY, USA, 1994; pp. 257–282. [Google Scholar]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar]
- Xueping, E.; Zhang, L.; Lu, J.; Tso, P.; Blaner, W.S.; Levin, M.S.; Li, E. Increased neonatal mortality in mice lacking cellular retinol-binding protein II. J. Biol. Chem. 2002, 277, 36617–36623. [Google Scholar]
- Theodosiou, M.; Laudet, V.; Schubert, M. From carrot to clinic: an overview of the retinoic acid signaling pathway. Cell. Mol. Life Sci. 2010, 67, 1423–1445. [Google Scholar]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996, 10, 940–954. [Google Scholar]
- Rochette-Egly, C.; Germain, P. Dynamic and combinatorial control of gene expression by nuclear retinoic acid receptors (RARs). Nucl. Recept. Signal. 2009, 7, e005. [Google Scholar]
- Repa, J.J.; Hanson, K.K.; Clagett-Dame, M. All-trans-retinol is a ligand for the retinoic acid receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 7293–7297. [Google Scholar]
- Mangelsdorf, D.; Umesono, K.; Evans, R.M. The Retinoid Receptors. In The Retinoids: Biology, Chemistry and Medicine, 2nd; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Raven Press: New York, NY, USA, 1994; pp. 319–350. [Google Scholar]
- Knutson, D.C.; Clagett-Dame, M. atRA Regulation of NEDD9, a gene involved in neurite outgrowth and cell adhesion. Arch. Biochem. Biophys. 2008, 477, 163–174. [Google Scholar]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar]
- Bastien, J.; Rochette-Egly, C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 2004, 328, 1–16. [Google Scholar]
- Balmer, J.E.; Blomhoff, R. Gene expression regulation by retinoic acid. J. Lipid Res. 2002, 43, 1773–1808. [Google Scholar]
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar]
- Mason, K.E. Differences in testis injury and repair after vitamin A-deficiency, vitamin E-deficiency, and inanitio. Am. J. Anat. 1933, 52, 153–239. [Google Scholar]
- Mitranond, V.; Sobhon, P.; Tosukhowong, P.; Chindaduangrat, W. Cytological changes in the testes of vitamin-A-deficient rats. I. Quantitation of germinal cells in the seminiferous tubules. Acta Anat. (Basel) 1979, 103, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.F.; Hembree, W.C. Spermatogenic response to vitamin A in vitamin A deficient rats. Biol. Reprod. 1979, 21, 891–904. [Google Scholar]
- Unni, E.; Rao, M.R.; Ganguly, J. Histological & ultrastructural studies on the effect of vitamin A depletion & subsequent repletion with vitamin A on germ cells & Sertoli cells in rat testis. Indian J. Exp. Biol. 1983, 21, 180–192. [Google Scholar]
- van Pelt, A.M.; de Rooij, D.G. Synchronization of the seminiferous epithelium after vitamin A replacement in vitamin A-deficient mice. Biol. Reprod. 1990, 43, 363–367. [Google Scholar]
- Morales, C.; Griswold, M.D. Retinol-induced stage synchronization in seminiferous tubules of the rat. Endocrinology 1987, 121, 432–434. [Google Scholar]
- van Pelt, A.M.; de Rooij, D.G. The origin of the synchronization of the seminiferous epithelium in vitamin A-deficient rats after vitamin A replacement. Biol. Reprod. 1990, 42, 677–682. [Google Scholar]
- Hogarth, C.A.; Griswold, M.D. The key role of vitamin A in spermatogenesis. J. Clin. Invest. 2010, 120, 956–962. [Google Scholar]
- Matson, C.K.; Murphy, M.W.; Griswold, M.D.; Yoshida, S.; Bardwell, V.J.; Zarkower, D. The mammalian doublesex homolog DMRT1 is a transcriptional gatekeeper that controls the mitosisversus meiosis decision in male germ cells. Dev. Cell 2010, 19, 612–624. [Google Scholar]
- Snyder, E.M.; Small, C.; Griswold, M.D. Retinoic acid availability drives the asynchronous initiation of spermatogonial differentiation in the mouse. Biol. Reprod. 2010, 83, 783–790. [Google Scholar]
- van Pelt, A.M.; de Rooij, D.G. Retinoic acid is able to reinitiate spermatogenesis in vitamin A-deficient rats and high replicate doses support the full development of spermatogenic cells. Endocrinology 1991, 128, 697–704. [Google Scholar]
- Vernet, N.; Dennefeld, C.; Rochette-Egly, C.; Oulad-Abdelghani, M.; Chambon, P.; Ghyselinck, N.B.; Mark, M. Retinoic acid metabolism and signaling pathways in the adult and developing mouse testis. Endocrinology 2006, 147, 96–110. [Google Scholar]
- Ghyselinck, N.B.; Vernet, N.; Dennefeld, C.; Giese, N.; Nau, H.; Chambon, P.; Viville, S.; Mark, M. Retinoids and spermatogenesis: lessons from mutant mice lacking the plasma retinol binding protein. Dev. Dyn. 2006, 235, 1608–1622. [Google Scholar]
- Zhai, Y.; Sperkova, Z.; Napoli, J.L. Cellular expression of retinal dehydrogenase types 1 and 2: effects of vitamin A status on testis mRNA. J. Cell. Physiol. 2001, 186, 220–232. [Google Scholar]
- Livera, G.; Rouiller-Fabre, V.; Pairault, C.; Levacher, C.; Habert, R. Regulation and perturbation of testicular functions by vitamin A. Reproduction 2002, 124, 173–180. [Google Scholar]
- Lohnes, D.; Kastner, P.; Dierich, A.; Mark, M.; LeMeur, M.; Chambon, P. Function of retinoic acid receptor gamma in the mouse. Cell 1993, 73, 643–658. [Google Scholar]
- Lufkin, T.; Lohnes, D.; Mark, M.; Dierich, A.; Gorry, P.; Gaub, M.P.; LeMeur, M.; Chambon, P. High postnatal lethality and testis degeneration in retinoic acid receptor alpha mutant mice. Proc. Natl. Acad. Sci. USA 1993, 90, 7225–7229. [Google Scholar]
- Vernet, N.; Dennefeld, C.; Guillou, F.; Chambon, P.; Ghyselinck, N.B.; Mark, M. Prepubertal testis development relies on retinoic acid but not rexinoid receptors in Sertoli cells. EMBO J. 2006, 25, 5816–5825. [Google Scholar]
- Doyle, T.J.; Braun, K.W.; McLean, D.J.; Wright, R.W.; Griswold, M.D.; Kim, K.H. Potential functions of retinoic acid receptor A in Sertoli cells and germ cells during spermatogenesis. Ann. N. Y. Acad. Sci. 2007, 1120, 114–130. [Google Scholar]
- Chung, S.S.; Choi, C.; Wang, X.; Hallock, L.; Wolgemuth, D.J. Aberrant distribution of junctional complex components in retinoic acid receptor alpha-deficient mice. Microsc. Res. Tech. 2010, 73, 583–596. [Google Scholar]
- Clagett-Dame, M.; DeLuca, H.F. The role of vitamin A in mammalian reproduction and embryonic development. Annu. Rev. Nutr. 2002, 22, 347–381. [Google Scholar]
- Evans, H.M.; Bishop, K.S. On an invariable and characteristic disturbance of reproductive function in animals reared on a diet poor in fat soluble vitamine A. Anat. Rec. 1922, 23, 17–18. [Google Scholar]
- Mason, K.E.; Ellison, E.T. Changes in the vaginal epithelium of the rat after vitamin A-deficiency. J. Nutr. 1935, 9, 735–755. [Google Scholar]
- Warkany, J.; Schraffenberger, E. Congenital malformations induced in rats by maternal vitamin A deficiency. I. Defects of the eye. Arch. Ophthalmol. 1946, 35, 150–169. [Google Scholar]
- Thompson, J.N.; Howell, J.M.; Pitt, G.A. Vitamin a and Reproduction in Rats. Proc. R. Soc. Lond. B Biol. Sci. 1964, 159, 510–535. [Google Scholar]
- White, J.C.; Shankar, V.N.; Highland, M.; Epstein, M.L.; DeLuca, H.F.; Clagett-Dame, M. Defects in embryonic hindbrain development and fetal resorption resulting from vitamin A deficiency in the rat are prevented by feeding pharmacological levels of all-trans-retinoic acid. Proc. Natl. Acad. Sci. USA 1998, 95, 13459–13464. [Google Scholar]
- White, J.C.; Highland, M.; Clagett-Dame, M. Abnormal development of the sinuatrial venous valve and posterior hindbrain may contribute to late fetal resorption of vitamin A-deficient rat embryos. Teratology 2000, 62, 374–384. [Google Scholar]
- White, J.C.; Highland, M.; Kaiser, M.; Clagett-Dame, M. Vitamin A deficiency results in the dose-dependent acquisition of anterior character and shortening of the caudal hindbrain of the rat embryo. Dev. Biol. 2000, 220, 263–284. [Google Scholar]
- Kaiser, M.E.; Merrill, R.A.; Stein, A.C.; Breburda, E.; Clagett-Dame, M. Vitamin A deficiency in the late gastrula stage rat embryo results in a one to two vertebral anteriorization that extends throughout the axial skeleton. Dev. Biol. 2003, 257, 14–29. [Google Scholar]
- See, A.W.; Kaiser, M.E.; White, J.C.; Clagett-Dame, M. A nutritional model of late embryonic vitamin A deficiency produces defects in organogenesis at a high penetrance and reveals new roles for the vitamin in skeletal development. Dev. Biol. 2008, 316, 171–190. [Google Scholar]
- Howell, J.M.; Thompson, J.N.; Pitt, G.A. Histology of the Lesions Produced in the Reproductive Tract of Animals Fed a Diet Deficient in Vitamin A Alcohol but Containing Vitamin A Acid. II. The Female Rat. J. Reprod. Fertil. 1964, 7, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Noback, C.R.; Takahashi, Y.I. Micromorphology of the placenta of rats reared on marginal vitamin-A-deficient diet. Acta Anat. (Basel) 1978, 102, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Koubova, J.; Menke, D.B.; Zhou, Q.; Capel, B.; Griswold, M.D.; Page, D.C. Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 2474–2479. [Google Scholar]
- Bowles, J.; Knight, D.; Smith, C.; Wilhelm, D.; Richman, J.; Mamiya, S.; Yashiro, K.; Chawengsaksophak, K.; Wilson, M.J.; Rossant, J.; Hamada, H.; Koopman, P. Retinoid signaling determines germ cell fate in mice. Science 2006, 312, 596–600. [Google Scholar]
- Bowles, J.; Koopman, P. Retinoic acid, meiosis and germ cell fate in mammals. Development 2007, 134, 3401–3411. [Google Scholar]
- MacLean, G.; Li, H.; Metzger, D.; Chambon, P.; Petkovich, M. Apoptotic extinction of germ cells in testes of Cyp26b1 knockout mice. Endocrinology 2007, 148, 4560–4567. [Google Scholar]
- Kumar, S.; Chatzi, C.; Brade, T.; Cunningham, T.J.; Zhao, X.; Duester, G. Sex-specific timing of meiotic initiation is regulated by Cyp26b1 independent of retinoic acid signalling. Nat. Commun. 2011, 2, 151. [Google Scholar]
- Livera, G.; Rouiller-Fabre, V.; Valla, J.; Habert, R. Effects of retinoids on the meiosis in the fetal rat ovary in culture. Mol. Cell. Endocrinol. 2000, 165, 225–231. [Google Scholar]
- Baltus, A.E.; Menke, D.B.; Hu, Y.C.; Goodheart, M.L.; Carpenter, A.E.; de Rooij, D.G.; Page, D.C. In germ cells of mouse embryonic ovaries, the decision to enter meiosis precedes premeiotic DNA replication. Nat. Genet. 2006, 38, 1430–1434. [Google Scholar]
- Barrios, F.; Filipponi, D.; Pellegrini, M.; Paronetto, M.P.; Di Siena, S.; Geremia, R.; Rossi, P.; De Felici, M.; Jannini, E.A.; Dolci, S. Opposing effects of retinoic acid and FGF9 on Nanos2 expression and meiotic entry of mouse germ cells. J. Cell Sci. 2010, 123, 871–880. [Google Scholar]
- Bowles, J.; Feng, C.W.; Spiller, C.; Davidson, T.L.; Jackson, A.; Koopman, P. FGF9 suppresses meiosis and promotes male germ cell fate in mice. Dev. Cell 2010, 19, 440–449. [Google Scholar]
- Li, H.; Clagett-Dame, M. Vitamin A deficiency blocks the initiation of meiosis of germ cells in the developing rat ovary in vivo. Biol. Reprod. 2009, 81, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Sirbu, I.O.; Mic, F.A.; Molotkova, N.; Molotkov, A.; Kumar, S.; Duester, G. Retinoic acid promotes limb induction through effects on body axis extension but is unnecessary for limb patterning. Curr. Biol. 2009, 19, 1050–1057. [Google Scholar] [PubMed]
- Bowles, J.; Feng, C.W.; Knight, D.; Smith, C.A.; Roeszler, K.N.; Bagheri-Fam, S.; Harley, V.R.; Sinclair, A.H.; Koopman, P. Male-specific expression of Aldh1a1 in mouse and chicken fetal testes: implications for retinoid balance in gonad development. Dev. Dyn. 2009, 238, 2073–2080. [Google Scholar]
- Li, H.; MacLean, G.; Cameron, D.; Clagett-Dame, M.; Petkovich, M. Cyp26b1 expression in murine Sertoli cells is required to maintain male germ cells in an undifferentiated state during embryogenesis. PLoS ONE 2009, 4, e7501. [Google Scholar]
- Li, H.; Palczewski, K.; Baehr, W.; Clagett-Dame, M. Vitamin A deficiency results in meiotic failure and accumulation of undifferentiated spermatogonia in prepubertal mouse testis. Biol. Reprod. 2011, 84, 336–341. [Google Scholar]
- Zhou, Q.; Nie, R.; Li, Y.; Friel, P.; Mitchell, D.; Hess, R.A.; Small, C.; Griswold, M.D. Expression of stimulated by retinoic acid gene 8 (Stra8) in spermatogenic cells induced by retinoic acid: an in vivo study in vitamin A-sufficient postnatal murine testes. Biol. Reprod. 2008, 79, 35–42. [Google Scholar]
- Bellve, A.R.; Millette, C.F.; Bhatnagar, Y.M.; O’Brien, D.A. Dissociation of the mouse testis and characterization of isolated spermatogenic cells. J. Histochem. Cytochem. 1977, 25, 480–494. [Google Scholar]
- Hale, F. Pigs born without eye balls. J. Hered. 1933, 24, 105–106. [Google Scholar]
- Hale, F. The relation of vitamin A to anophthalmos in pigs. Am. J. Ophthalmol. 1935, 18, 1087–1093. [Google Scholar]
- Warkany, J.; Schraffenberger, E. Congenital malformations of the eyes induced in rats by maternal vitamin A deficiency. Proc. Soc. Exp. Biol. Med. 1944, 57, 49–52. [Google Scholar]
- Warkany, J.; Roth, C.B. Congenital malformations induced in rats by maternal vitamin A deficiency. II. Effect of varying the preparatory diet upon the yield of abnormal young. J. Nutr. 1948, 35, 1–11. [Google Scholar]
- Warkany, J.; Roth, C.B.; Wilson, J.G. Multiple congenital malformations: a consideration of etiologic factors. Pediatrics 1948, 1, 462–471. [Google Scholar]
- Wilson, J.G.; Warkany, J. Malformations in the genito-urinary tract induced by maternal vitamin A deficiency in the rat. Am. J. Anat. 1948, 83, 357–407. [Google Scholar]
- Wilson, J.G.; Barch, S. Fetal death and maldevelopment resulting from maternal vitamin A deficiency in the rat. Proc. Soc. Exp. Biol. Med. 1949, 72, 687–693, illust. [Google Scholar] [PubMed]
- Wilson, J.G.; Warkany, J. Aortic-arch and cardiac anomalies in the offspring of vitamin A deficient rats. Am. J. Anat. 1949, 85, 113–155. [Google Scholar]
- Wilson, J.G.; Warkany, J. Cardiac and aortic arch anomalies in the offspring of vitamin A deficient rats correlated with similar human anomalies. Pediatrics 1950, 5, 708–725. [Google Scholar]
- Wilson, J.G.; Roth, C.B.; Warkany, J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am. J. Anat. 1953, 92, 189–217. [Google Scholar] [CrossRef] [PubMed]
- Dickman, E.D.; Thaller, C.; Smith, S.M. Temporally-regulated retinoic acid depletion produces specific neural crest, ocular and nervous system defects. Development 1997, 124, 3111–3121. [Google Scholar]
- Lohnes, D.; Mark, M.; Mendelsohn, C.; Dolle, P.; Dierich, A.; Gorry, P.; Gansmuller, A.; Chambon, P. Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development 1994, 120, 2723–2748. [Google Scholar] [PubMed]
- Mendelsohn, C.; Lohnes, D.; Decimo, D.; Lufkin, T.; LeMeur, M.; Chambon, P.; Mark, M. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 1994, 120, 2749–2771. [Google Scholar] [PubMed]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoic acid receptors during embryonic development. Nucl. Recept. Signal. 2009, 7, e002. [Google Scholar]
- Dolle, P. Developmental expression of retinoic acid receptors (RARs). Nucl. Recept. Signal. 2009, 7, e006. [Google Scholar]
- Lohnes, D.; Mark, M.; Mendelsohn, C.; Dolle, P.; Decimo, D.; LeMeur, M.; Dierich, A.; Gorry, P.; Chambon, P. Developmental roles of the retinoic acid receptors. J. Steroid Biochem. Mol. Biol. 1995, 53, 475–486. [Google Scholar]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoid nuclear receptors: lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 451–480. [Google Scholar]
- Warkany, J. Disturbance of embryonic development by maternal vitamin deficiencies. J. Cell. Physiol. 1954, 43, 207–236. [Google Scholar]
- See, A.W.; Clagett-Dame, M. The temporal requirement for vitamin A in the developing eye: mechanism of action in optic fissure closure and new roles for the vitamin in regulating cell proliferation and adhesion in the embryonic retina. Dev. Biol. 2009, 325, 94–105. [Google Scholar]
- Krezel, W.; Dupe, V.; Mark, M.; Dierich, A.; Kastner, P.; Chambon, P. RXR gamma null mice are apparently normal and compound RXR alpha +/−/RXR beta −/−/RXR gamma −/− mutant mice are viable. Proc. Natl. Acad. Sci. USA 1996, 93, 9010–9014. [Google Scholar]
- Mascrez, B.; Ghyselinck, N.B.; Chambon, P.; Mark, M. A transcriptionally silent RXRalpha supports early embryonic morphogenesis and heart development. Proc. Natl. Acad. Sci. USA 2009, 106, 4272–4277. [Google Scholar]
- Mascrez, B.; Mark, M.; Dierich, A.; Ghyselinck, N.B.; Kastner, P.; Chambon, P. The RXRalpha ligand-dependent activation function 2 (AF-2) is important for mouse development. Development 1998, 125, 4691–4707. [Google Scholar]
- Mic, F.A.; Molotkov, A.; Benbrook, D.M.; Duester, G. Retinoid activation of retinoic acid receptor but not retinoid X receptor is sufficient to rescue lethal defect in retinoic acid synthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 7135–7140. [Google Scholar]
- Morriss-Kay, G.M.; Ward, S.J. Retinoids and mammalian development. Int. Rev. Cytol. 1999, 188, 73–131. [Google Scholar]
- Quadro, L.; Hamberger, L.; Gottesman, M.E.; Wang, F.; Colantuoni, V.; Blaner, W.S.; Mendelsohn, C.L. Pathways of vitamin A delivery to the embryo: insights from a new tunable model of embryonic vitamin A deficiency. Endocrinology 2005, 146, 4479–4490. [Google Scholar]
- Soprano, D.R.; Soprano, K.J.; Goodman, D.S. Retinol-binding protein and transthyretin mRNA levels in visceral yolk sac and liver during fetal development in the rat. Proc. Natl. Acad. Sci. USA 1986, 83, 7330–7334. [Google Scholar]
- Sapin, V.; Ward, S.J.; Bronner, S.; Chambon, P.; Dolle, P. Differential expression of transcripts encoding retinoid binding proteins and retinoic acid receptors during placentation of the mouse. Dev. Dyn. 1997, 208, 199–210. [Google Scholar]
- Quadro, L.; Hamberger, L.; Gottesman, M.E.; Colantuoni, V.; Ramakrishnan, R.; Blaner, W.S. Transplacental delivery of retinoid: the role of retinol-binding protein and lipoprotein retinyl ester. Am. J. Physiol. Endocrinol. Metab 2004, 286, E844–E851. [Google Scholar]
- Bavik, C.; Ward, S.J.; Chambon, P. Developmental abnormalities in cultured mouse embryos deprived of retinoic by inhibition of yolk-sac retinol binding protein synthesis. Proc. Natl. Acad. Sci. USA 1996, 93, 3110–3114. [Google Scholar]
- Farese, R.V., Jr.; Cases, S.; Ruland, S.L.; Kayden, H.J.; Wong, J.S.; Young, S.G.; Hamilton, R.L. A novel function for apolipoprotein B: lipoprotein synthesis in the yolk sac is critical for maternal-fetal lipid transport in mice. J. Lipid Res. 1996, 37, 347–360. [Google Scholar]
- Sapin, V.; Chaib, S.; Blanchon, L.; Alexandre-Gouabau, M.C.; Lemery, D.; Charbonne, F.; Gallot, D.; Jacquetin, B.; Dastugue, B.; Azais-Braesco, V. Esterification of vitamin A by the human placenta involves villous mesenchymal fibroblasts. Pediatr. Res. 2000, 48, 565–572. [Google Scholar]
- Kochhar, D.M. Teratogenic activity of retinoic acid. Acta Pathol. Microbiol. Scand. 1967, 70, 398–404. [Google Scholar]
- Conlon, R.A. Retinoic acid and pattern formation in vertebrates. Trends Genet. 1995, 11, 314–319. [Google Scholar]
- Shenefelt, R.E. Morphogenesis of malformations in hamsters caused by retinoic acid: relation to dose and stage at treatment. Teratology 1972, 5, 103–118. [Google Scholar]
- Collins, M.D.; Mao, G.E. Teratology of retinoids. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 399–430. [Google Scholar]
- Reijntjes, S.; Blentic, A.; Gale, E.; Maden, M. The control of morphogen signalling: regulation of the synthesis and catabolism of retinoic acid in the developing embryo. Dev. Biol. 2005, 285, 224–237. [Google Scholar]
- Sandell, L.L.; Sanderson, B.W.; Moiseyev, G.; Johnson, T.; Mushegian, A.; Young, K.; Rey, J.P.; Ma, J.X.; Staehling-Hampton, K.; Trainor, P.A. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ developmen. Genes Dev. 2007, 21, 1113–1124. [Google Scholar]
- Niederreither, K.; Subbarayan, V.; Dolle, P.; Chambon, P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet. 1999, 21, 444–448. [Google Scholar]
- Reijntjes, S.; Zile, M.H.; Maden, M. The expression of Stra6 and Rdh10 in the avian embryo and their contribution to the generation of retinoid signatures. Int. J. Dev. Biol. 2010, 54, 1267–1275. [Google Scholar]
- Strate, I.; Min, T.H.; Iliev, D.; Pera, E.M. Retinol dehydrogenase 10 is a feedback regulator of retinoic acid signalling during axis formation and patterning of the central nervous system. Development 2009, 136, 461–472. [Google Scholar]
- Ang, H.L.; Deltour, L.; Hayamizu, T.F.; Zgombic-Knight, M.; Duester, G. Retinoic acid synthesis in mouse embryos during gastrulation and craniofacial development linked to class IV alcohol dehydrogenase gene expression. J. Biol. Chem. 1996, 271, 9526–9534. [Google Scholar]
- Ang, H.L.; Deltour, L.; Zgombic-Knight, M.; Wagner, M.A.; Duester, G. Expression patterns of class I and class IV alcohol dehydrogenase genes in developing epithelia suggest a role for alcohol dehydrogenase in local retinoic acid synthesis. Alcohol. Clin. Exp. Res. 1996, 20, 1050–1064. [Google Scholar]
- Molotkov, A.; Deltour, L.; Foglio, M.H.; Cuenca, A.E.; Duester, G. Distinct retinoid metabolic functions for alcohol dehydrogenase genes Adh1 and Adh4 in protection against vitamin A toxicity or deficiency revealed in double null mutant mice. J. Biol. Chem. 2002, 277, 13804–13811. [Google Scholar]
- Molotkov, A.; Fan, X.; Deltour, L.; Foglio, M.H.; Martras, S.; Farres, J.; Pares, X.; Duester, G. Stimulation of retinoic acid production and growth by ubiquitously expressed alcohol dehydrogenase Adh3. Proc. Natl. Acad. Sci. USA 2002, 99, 5337–5342. [Google Scholar]
- Mic, F.A.; Haselbeck, R.J.; Cuenca, A.E.; Duester, G. Novel retinoic acid generating activities in the neural tube and heart identified by conditional rescue of Raldh2 null mutant mice. Development 2002, 129, 2271–2282. [Google Scholar]
- Dupe, V.; Matt, N.; Garnier, J.M.; Chambon, P.; Mark, M.; Ghyselinck, N.B. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc. Natl. Acad. Sci. USA 2003, 100, 14036–14041. [Google Scholar]
- Mic, F.A.; Molotkov, A.; Molotkova, N.; Duester, G. Raldh2 expression in optic vesicle generates a retinoic acid signal needed for invagination of retina during optic cup formation. Dev. Dyn. 2004, 231, 270–277. [Google Scholar]
- Fan, X.; Molotkov, A.; Manabe, S.; Donmoyer, C.M.; Deltour, L.; Foglio, M.H.; Cuenca, A.E.; Blaner, W.S.; Lipton, S.A.; Duester, G. Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol. Cell. Biochem. 2003, 23, 4637–4648. [Google Scholar]
- Matt, N.; Dupe, V.; Garnier, J.M.; Dennefeld, C.; Chambon, P.; Mark, M.; Ghyselinck, N.B. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development 2005, 132, 4789–4800. [Google Scholar]
- Molotkov, A.; Molotkova, N.; Duester, G. Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development 2006, 133, 1901–1910. [Google Scholar]
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar]
- Buters, J.T.; Sakai, S.; Richter, T.; Pineau, T.; Alexander, D.L.; Savas, U.; Doehmer, J.; Ward, J.M.; Jefcoate, C.R.; Gonzalez, F.J. Cytochrome P450 CYP1B1 determines susceptibility to 7,12-dimethylbenz[a]anthracene-induced lymphomas. Proc. Natl. Acad. Sci. USA 1999, 96, 1977–1982. [Google Scholar]
- Feng, L.; Hernandez, R.E.; Waxman, J.S.; Yelon, D.; Moens, C.B. Dhrs3a regulates retinoic acid biosynthesis through a feedback inhibition mechanism. Dev. Biol. 2010, 338, 1–14. [Google Scholar]
- Abu-Abed, S.; Dolle, P.; Metzger, D.; Beckett, B.; Chambon, P.; Petkovich, M. The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structu. Genes Dev. 2001, 15, 226–240. [Google Scholar]
- Sakai, Y.; Meno, C.; Fujii, H.; Nishino, J.; Shiratori, H.; Saijoh, Y.; Rossant, J.; Hamada, H. The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 2001, 15, 213–225. [Google Scholar]
- Pennimpede, T.; Cameron, D.A.; Maclean, G.A.; Li, H.; Abu-Abed, S.; Petkovich, M. The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, K.; Zhao, X.; Uehara, M.; Yamashita, K.; Nishijima, M.; Nishino, J.; Saijoh, Y.; Sakai, Y.; Hamada, H. Regulation of retinoic acid distribution is required for proximodistal patterning and outgrowth of the developing mouse limb. Dev. Cell 2004, 6, 411–422. [Google Scholar]
- MacLean, G.; Dolle, P.; Petkovich, M. Genetic disruption of CYP26B1 severely affects development of neural crest derived head structures, but does not compromise hindbrain patterning. Dev. Dyn. 2009, 238, 732–745. [Google Scholar]
- Uehara, M.; Yashiro, K.; Mamiya, S.; Nishino, J.; Chambon, P.; Dolle, P.; Sakai, Y. CYP26A1 and CYP26C1 cooperatively regulate anterior-posterior patterning of the developing brain and the production of migratory cranial neural crest cells in the mouse. Dev. Biol. 2007, 302, 399–411. [Google Scholar]
- Uehara, M.; Yashiro, K.; Takaoka, K.; Yamamoto, M.; Hamada, H. Removal of maternal retinoic acid by embryonic CYP26 is required for correct Nodal expression during early embryonic patterning. Genes Dev. 2009, 23, 1689–1698. [Google Scholar]
- Ulven, S.M.; Gundersen, T.E.; Weedon, M.S.; Landaas, V.O.; Sakhi, A.K.; Fromm, S.H.; Geronimo, B.A.; Moskaug, J.O.; Blomhoff, R. Identification of endogenous retinoids, enzymes, binding proteins, and receptors during early postimplantation development in mouse: important role of retinal dehydrogenase type 2 in synthesis of all-trans-retinoic acid. Dev. Biol. 2000, 220, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Rossant, J.; Zirngibl, R.; Cado, D.; Shago, M.; Giguere, V. Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes Dev. 1991, 5, 1333–1344. [Google Scholar]
- Niederreither, K.; McCaffery, P.; Drager, U.C.; Chambon, P.; Dolle, P. Restricted expression and retinoic acid-induced downregulation of the retinaldehyde dehydrogenase type 2 (RALDH-2) gene during mouse development. Mech. Dev. 1997, 62, 67–78. [Google Scholar]
- Ribes, V.; Le Roux, I.; Rhinn, M.; Schuhbaur, B.; Dolle, P. Early mouse caudal development relies on crosstalk between retinoic acid, Shh and Fgf signalling pathways. Development 2009, 136, 665–676. [Google Scholar]
- Lai, L.; Bohnsack, B.L.; Niederreither, K.; Hirschi, K.K. Retinoic acid regulates endothelial cell proliferation during vasculogenesis. Development 2003, 130, 6465–6474. [Google Scholar]
- Bohnsack, B.L.; Hirschi, K.K. Red light, green light: signals that control endothelial cell proliferation during embryonic vascular development. Cell Cycle 2004, 3, 1506–1511. [Google Scholar]
- Bohnsack, B.L.; Lai, L.; Dolle, P.; Hirschi, K.K. Signaling hierarchy downstream of retinoic acid that independently regulates vascular remodeling and endothelial cell proliferation. Genes Dev. 2004, 18, 1345–1358. [Google Scholar]
- Satre, M.A.; Kochhar, D.M. Elevations in the endogenous levels of the putative morphogen retinoic acid in embryonic mouse limb-buds associated with limb dysmorphogenesis. Dev. Biol. 1989, 133, 529–536. [Google Scholar]
- Horton, C.; Maden, M. Endogenous distribution of retinoids during normal development and teratogenesis in the mouse embryo. Dev. Dyn. 1995, 202, 312–323. [Google Scholar]
- Scott, W.J., Jr.; Walter, R.; Tzimas, G.; Sass, J.O.; Nau, H.; Collins, M.D. Endogenous status of retinoids and their cytosolic binding proteins in limb buds of chick vs. mouse embryos. Dev. Biol. 1994, 165, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Vermot, J.; Fraulob, V.; Dolle, P.; Niederreither, K. Expression of enzymes synthesizing (aldehyde dehydrogenase 1 and reinaldehyde dehydrogenase 2) and metabolizaing (Cyp26) retinoic acid in the mouse female reproductive system. Endocrinology 2000, 141, 3638–3645. [Google Scholar]
- Clagett-Dame, M.; McNeill, E.M.; Muley, P.D. Role of all-trans retinoic acid in neurite outgrowth and axonal elongation. J. Neurobiol. 2006, 66, 739–756. [Google Scholar]
- Maden, M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 2007, 8, 755–765. [Google Scholar]
- Glover, J.C.; Renaud, J.S.; Rijli, F.M. Retinoic acid and hindbrain patterning. J. Neurobiol. 2006, 66, 705–725. [Google Scholar]
- Maden, M. Retinoids and spinal cord development. J. Neurobiol. 2006, 66, 726–738. [Google Scholar]
- Gavalas, A.; Krumlauf, R. Retinoid signalling and hindbrain patterning. Curr. Opin. Genet. Dev. 2000, 10, 380–386. [Google Scholar]
- Maden, M.; Gale, E.; Kostetskii, I.; Zile, M. Vitamin A-deficient quail embryos have half a hindbrain and other neural defects. Curr. Biol. 1996, 6, 417–426. [Google Scholar]
- Niederreither, K.; Vermot, J.; Schuhbaur, B.; Chambon, P.; Dolle, P. Retinoic acid synthesis and hindbrain patterning in the mouse embryo. Development 2000, 127, 75–85. [Google Scholar]
- Begemann, G.; Schilling, T.F.; Rauch, G.J.; Geisler, R.; Ingham, P.W. The zebrafish neckless mutation reveals a requirement for raldh2 in mesodermal signals that pattern the hindbrain. Development 2001, 128, 3081–3094. [Google Scholar]
- Grandel, H.; Lun, K.; Rauch, G.J.; Rhinn, M.; Piotrowski, T.; Houart, C.; Sordino, P.; Kuchler, A.M.; Schulte-Merker, S.; Geisler, R.; Holder, N.; Wilson, S.W.; Brand, M. Retinoic acid signalling in the zebrafish embryo is necessary during pre-segmentation stages to pattern the anterior-posterior axis of the CNS and to induce a pectoral fin bud. Development 2002, 129, 2851–2865. [Google Scholar]
- Wendling, O.; Ghyselinck, N.B.; Chambon, P.; Mark, M. Roles of retinoic acid receptors in early embryonic morphogenesis and hindbrain patterning. Development 2001, 128, 2031–2038. [Google Scholar]
- Dupe, V.; Ghyselinck, N.B.; Wendling, O.; Chambon, P.; Mark, M. Key roles of retinoic acid receptors alpha and beta in the patterning of the caudal hindbrain, pharyngeal arches and otocyst in the mouse. Development 1999, 126, 5051–5059. [Google Scholar]
- Linville, A.; Radtke, K.; Waxman, J.S.; Yelon, D.; Schilling, T.F. Combinatorial roles for zebrafish retinoic acid receptors in the hindbrain, limbs and pharyngeal arches. Dev. Biol. 2009, 325, 60–70. [Google Scholar]
- Dupe, V.; Lumsden, A. Hindbrain patterning involves graded responses to retinoic acid signalling. Development 2001, 128, 2199–2208. [Google Scholar]
- Sirbu, I.O.; Duester, G. Retinoic-acid signalling in node ectoderm and posterior neural plate directs left-right patterning of somitic mesoderm. Nat. Cell Biol. 2006, 8, 271–277. [Google Scholar]
- MacLean, G.; Abu-Abed, S.; Dolle, P.; Tahayato, A.; Chambon, P.; Petkovich, M. Cloning of a novel retinoic-acid metabolizing cytochrome P450, Cyp26B1, and comparative expression analysis with Cyp26A1 during early murine developmen. Mech. Dev. 2001, 107, 195–201. [Google Scholar]
- Sirbu, I.O.; Gresh, L.; Barra, J.; Duester, G. Shifting boundaries of retinoic acid activity control hindbrain segmental gene expression. Development 2005, 132, 2611–2622. [Google Scholar]
- Hernandez, R.E.; Putzke, A.P.; Myers, J.P.; Margaretha, L.; Moens, C.B. Cyp26 enzymes generate the retinoic acid response pattern necessary for hindbrain development. Development 2007, 134, 177–187. [Google Scholar]
- Ribes, V.; Fraulob, V.; Petkovich, M.; Dolle, P. The oxidizing enzyme CYP26a1 tightly regulates the availability of retinoic acid in the gastrulating mouse embryo to ensure proper head development and vasculogenesis. Dev. Dyn. 2007, 236, 644–653. [Google Scholar]
- Koide, T.; Downes, M.; Chandraratna, R.A.; Blumberg, B.; Umesono, K. Active repression of RAR signaling is required for head formation. Genes Dev. 2001, 15, 2111–2121. [Google Scholar]
- Schneider, R.A.; Hu, D.; Rubenstein, J.L.; Maden, M.; Helms, J.A. Local retinoid signaling coordinates forebrain and facial morphogenesis by maintaining FGF8 and SHH. Development 2001, 128, 2755–2767. [Google Scholar]
- Halilagic, A.; Zile, M.H.; Studer, M. A novel role for retinoids in patterning the avian forebrain during presomite stages. Development 2003, 130, 2039–2050. [Google Scholar]
- Halilagic, A.; Ribes, V.; Ghyselinck, N.B.; Zile, M.H.; Dolle, P.; Studer, M. Retinoids control anterior and dorsal properties in the developing forebrain. Dev. Biol. 2007, 303, 362–375. [Google Scholar]
- Marklund, M.; Sjodal, M.; Beehler, B.C.; Jessell, T.M.; Edlund, T.; Gunhaga, L. Retinoic acid signalling specifies intermediate character in the developing telencephalon. Development 2004, 131, 4323–4332. [Google Scholar]
- Molotkova, N.; Molotkov, A.; Duester, G. Role of retinoic acid during forebrain development begins late when Raldh3 generates retinoic acid in the ventral subventricular zone. Dev. Biol. 2007, 303, 601–610. [Google Scholar]
- Luo, T.; Wagner, E.; Drager, U.C. Integrating retinoic acid signaling with brain function. Dev. Psychol. 2009, 45, 139–150. [Google Scholar]
- Zhang, J.; Smith, D.; Yamamoto, M.; Ma, L.; McCaffery, P. The meninges is a source of retinoic acid for the late-developing hindbrain. J. Neurosci. 2003, 23, 7610–7620. [Google Scholar]
- Yamamoto, M.; Fujinuma, M.; Hirano, S.; Hayakawa, Y.; Clagett-Dame, M.; Zhang, J.; McCaffery, P. Retinoic acid influences the development of the inferior olivary nucleus in the rodent. Dev. Biol. 2005, 280, 421–433. [Google Scholar]
- Smith, D.; Wagner, E.; Koul, O.; McCaffery, P.; Drager, U.C. Retinoic acid synthesis for the developing telencephalon. Cereb. Cortex 2001, 11, 894–905. [Google Scholar]
- Siegenthaler, J.A.; Ashique, A.M.; Zarbalis, K.; Patterson, K.P.; Hecht, J.H.; Kane, M.A.; Folias, A.E.; Choe, Y.; May, S.R.; Kume, T.; Napoli, J.L.; Peterson, A.S.; Pleasure, S.J. Retinoic acid from the meninges regulates cortical neuron generation. Cell 2009, 139, 597–609. [Google Scholar]
- Wilson, V.; Olivera-Martinez, I.; Storey, K.G. Stem cells, signals and vertebrate body axis extension. Development 2009, 136, 1591–1604. [Google Scholar]
- Molotkova, N.; Molotkov, A.; Sirbu, I.O.; Duester, G. Requirement of mesodermal retinoic acid generated by Raldh2 for posterior neural transformation. Mech. Dev. 2005, 122, 145–155. [Google Scholar]
- Diez del Corral, R.; Olivera-Martinez, I.; Goriely, A.; Gale, E.; Maden, M.; Storey, K. Opposing FGF and retinoid pathways control ventral neural pattern, neuronal differentiation, and segmentation during body axis extensio. Neuron 2003, 40, 65–79. [Google Scholar]
- Diez del Corral, R.; Storey, K.G. Opposing FGF and retinoid pathways: a signalling switch that controls differentiation and patterning onset in the extending vertebrate body axis. Bioessays 2004, 26, 857–869. [Google Scholar]
- Pierani, A.; Brenner-Morton, S.; Chiang, C.; Jessell, T.M. A sonic hedgehog-independent, retinoid-activated pathway of neurogenesis in the ventral spinal cord. Cell 1999, 97, 903–915. [Google Scholar]
- Novitch, B.G.; Wichterle, H.; Jessell, T.M.; Sockanathan, S. A requirement for retinoic acid-mediated transcriptional activation in ventral neural patterning and motor neuron specification. Neuron 2003, 40, 81–95. [Google Scholar]
- Sockanathan, S.; Perlmann, T.; Jessell, T.M. Retinoid receptor signaling in postmitotic motor neurons regulates rostrocaudal positional identity and axonal projection pattern. Neuron 2003, 40, 97–111. [Google Scholar]
- Vermot, J.; Schuhbaur, B.; Le Mouellic, H.; McCaffery, P.; Garnier, J.M.; Hentsch, D.; Brulet, P.; Niederreither, K.; Chambon, P.; Dolle, P.; Le Roux, I. Retinaldehyde dehydrogenase 2 and Hoxc8 are required in the murine brachial spinal cord for the specification of Lim1+ motoneurons and the correct distribution of Islet1+ motoneurons. Development 2005, 132, 1611–1621. [Google Scholar]
- Misra, M.; Shah, V.; Carpenter, E.; McCaffery, P.; Lance-Jones, C. Restricted patterns of Hoxd10 and Hoxd11 set segmental differences in motoneuron subtype complement in the lumbosacral spinal cord. Dev. Biol. 2009, 330, 54–72. [Google Scholar]
- Rodriguez-Tebar, A.; Rohrer, H. Retinoic acid induces NGF-dependent survival response and high-affinity NGF receptors in immature chick sympathetic neurons. Development 1991, 112, 813–820. [Google Scholar]
- Plum, L.A.; Clagett-Dame, M. All-trans retinoic acid stimulates and maintains neurite outgrowth in nerve growth factor-supported developing chick embryonic sympathetic neurons. Dev. Biol. 1996, 205, 52–63. [Google Scholar]
- Plum, L.A.; Parada, L.F.; Tsoulfas, P.; Clagett-Dame, M. Retinoic acid combined with neurotrophin-3 enhances the survival and neurite outgrowth of embryonic sympathetic neurons. Exp. Biol. Med. 2001, 226, 766–775. [Google Scholar]
- Merrill, R.A.; Plum, L.A.; Kaiser, M.E.; Clagett-Dame, M. A mammalian homolog of unc-53 is regulated by all-trans retinoic acid in neuroblastoma cells and embryos. Proc. Natl. Acad. Sci. USA 2002, 99, 3422–3427. [Google Scholar]
- Merrill, R.A.; Ahrens, J.M.; Kaiser, M.E.; Federhart, K.S.; Poon, V.Y.; Clagett-Dame, M. All-trans retinoic acid-responsive genes identified in the human SH-SY5Y neuroblastoma cell line and their regulated expression in the nervous system of early embryos. Biol. Chem. 2004, 385, 605–614. [Google Scholar]
- Merrill, R.A.; See, A.W.; Wertheim, M.L.; Clagett-Dame, M. Crk-associated substrate (Cas) family member, NEDD9, is regulated in human neuroblastoma cells and in the embryonic hindbrain by all-trans retinoic aci. Dev. Dyn. 2004, 231, 564–575. [Google Scholar]
- Muley, P.D.; McNeill, E.M.; Marzinke, M.A.; Knobel, K.M.; Barr, M.M.; Clagett-Dame, M. The atRA-responsive gene neuron navigator 2 functions in neurite outgrowth and axonal elongation. Dev. Neurobiol. 2008, 68, 1441–1453. [Google Scholar]
- McNeill, E.M.; Roos, K.P.; Moechars, D.; Clagett-Dame, M. Nav2 is necessary for cranial nerve development and blood pressure regulation. Neural Dev. 2010, 5, 6. [Google Scholar]
- Wagner, E.; McCaffery, P.; Drager, U.C. Retinoic acid in the formation of the dorsoventral retina and its central projections. Dev. Biol. 2000, 222, 460–470. [Google Scholar]
- Drager, U.C.; Li, H.; Wagner, E.; McCaffery, P. Retinoic acid synthesis and breakdown in the developing mouse retina. Prog. Brain Res. 2001, 131, 579–587. [Google Scholar]
- Mic, F.A.; Molotkov, A.; Fan, X.; Cuenca, A.E.; Duester, G. RALDH3, a retinaldehyde dehydrogenase that generates retinoic acid, is expressed in the ventral retina, otic vesicle and olfactory pit during mouse development. Mech. Dev. 2000, 97, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wagner, E.; McCaffery, P.; Smith, D.; Andreadis, A.; Drager, U.C. A retinoic acid synthesizing enzyme in ventral retina and telencephalon of the embryonic mouse. Mech. Dev. 2000, 95, 283–289. [Google Scholar]
- McCaffery, P.; Tempst, P.; Lara, G.; Drager, U.C. Aldehyde dehydrogenase is a positional marker in the retina. Development 1991, 112, 693–702. [Google Scholar]
- Mori, M.; Ghyselinck, N.B.; Chambon, P.; Mark, M. Systematic immunolocalization of retinoid receptors in developing and adult mouse eyes. Invest. Ophthalmol. Vis. Sci. 2001, 42, 1312–1318. [Google Scholar]
- Ghyselinck, N.B.; Dupe, V.; Dierich, A.; Messaddeq, N.; Garnier, J.M.; Rochette-Egly, C.; Chambon, P.; Mark, M. Role of the retinoic acid receptor beta (RARbeta) during mouse development. Int. J. Dev. Biol. 1997, 41, 425–447. [Google Scholar]
- Kastner, P.; Grondona, J.M.; Mark, M.; Gansmuller, A.; LeMeur, M.; Decimo, D.; Vonesch, J.L.; Dolle, P.; Chambon, P. Genetic analysis of RXR alpha developmental function: convergence of RXR and RAR signaling pathways in heart and eye morphogenesis. Cell 1994, 78, 987–1003. [Google Scholar]
- Kastner, P.; Mark, M.; Ghyselinck, N.; Krezel, W.; Dupe, V.; Grondona, J.M.; Chambon, P. Genetic evidence that the retinoid signal is transduced by heterodimeric RXR/RAR functional units during mouse development. Development 1997, 124, 313–326. [Google Scholar]
- Matt, N.; Ghyselinck, N.B.; Pellerin, I.; Dupe, V. Impairing retinoic acid signalling in the neural crest cells is sufficient to alter entire eye morphogenesis. Dev. Biol. 2008, 320, 140–148. [Google Scholar]
- Cammas, L.; Trensz, F.; Jellali, A.; Ghyselinck, N.B.; Roux, M.J.; Dolle, P. Retinoic acid receptor (RAR)-alpha is not critically required for mediating retinoic acid effects in the developing mouse retina. Invest. Ophthalmol. Vis. Sci. 2010, 51, 3281–3290. [Google Scholar]
- Kumar, S.; Duester, G. Retinoic acid signaling in perioptic mesenchyme represses Wnt signaling via induction of Pitx2 and Dkk2. Dev. Biol. 2010, 340, 67–74. [Google Scholar]
- Gage, P.J.; Suh, H.; Camper, S.A. Dosage requirement of Pitx2 for development of multiple organs. Development 1999, 126, 4643–4651. [Google Scholar]
- Zacharias, A.L.; Gage, P.J. Canonical Wnt/beta-catenin signaling is required for maintenance but not activation of Pitx2 expression in neural crest during eye development. Dev. Dyn. 2010, 239, 3215–3225. [Google Scholar]
- Aulehla, A.; Pourquie, O. Signaling gradients during paraxial mesoderm development. Cold Spring Harb. Perspect. Biol. 2010, 2, a000869. [Google Scholar]
- Dubrulle, J.; McGrew, M.J.; Pourquie, O. FGF signaling controls somite boundary position and regulates segmentation clock control of spatiotemporal Hox gene activation. Cell 2001, 106, 219–232. [Google Scholar]
- Sawada, A.; Shinya, M.; Jiang, Y.J.; Kawakami, A.; Kuroiwa, A.; Takeda, H. Fgf/MAPK signalling is a crucial positional cue in somite boundary formation. Development 2001, 128, 4873–4880. [Google Scholar]
- Tenin, G.; Wright, D.; Ferjentsik, Z.; Bone, R.; McGrew, M.J.; Maroto, M. The chick somitogenesis oscillator is arrested before all paraxial mesoderm is segmented into somites. BMC Dev. Biol. 2010, 10, 24. [Google Scholar]
- Kieny, M.; Mauger, A.; Sengel, P. Early regionalization of somitic mesoderm as studied by the development of axial skeleton of the chick embryo. Dev. Biol. 1972, 28, 142–161. [Google Scholar]
- Gruss, P.; Kessel, M. Axial specification in higher vertebrates. Curr. Opin. Genet. Dev. 1991, 1, 204–210. [Google Scholar]
- Kessel, M. Respecification of vertebral identities by retinoic acid. Development 1992, 115, 487–501. [Google Scholar]
- Kessel, M.; Gruss, P. Homeotic transformations of murine vertebrae and concomitant alteration of Hox codes induced by retinoic acid. Cell 1991, 67, 89–104. [Google Scholar]
- Marshall, H.; Nonchev, S.; Sham, M.H.; Muchamore, I.; Lumsden, A.; Krumlauf, R. Retinoic acid alters hindbrain Hox code and induces transformation of rhombomeres 2/3 into a 4/5 identity. Nature 1992, 360, 737–741. [Google Scholar]
- Hall, B.K.; Horstadius, S. The Neural Crest; Oxford University Press: New York, NY, USA, 1988; pp. 1–303. [Google Scholar]
- Dupe, V.; Pellerin, I. Retinoic acid receptors exhibit cell-autonomous functions in cranial neural crest cells. Dev. Dyn. 2009, 238, 2701–2711. [Google Scholar]
- Dersch, H.; Zile, M.H. Induction of normal cardiovascular development in the vitamin A-deprived quail embryo by natural retinoids. Dev. Biol. 1993, 160, 424–433. [Google Scholar]
- Zile, M.H. Vitamin A-Not for Your Eyes Only: Requirement for Heart Formation Begins Early in Embryogenesis. Nutrients 2010, 2, 532–550. [Google Scholar]
- Niederreither, K.; Vermot, J.; Messaddeq, N.; Schuhbaur, B.; Chambon, P.; Dolle, P. Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development 2001, 128, 1019–1031. [Google Scholar]
- Wagner, M.; Siddiqui, M.A. Signal transduction in early heart development (II): ventricular chamber specification, trabeculation, and heart valve formation. Exp. Biol. Med. (Maywood) 2007, 232, 866–880. [Google Scholar] [PubMed]
- Hoover, L.L.; Burton, E.G.; Brooks, B.A.; Kubalak, S.W. The expanding role for retinoid signaling in heart development. Sci. World J. 2008, 8, 194–211. [Google Scholar]
- Lin, S.C.; Dolle, P.; Ryckebusch, L.; Noseda, M.; Zaffran, S.; Schneider, M.D.; Niederreither, K. Endogenous retinoic acid regulates cardiac progenitor differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 9234–9239. [Google Scholar]
- Rochais, F.; Mesbah, K.; Kelly, R.G. Signaling pathways controlling second heart field development. Circ. Res. 2009, 104, 933–942. [Google Scholar]
- Lelievre-Pegorier, M.; Vilar, J.; Ferrier, M.L.; Moreau, E.; Freund, N.; Gilbert, T.; Merlet-Benichou, C. Mild vitamin A deficiency leads to inborn nephron deficit in the rat. Kidney Int. 1998, 54, 1455–1462. [Google Scholar]
- Mendelsohn, C.; Batourina, E.; Fung, S.; Gilbert, T.; Dodd, J. Stromal cells mediate retinoid-dependent functions essential for renal development. Development 1999, 126, 1139–1148. [Google Scholar]
- Batourina, E.; Gim, S.; Bello, N.; Shy, M.; Clagett-Dame, M.; Srinivas, S.; Costantini, F.; Mendelsohn, C. Vitamin A controls epithelial/mesenchymal interactions through Ret expression. Nat. Genet. 2001, 27, 74–78. [Google Scholar]
- Niederreither, K.; Fraulob, V.; Garnier, J.M.; Chambon, P.; Dolle, P. Differential expression of retinoic acid-synthesizing (RALDH) enzymes during fetal development and organ differentiation in the mouse. Mech. Dev. 2002, 110, 165–171. [Google Scholar]
- Schuchardt, A.; D’Agati, V.; Larsson-Blomberg, L.; Costantini, F.; Pachnis, V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 1994, 367, 380–383. [Google Scholar]
- Schuchardt, A.; D’Agati, V.; Pachnis, V.; Costantini, F. Renal agenesis and hypodysplasia in ret-k- mutant mice result from defects in ureteric bud development. Development 1996, 122, 1919–1929. [Google Scholar]
- Batourina, E.; Choi, C.; Paragas, N.; Bello, N.; Hensle, T.; Costantini, F.D.; Schuchardt, A.; Bacallao, R.L.; Mendelsohn, C.L. Distal ureter morphogenesis depends on epithelial cell remodeling mediated by vitamin A and Ret. Nat. Genet. 2002, 32, 109–115. [Google Scholar]
- Rosselot, C.; Spraggon, L.; Chia, I.; Batourina, E.; Riccio, P.; Lu, B.; Niederreither, K.; Dolle, P.; Duester, G.; Chambon, P.; Costantini, F.; Gilbert, T.; Molotkov, A.; Mendelsohn, C. Non-cell-autonomous retinoid signaling is crucial for renal development. Development 2010, 137, 283–292. [Google Scholar]
- Hoy, W.E.; Hughson, M.D.; Bertram, J.F.; Douglas-Denton, R.; Amann, K. Nephron number, hypertension, renal disease, and renal failu. J. Am. Soc. Nephrol. 2005, 16, 2557–2564. [Google Scholar]
- Brenner, B.M.; Mackenzie, H.S. Nephron mass as a risk factor for progression of renal disease. Kidney Int. Suppl. 1997, 63, S124–S127. [Google Scholar]
- Poladia, D.P.; Kish, K.; Kutay, B.; Bauer, J.; Baum, M.; Bates, C.M. Link between reduced nephron number and hypertension: studies in a mutant mouse model. Pediatr. Res. 2006, 59, 489–493. [Google Scholar]
- Keller, G.; Zimmer, G.; Mall, G.; Ritz, E.; Amann, K. Nephron number in patients with primary hypertension. New Engl. J. Med. 2003, 348, 101–108. [Google Scholar]
- Makrakis, J.; Zimanyi, M.A.; Black, M.J. Retinoic acid enhances nephron endowment in rats exposed to maternal protein restriction. Pediatr. Nephrol. 2007, 22, 1861–1867. [Google Scholar]
- Torfs, C.P.; Curry, C.J.; Bateson, T.F.; Honore, L.H. A population-based study of congenital diaphragmatic hernia. Teratology 1992, 46, 555–565. [Google Scholar]
- Beurskens, L.W.; Tibboel, D.; Lindemans, J.; Duvekot, J.J.; Cohen-Overbeek, T.E.; Veenma, D.C.; de Klein, A.; Greer, J.J.; Steegers-Theunissen, R.P. Retinol status of newborn infants is associated with congenital diaphragmatic hernia. Pediatrics 2010, 126, 712–720. [Google Scholar]
- Pasutto, F.; Sticht, H.; Hammersen, G.; Gillessen-Kaesbach, G.; Fitzpatrick, D.R.; Nurnberg, G.; Brasch, F.; Schirmer-Zimmermann, H.; Tolmie, J.L.; Chitayat, D.; et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retarda. Am. J. Hum. Genet. 2007, 80, 550–560. [Google Scholar]
- Goumy, C.; Gouas, L.; Marceau, G.; Coste, K.; Veronese, L.; Gallot, D.; Sapin, V.; Vago, P.; Tchirkov, A. Retinoid pathway and congenital diaphragmatic hernia: hypothesis from the analysis of chromosomal abnormalities. Fetal Diagn. Ther. 2010, 28, 129–139. [Google Scholar]
- Clugston, R.D.; Klattig, J.; Englert, C.; Clagett-Dame, M.; Martinovic, J.; Benachi, A.; Greer, J.J. Teratogen-induced, dietary and genetic models of congenital diaphragmatic hernia share a common mechanism of pathogenesis. Am. J. Pathol. 2006, 169, 1541–1549. [Google Scholar]
- Mey, J.; Babiuk, R.P.; Clugston, R.; Zhang, W.; Greer, J.J. Retinal dehydrogenase-2 is inhibited by compounds that induce congenital diaphragmatic hernias in rodents. Am. J. Pathol. 2003, 162, 673–679. [Google Scholar]
- Clugston, R.D.; Zhang, W.; Alvarez, S.; de Lera, A.R.; Greer, J.J. Understanding abnormal retinoid signaling as a causative mechanism in congenital diaphragmatic hernia. Am. J. Respir. Cell Mol. Biol. 2010, 42, 276–285. [Google Scholar]
- Hind, M.; Corcoran, J.; Maden, M. Temporal/spatial expression of retinoid binding proteins and RAR isoforms in the postnatal lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L468–L476. [Google Scholar]
- Malpel, S.; Mendelsohn, C.; Cardoso, W.V. Regulation of retinoic acid signaling during lung morphogenesis. Development 2000, 127, 3057–3067. [Google Scholar]
- Mollard, R.; Viville, S.; Ward, S.J.; Decimo, D.; Chambon, P.; Dolle, P. Tissue-specific expression of retinoic acid receptor isoform transcripts in the mouse embryo. Mech. Dev. 2000, 94, 223–232. [Google Scholar]
- Wang, Z.; Dolle, P.; Cardoso, W.V.; Niederreither, K. Retinoic acid regulates morphogenesis and patterning of posterior foregut derivatives. Dev. Biol. 2006, 297, 433–445. [Google Scholar]
- Desai, T.J.; Chen, F.; Lu, J.; Qian, J.; Niederreither, K.; Dolle, P.; Chambon, P.; Cardoso, W.V. Distinct roles for retinoic acid receptors alpha and beta in early lung morphogenesis. Dev. Biol. 2006, 291, 12–24. [Google Scholar]
- Hind, M.; Gilthorpe, A.; Stinchcombe, S.; Maden, M. Retinoid induction of alveolar regeneration: from mice to man? Thorax 2009, 64, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Mollard, R.; Ghyselinck, N.B.; Wendling, O.; Chambon, P.; Mark, M. Stage-dependent responses of the developing lung to retinoic acid signaling. Int. J. Dev. Biol. 2000, 44, 457–462. [Google Scholar]
- Chen, F.; Cao, Y.; Qian, J.; Shao, F.; Niederreither, K.; Cardoso, W.V. A retinoic acid-dependent network in the foregut controls formation of the mouse lung primordium. J. Clin. Invest. 2010, 120, 2040–2048. [Google Scholar]
- Wongtrakool, C.; Malpel, S.; Gorenstein, J.; Sedita, J.; Ramirez, M.I.; Underhill, T.M.; Cardoso, W.V. Down-regulation of retinoic acid receptor alpha signaling is required for sacculation and type I cell formation in the developing lung. J. Biol. Chem. 2003, 278, 46911–46918. [Google Scholar]
- Massaro, D.; Massaro, G.D. Retinoids, alveolus formation, and alveolar deficiency: clinical implication. Am. J. Respir. Cell Mol. Biol. 2003, 28, 271–274. [Google Scholar]
- Massaro, G.D.; Massaro, D. Retinoic acid treatment partially rescues failed septation in rats and in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 278, L955–L960. [Google Scholar]
- Massaro, G.D.; Massaro, D. Postnatal treatment with retinoic acid increases the number of pulmonary alveoli in rats. Am. J. Physiol. 1996, 270, L305–L310. [Google Scholar]
- Massaro, G.D.; Massaro, D.; Chambon, P. Retinoic acid receptor-alpha regulates pulmonary alveolus formation in mice after, but not during, perinatal perio. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L431–L433. [Google Scholar]
- Massaro, G.D.; Massaro, D.; Chan, W.Y.; Clerch, L.B.; Ghyselinck, N.; Chambon, P.; Chandraratna, R.A. Retinoic acid receptor-beta: an endogenous inhibitor of the perinatal formation of pulmonary alveoli. Physiol. Genomics 2000, 4, 51–57. [Google Scholar]
- McGowan, S.; Jackson, S.K.; Jenkins-Moore, M.; Dai, H.H.; Chambon, P.; Snyder, J.M. Mice bearing deletions of retinoic acid receptors demonstrate reduced lung elastin and alveolar numbers. Am. J. Respir. Cell Mol. Biol. 2000, 23, 162–167. [Google Scholar]
- Checkley, W.; West, K.P., Jr.; Wise, R.A.; Baldwin, M.R.; Wu, L.; LeClerq, S.C.; Christian, P.; Katz, J.; Tielsch, J.M.; Khatry, S.; Sommer, A. Maternal vitamin A supplementation and lung function in offspring. New Engl. J. Med. 2010, 362, 1784–1794. [Google Scholar]
- Martin, M.; Gallego-Llamas, J.; Ribes, V.; Kedinger, M.; Niederreither, K.; Chambon, P.; Dolle, P.; Gradwohl, G. Dorsal pancreas agenesis in retinoic acid-deficient Raldh2 mutant mice. Dev. Biol. 2005, 284, 399–411. [Google Scholar]
- Molotkov, A.; Molotkova, N.; Duester, G. Retinoic acid generated by Raldh2 in mesoderm is required for mouse dorsal endodermal pancreas development. Dev. Dyn. 2005, 232, 950–957. [Google Scholar]
- Stafford, D.; Hornbruch, A.; Mueller, P.R.; Prince, V.E. A conserved role for retinoid signaling in vertebrate pancreas development. Dev. Genes Evol. 2004, 214, 432–441. [Google Scholar]
- Stafford, D.; Prince, V.E. Retinoic acid signaling is required for a critical early step in zebrafish pancreatic development. Curr. Biol. 2002, 12, 1215–1220. [Google Scholar]
- Chen, Y.; Pan, F.C.; Brandes, N.; Afelik, S.; Solter, M.; Pieler, T. Retinoic acid signaling is essential for pancreas development and promotes endocrine at the expense of exocrine cell differentiation in Xenopus. Dev. Biol. 2004, 271, 144–160. [Google Scholar]
- Alexa, K.; Choe, S.K.; Hirsch, N.; Etheridge, L.; Laver, E.; Sagerstrom, C.G. Maternal and zygotic aldh1a2 activity is required for pancreas development in zebrafish. PLoS ONE 2009, 4, e8261. [Google Scholar]
- Pan, F.C.; Chen, Y.; Bayha, E.; Pieler, T. Retinoic acid-mediated patterning of the pre-pancreatic endoderm in Xenopus operates via direct and indirect mechanisms. Mech. Dev. 2007, 124, 518–531. [Google Scholar]
- Tulachan, S.S.; Doi, R.; Kawaguchi, Y.; Tsuji, S.; Nakajima, S.; Masui, T.; Koizumi, M.; Toyoda, E.; Mori, T.; Ito, D.; Kami, K.; Fujimoto, K.; Imamura, M. All-trans retinoic acid induces differentiation of ducts and endocrine cells by mesenchymal/epithelial interactions in embryonic pancreas. Diabetes 2003, 52, 76–84. [Google Scholar]
- Ahlgren, U.; Jonsson, J.; Edlund, H. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development 1996, 122, 1409–1416. [Google Scholar]
- Jonsson, J.; Carlsson, L.; Edlund, T.; Edlund, H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 1994, 371, 606–609. [Google Scholar]
- Offield, M.F.; Jetton, T.L.; Labosky, P.A.; Ray, M.; Stein, R.W.; Magnuson, M.A.; Hogan, B.L.; Wright, C.V. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 1996, 122, 983–995. [Google Scholar]
- Kinkel, M.D.; Sefton, E.M.; Kikuchi, Y.; Mizoguchi, T.; Ward, A.B.; Prince, V.E. Cyp26 enzymes function in endoderm to regulate pancreatic field size. Proc. Natl. Acad. Sci. USA 2009, 106, 7864–7869. [Google Scholar]
- Gittes, G.K. Developmental biology of the pancreas: a comprehensive review. Dev. Biol. 2009, 326, 4–35. [Google Scholar]
- Pearl, E.J.; Bilogan, C.K.; Mukhi, S.; Brown, D.D.; Horb, M.E. Xenopus pancreas development. Dev. Dyn. 2009, 238, 1271–1286. [Google Scholar]
- Stratford, T.; Horton, C.; Maden, M. Retinoic acid is required for the initiation of outgrowth in the chick limb bud. Curr. Biol. 1996, 6, 1124–1133. [Google Scholar]
- Stratford, T.; Logan, C.; Zile, M.; Maden, M. Abnormal anteroposterior and dorsoventral patterning of the limb bud in the absence of retinoids. Mech. Dev. 1999, 81, 115–125. [Google Scholar]
- Niederreither, K.; Vermot, J.; Schuhbaur, B.; Chambon, P.; Dolle, P. Embryonic retinoic acid synthesis is required for forelimb growth and anteroposterior patterning in the mouse. Development 2002, 129, 3563–3574. [Google Scholar]
- Mic, F.A.; Sirbu, I.O.; Duester, G. Retinoic acid synthesis controlled by Raldh2 is required early for limb bud initiation and then later as a proximodistal signal during apical ectodermal ridge formation. J. Biol. Chem. 2004, 279, 26698–26706. [Google Scholar]
- Gibert, Y.; Gajewski, A.; Meyer, A.; Begemann, G. Induction and prepatterning of the zebrafish pectoral fin bud requires axial retinoic acid signaling. Development 2006, 133, 2649–2659. [Google Scholar]
- Mercader, N.; Leonardo, E.; Piedra, M.E.; Martinez, A.C.; Ros, M.A.; Torres, M. Opposing RA and FGF signals control proximodistal vertebrate limb development through regulation of Meis genes. Development 2000, 127, 3961–3970. [Google Scholar]
- Benazet, J.D.; Zeller, R. Vertebrate limb development: moving from classical morphogen gradients to an integrated 4-dimensional patterning system. Cold Spring Harb. Perspect. Biol. 2009, 1, a001339. [Google Scholar]
- Zeller, R.; Lopez-Rios, J.; Zuniga, A. Vertebrate limb bud development: moving towards integrative analysis of organogenesis. Nat. Rev. Genet. 2009, 10, 845–858. [Google Scholar]
- Lewandoski, M.; Mackem, S. Limb development: the rise and fall of retinoic acid. Curr. Biol. 2009, 19, R558–R561. [Google Scholar]
- Dupe, V.; Ghyselinck, N.B.; Thomazy, V.; Nagy, L.; Davies, P.J.; Chambon, P.; Mark, M. Essential roles of retinoic acid signaling in interdigital apoptosis and control of BMP-7 expression in mouse autopods. Dev. Biol. 1999, 208, 30–43. [Google Scholar]
- Zhao, X.; Brade, T.; Cunningham, T.J.; Duester, G. Retinoic acid controls expression of tissue remodeling genes Hmgn1 and Fgf18 at the digit-interdigit junction. Dev. Dyn. 2010, 239, 665–671. [Google Scholar]
- Rodriguez-Leon, J.; Merino, R.; Macias, D.; Ganan, Y.; Santesteban, E.; Hurle, J.M. Retinoic acid regulates programmed cell death through BMP signalling. Nat. Cell Biol. 1999, 1, 125–126. [Google Scholar]
- Hernandez-Martinez, R.; Castro-Obregon, S.; Covarrubias, L. Progressive interdigital cell death: regulation by the antagonistic interaction between fibroblast growth factor 8 and retinoic acid. Development 2009, 136, 3669–3678. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).