
Curcumin and Cancer-Related Inflammation


Abstract
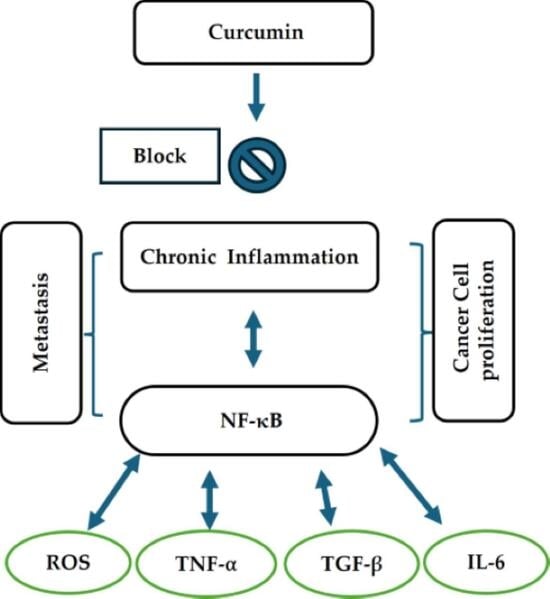
1. Introduction
2. Role of Cytokines
2.1. Interleukin-6
2.2. Transforming Growth Factor-β
2.3. Tumor Necrosis Factor Alpha
3. Reactive Oxygen Species
4. Nuclear Factor Kappa B
5. Curcumin
6. Summary
7. Future Prospectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| IL-6 | Interleukin-6 |
| TNF-α | Tumor necrosis factor-α |
| TGF-β | Transforming growth factor-β |
| TME | Tumor microenvironment |
| NF-κB | Nuclear factor kappa B |
| COX-2 | Cyclooxygenase-2 |
| iNOS | Inducible nitric oxide synthase |
| STAT3 | Signal transducer and activator of transcription 3 |
| ROS | Reactive oxygen species |
| TRADD | TNFR-associated death domain |
| TNFR | Tumor necrosis factor (TNF) receptor |
| TRAF | TNF receptor-associated factor |
| OSCC | Oral squamous cell carcinoma |
| PLGA | poly(lactic-co-glycolic acid) |
References
- Adrian, K.; Ghaib, H.; Ali, I. Tumour necrosis factor-alpha levels as predictor factor on clinical response of anthracycline-based neoadjuvant chemotherapy in locally advance breast cancer patients: Experimental research. Ann. Med. Surg. 2023, 85, 807–811. [Google Scholar]
- Zhang, M.; Zhang, Y.Y.; Chen, Y.; Wang, J.; Wang, Q.; Lu, H. TGF-beta Signaling and Resistance to Cancer Therapy. Front. Cell Dev. Biol. 2021, 9, 786728. [Google Scholar]
- Liang, Y.; Shao, Y.; Gu, W. Role of interleukin-6 in resistance to tumor therapy. Discov. Oncol. 2025, 16, 1791. [Google Scholar] [CrossRef]
- Woo, D.K.; Green, P.D.; Santos, J.H.; D’Souza, A.D.; Walther, Z.; Martin, W.D.; Christian, B.E.; Chandel, N.S.; Shadel, G.S. Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC(Min/+) mice. Am. J. Pathol. 2012, 180, 24–31. [Google Scholar] [PubMed]
- Chen, A.; Huang, H.; Fang, S.; Hang, Q. ROS: A “booster” for chronic inflammation and tumor metastasis. Biochim. Biophys. Acta Rev. Cancer 2024, 1879, 189175. [Google Scholar]
- He, G.; Karin, M. NF-kappaB and STAT3–key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor. Rev. 2010, 21, 11–19. [Google Scholar]
- Thongpon, P.; Intuyod, K.; Chomwong, S.; Pongking, T.; Klungsaeng, S.; Muisuk, K.; Charoenram, N.; Sitthirach, C.; Thanan, R.; Pinlaor, P.; et al. Curcumin synergistically enhances the efficacy of gemcitabine against gemcitabine-resistant cholangiocarcinoma via the targeting LAT2/glutamine pathway. Sci. Rep. 2024, 14, 16059. [Google Scholar]
- Kakarala, M.; Brenner, D.E.; Korkaya, H.; Cheng, C.; Tazi, K.; Ginestier, C.; Liu, S.; Dontu, G.; Wicha, M.S. Targeting breast stem cells with the cancer preventive compounds curcumin and piperine. Breast Cancer Res. Treat. 2010, 122, 777–785. [Google Scholar]
- Zoi, V.; Galani, V.; Lianos, G.D.; Voulgaris, S.; Kyritsis, A.P.; Alexiou, G.A. The Role of Curcumin in Cancer Treatment. Biomedicines 2021, 9, 1086. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. 2008, 14, 4491–4499. [Google Scholar] [CrossRef]
- Puri, R.; Arora, V. Synergistic anticancer efficacy of optimized curcumin-piperine loaded magnetic nanoparticles for the treatment of colorectal cancer. Explor. Target. Antitumor Ther. 2025, 6, 1002340. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef]
- Rose-John, S. Local and systemic effects of interleukin-6 (IL-6) in inflammation and cancer. FEBS Lett. 2022, 596, 557–566. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef]
- Manore, S.G.; Doheny, D.L.; Wong, G.L.; Lo, H.W. IL-6/JAK/STAT3 Signaling in Breast Cancer Metastasis: Biology and Treatment. Front. Oncol. 2022, 12, 866014. [Google Scholar] [CrossRef]
- Heichler, C.; Scheibe, K.; Schmied, A.; Geppert, C.I.; Schmid, B.; Wirtz, S.; Thoma, O.M.; Kramer, V.; Waldner, M.J.; Buttner, C.; et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut 2020, 69, 1269–1282. [Google Scholar] [CrossRef]
- Bharadwaj, U.; Marin-Muller, C.; Li, M.; Chen, C.; Yao, Q. Mesothelin overexpression promotes autocrine IL-6/sIL-6R trans-signaling to stimulate pancreatic cancer cell proliferation. Carcinogenesis 2011, 32, 1013–1024. [Google Scholar] [CrossRef]
- Fu, Z.Z.; Peng, Y.; Cao, L.Y.; Chen, Y.S.; Li, K.; Fu, B.H. Correlations Between Serum IL-6 Levels and Radiation Pneumonitis in Lung Cancer Patients: A Meta-Analysis. J. Clin. Lab. Anal. 2016, 30, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, J.; Luo, J. Decreased IL-6 induces sensitivity of hepatocellular carcinoma cells to sorafenib. Pathol. Res. Pract. 2019, 215, 152565. [Google Scholar] [CrossRef]
- Thuya, W.L.; Cao, Y.; Ho, P.C.; Wong, A.L.; Wang, L.; Zhou, J.; Nicot, C.; Goh, B.C. Insights into IL-6/JAK/STAT3 signaling in the tumor microenvironment: Implications for cancer therapy. Cytokine Growth Factor Rev. 2025, 85, 26–42. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef]
- Saito, A.; Horie, M.; Micke, P.; Nagase, T. The Role of TGF-beta Signaling in Lung Cancer Associated with Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2018, 19, 361. [Google Scholar] [CrossRef] [PubMed]
- Boguslawska, J.; Kryst, P.; Poletajew, S.; Piekielko-Witkowska, A. TGF-beta and microRNA Interplay in Genitourinary Cancers. Cells 2019, 8, 1619. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Suzuki, S.; Takahara, T.; Ono, S.; Goto, M.; Miyabe, S.; Sugita, Y.; Ogawa, T.; Ito, H.; Satou, A.; et al. Improving function of cytotoxic T-lymphocytes by transforming growth factor-beta inhibitor in oral squamous cell carcinoma. Cancer Sci. 2021, 112, 4037–4049. [Google Scholar] [CrossRef]
- Li, C.; Wan, L.; Liu, Z.; Xu, G.; Wang, S.; Su, Z.; Zhang, Y.; Zhang, C.; Liu, X.; Lei, Z.; et al. Long non-coding RNA XIST promotes TGF-beta-induced epithelial-mesenchymal transition by regulating miR-367/141-ZEB2 axis in non-small-cell lung cancer. Cancer Lett. 2018, 418, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Bai, J.; Long, Q.; Wei, Y.; Pan, J.; Li, X.; Tang, Q. TCF-3-mediated transcription of lncRNA HNF1A-AS1 targeting oncostatin M expression inhibits epithelial-mesenchymal transition via TGFbeta signaling in gastroenteropancreatic neuroendocrine neoplasms. Aging 2021, 13, 14065–14077. [Google Scholar] [CrossRef]
- Aoyagi, Y.; Oda, T.; Kinoshita, T.; Nakahashi, C.; Hasebe, T.; Ohkohchi, N.; Ochiai, A. Overexpression of TGF-beta by infiltrated granulocytes correlates with the expression of collagen mRNA in pancreatic cancer. Br. J. Cancer 2004, 91, 1316–1326. [Google Scholar] [CrossRef]
- Annibaldi, A.; Meier, P. Checkpoints in TNF-Induced Cell Death: Implications in Inflammation and Cancer. Trends Mol. Med. 2018, 24, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Benedict, C.A.; Ware, C.F. Clinical targeting of the TNF and TNFR superfamilies. Nat. Rev. Drug Discov. 2013, 12, 147–168. [Google Scholar] [CrossRef] [PubMed]
- Riches, D.W.; Chan, E.D.; Winston, B.W. TNF-alpha-induced regulation and signalling in macrophages. Immunobiology 1996, 195, 477–490. [Google Scholar] [CrossRef]
- Kirchner, S.; Boldt, S.; Kolch, W.; Haffner, S.; Kazak, S.; Janosch, P.; Holler, E.; Andreesen, R.; Eissner, G. LPS resistance in monocytic cells caused by reverse signaling through transmembrane TNF (mTNF) is mediated by the MAPK/ERK pathway. J. Leukoc. Biol. 2004, 75, 324–331. [Google Scholar]
- Hehlgans, T.; Pfeffer, K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: Players, rules and the games. Immunology 2005, 115, 1–20. [Google Scholar] [CrossRef]
- Hsu, H.; Huang, J.; Shu, H.B.; Baichwal, V.; Goeddel, D.V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 1996, 4, 387–396. [Google Scholar] [CrossRef]
- Siakavellas, S.I.; Bamias, G. Tumor Necrosis Factor-like Cytokine TL1A and Its Receptors DR3 and DcR3: Important New Factors in Mucosal Homeostasis and Inflammation. Inflamm. Bowel Dis. 2015, 21, 2441–2452. [Google Scholar] [CrossRef]
- Aggarwal, S.; Takada, Y.; Singh, S.; Myers, J.N.; Aggarwal, B.B. Inhibition of growth and survival of human head and neck squamous cell carcinoma cells by curcumin via modulation of nuclear factor-kappaB signaling. Int. J. Cancer 2004, 111, 679–692. [Google Scholar]
- Zhou, X.; Zhou, S.; Li, B.; Li, Q.; Gao, L.; Li, D.; Gong, Q.; Zhu, L.; Wang, J.; Wang, N.; et al. Transmembrane TNF-alpha preferentially expressed by leukemia stem cells and blasts is a potent target for antibody therapy. Blood 2015, 126, 1433–1442. [Google Scholar]
- Zhao, X.; Fan, W.; Xu, Z.; Chen, H.; He, Y.; Yang, G.; Yang, G.; Hu, H.; Tang, S.; Wang, P.; et al. Inhibiting tumor necrosis factor-alpha diminishes desmoplasia and inflammation to overcome chemoresistance in pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 81110–81122. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zhou, X.; Niu, L.; Lin, G.; Huang, J.; Zhou, W.; Gan, H.; Wang, J.; Jiang, X.; Yin, B.; et al. Targeting transmembrane TNF-alpha suppresses breast cancer growth. Cancer Res. 2013, 73, 4061–4074. [Google Scholar] [CrossRef]
- Prisco, A.R.; Hoffmann, B.R.; Kaczorowski, C.C.; McDermott-Roe, C.; Stodola, T.J.; Exner, E.C.; Greene, A.S. Tumor Necrosis Factor alpha Regulates Endothelial Progenitor Cell Migration via CADM1 and NF-kB. Stem Cells 2016, 34, 1922–1933. [Google Scholar] [CrossRef]
- Mitoma, H.; Horiuchi, T.; Tsukamoto, H.; Tamimoto, Y.; Kimoto, Y.; Uchino, A.; To, K.; Harashima, S.; Hatta, N.; Harada, M. Mechanisms for cytotoxic effects of anti-tumor necrosis factor agents on transmembrane tumor necrosis factor alpha-expressing cells: Comparison among infliximab, etanercept, and adalimumab. Arthritis Rheum. 2008, 58, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in Biology and Medicine. React. Oxyg. Species 2016, 1, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Arner, E.S.; Holmgren, A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006, 16, 420–426. [Google Scholar] [CrossRef]
- Noubade, R.; Wong, K.; Ota, N.; Rutz, S.; Eidenschenk, C.; Valdez, P.A.; Ding, J.; Peng, I.; Sebrell, A.; Caplazi, P.; et al. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature 2014, 509, 235–239. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Matsui, A.; Kobayashi, J.; Kanno, S.I.; Hashiguchi, K.; Miyaji, M.; Yoshikawa, Y.; Yasui, A.; Zhang-Akiyama, Q.M. Oxidation resistance 1 prevents genome instability through maintenance of G2/M arrest in gamma-ray-irradiated cells. J. Radiat. Res. 2020, 61, 1–13. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Podsednik, A.; Jacob, A.; Li, L.Z.; Xu, H.N. Relationship between Optical Redox Status and Reactive Oxygen Species in Cancer Cells. React. Oxyg. Species 2020, 9, 95–108. [Google Scholar] [CrossRef]
- Huang, R.; Chen, H.; Liang, J.; Li, Y.; Yang, J.; Luo, C.; Tang, Y.; Ding, Y.; Liu, X.; Yuan, Q.; et al. Dual Role of Reactive Oxygen Species and their Application in Cancer Therapy. J. Cancer 2021, 12, 5543–5561. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Guo, J.; Rao, H.; Xin, J.; Song, X.; Liu, R.; Shao, S.; Hou, J.; Kong, L.; Hu, Z.; et al. The 8-oxoguanine DNA glycosylase-synaptotagmin 7 pathway increases extracellular vesicle release and promotes tumour metastasis during oxidative stress. J. Extracell. Vesicles 2024, 13, e12505. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Ciccarese, F.; Ciminale, V. Escaping Death: Mitochondrial Redox Homeostasis in Cancer Cells. Front. Oncol. 2017, 7, 117. [Google Scholar] [CrossRef]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- van Ravensteijn, S.G.; Amir, A.L.; Tauriello, D.V.F.; van Herpen, C.M.L.; Boers-Sonderen, M.J.; Wesseling, Y.J.W.; van Brussel, A.G.C.; Keizer, D.M.; Verheul, H.M.W.; Bol, K.F. Exploring the relation between TGF-beta pathway activity and response to checkpoint inhibition in patients with metastatic melanoma. Clin. Exp. Immunol. 2025, 219, uxae108. [Google Scholar] [CrossRef]
- Vyas, D.; Lopez-Hisijos, N.; Gandhi, S.; El-Dakdouki, M.; Basson, M.D.; Walsh, M.F.; Huang, X.; Vyas, A.K.; Chaturvedi, L.S. Doxorubicin-Hyaluronan Conjugated Super-Paramagnetic Iron Oxide Nanoparticles (DOX-HA-SPION) Enhanced Cytoplasmic Uptake of Doxorubicin and Modulated Apoptosis, IL-6 Release and NF-kappaB Activity in Human MDA-MB-231 Breast Cancer Cells. J. Nanosci. Nanotechnol. 2015, 15, 6413–6422. [Google Scholar] [CrossRef] [PubMed]
- Shirmohammadi, E.; Ebrahimi, S.S.; Farshchi, A.; Salimi, M. The efficacy of etanercept as anti-breast cancer treatment is attenuated by residing macrophages. BMC Cancer 2020, 20, 836. [Google Scholar] [PubMed]
- Jensen-Jarolim, E.; Fazekas, J.; Singer, J.; Hofstetter, G.; Oida, K.; Matsuda, H.; Tanaka, A. Crosstalk of carcinoembryonic antigen and transforming growth factor-beta via their receptors: Comparing human and canine cancer. Cancer Immunol. Immunother. 2015, 64, 531–537. [Google Scholar] [CrossRef]
- Ma, N.; Wang, Y.; Li, X.; Xu, M.; Tan, D. Reactive oxygen species in cancer: Mechanistic insights and therapeutic innovations. Cell Stress Chaperones 2025, 30, 100108. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Sahu, A.; Shekhar, H.; Sharma, B.; Haque, S.; Kaur, D.; Tuli, H.S.; Mishra, A.; Ahmad, F. The heat of the battle: Inflammation’s role in prostate cancer development and inflammation-targeted therapies. Discov. Oncol. 2025, 16, 108. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Chen, D.D.; Li, X.H.; Zhang, H.W.; Lu, Y.Q.; Ye, C.L.; Ren, X.D. Changes of NF-kB, p53, Bcl-2 and caspase in apoptosis induced by JTE-522 in human gastric adenocarcinoma cell line AGS cells: Role of reactive oxygen species. World J. Gastroenterol. 2002, 8, 431–435. [Google Scholar] [CrossRef]
- Shen, K.; Zhang, Q. Literature review: Nuclear factor kappa B (NF-kappaB) regulation in human cancers mediated by ubiquitin-specific proteases (USPs). Ann. Transl. Med. 2024, 12, 90. [Google Scholar] [CrossRef]
- Daniels, M.A.; Teixeiro, E. The NF-kappaB signaling network in the life of T cells. Front. Immunol. 2025, 16, 1559494. [Google Scholar] [CrossRef]
- Anilkumar, S.; Wright-Jin, E. NF-kappaB as an Inducible Regulator of Inflammation in the Central Nervous System. Cells 2024, 13, 485. [Google Scholar] [CrossRef]
- Baud, V.; Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef]
- Islam, M.T.; Chen, F.Z.; Chen, H.C.; Wahid, A. Knockdown of USP8 inhibits prostate cancer cell growth, proliferation, and metastasis and promotes docetaxel’s activity by suppressing the NF-kB signaling pathway. Front. Oncol. 2022, 12, 923270. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, X.G.; Xu, J.H.; Wu, X.P.; Qiu, H.L.; Yi, H.; Li, W.X. Overexpression of the human ZNF300 gene enhances growth and metastasis of cancer cells through activating NF-kB pathway. J. Cell. Mol. Med. 2012, 16, 1134–1145. [Google Scholar] [CrossRef]
- Mu, G.; Zhang, W.; Gu, Y.; Wang, H.; Huang, J.; Bian, C.; Wei, K.; Xia, Y.; Wang, J. The P4HB/LGALS3BP/NF-kB axis promotes the progression of MIA to IAC in lung adenocarcinoma. Sci. Rep. 2025, 15, 26672. [Google Scholar] [CrossRef]
- Mitrovic Ajtic, O.; Suboticki, T.; Diklic, M.; Dikic, D.; Vukotic, M.; Dragojevic, T.; Zivkovic, E.; Antic, D.; Cokic, V. Regulation of S100As Expression by Inflammatory Cytokines in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2022, 23, 6952. [Google Scholar] [CrossRef]
- Burstein, E.; Duckett, C.S. Dying for NF-kappaB? Control of cell death by transcriptional regulation of the apoptotic machinery. Curr. Opin. Cell Biol. 2003, 15, 732–737. [Google Scholar] [CrossRef]
- Huang, C.P.; Chen, J.; Chen, C.C.; Liu, G.; Zhang, Y.; Messing, E.; Yeh, S.; Chang, C. ASC-J9(R) increases the bladder cancer chemotherapy efficacy via altering the androgen receptor (AR) and NF-kappaB survival signals. J. Exp. Clin. Cancer Res. 2019, 38, 275. [Google Scholar] [CrossRef]
- Sun, Y.; St Clair, D.K.; Fang, F.; Warren, G.W.; Rangnekar, V.M.; Crooks, P.A.; St Clair, W.H. The radiosensitization effect of parthenolide in prostate cancer cells is mediated by nuclear factor-kappaB inhibition and enhanced by the presence of PTEN. Mol. Cancer Ther. 2007, 6, 2477–2486. [Google Scholar] [CrossRef]
- Alam, M.M.; Naeem, M.; Khan, M.M.A.; Uddin, M. Vincristine and Vinblastine Anticancer Catharanthus Alkaloids: Pharmacological Applications and Strategies for Yield Improvement. In Catharanthus Roseus: Current Research and Future Prospects; Naeem, M., Aftab, T., Khan, M.M.A., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 277–307. [Google Scholar]
- Aggarwal, S.; Ichikawa, H.; Takada, Y.; Sandur, S.K.; Shishodia, S.; Aggarwal, B.B. Curcumin (diferuloylmethane) down-regulates expression of cell proliferation and antiapoptotic and metastatic gene products through suppression of IkappaBalpha kinase and Akt activation. Mol. Pharmacol. 2006, 69, 195–206. [Google Scholar] [CrossRef]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The Essential Medicinal Chemistry of Curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Nagpal, M.; Vashistha, V.K.; Arora, R.; Issar, U. Recent advances in the antioxidant activity of metal-curcumin complexes: A combined computational and experimental review. Free Radic. Res. 2024, 58, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Bharti, A.C.; Takada, Y.; Aggarwal, B.B. Curcumin (diferuloylmethane) inhibits receptor activator of NF-kappa B ligand-induced NF-kappa B activation in osteoclast precursors and suppresses osteoclastogenesis. J. Immunol. 2004, 172, 5940–5947. [Google Scholar] [CrossRef] [PubMed]
- Hewlings, S.J.; Kalman, D.S. Curcumin: A Review of Its Effects on Human Health. Foods 2017, 6, 92. [Google Scholar] [CrossRef]
- He, Y.; Liao, Y.; Fang, S.; Zhu, L.; Zhao, Z.; Chen, T.; Zhang, Z. Mechanism of Curcumin in Inhibiting Proliferation of Head and Neck Squamous Cell Carcinoma: A Network Pharmacology and Cellular Experimental Study. BioMed Res. Int. 2025, 2025, 4318115. [Google Scholar] [CrossRef]
- Sala de Oyanguren, F.J.; Rainey, N.E.; Moustapha, A.; Saric, A.; Sureau, F.; O’Connor, J.E.; Petit, P.X. Highlighting Curcumin-Induced Crosstalk between Autophagy and Apoptosis as Supported by Its Specific Subcellular Localization. Cells 2020, 9, 361. [Google Scholar] [CrossRef]
- Guneydas, G.; Topcul, M.R. Antiproliferative Effects of Curcumin Different Types of Breast Cancer. Asian Pac. J. Cancer Prev. 2022, 23, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.N.; Haggag, Y.A.; Lane, M.E.; McCarron, P.A.; Tambuwala, M.M. Polymeric Nano-Encapsulation of Curcumin Enhances its Anti-Cancer Activity in Breast (MDA-MB231) and Lung (A549) Cancer Cells Through Reduction in Expression of HIF-1alpha and Nuclear p65 (Rel A). Curr. Drug Deliv. 2018, 15, 286–295. [Google Scholar] [CrossRef]
- Starvaggi Cucuzza, L.; Motta, M.; Miretti, S.; Accornero, P.; Baratta, M. Curcuminoid-phospholipid complex induces apoptosis in mammary epithelial cells by STAT-3 signaling. Exp. Mol. Med. 2008, 40, 647–657. [Google Scholar] [CrossRef]
- Ramachandran, C.; Fonseca, H.B.; Jhabvala, P.; Escalon, E.A.; Melnick, S.J. Curcumin inhibits telomerase activity through human telomerase reverse transcritpase in MCF-7 breast cancer cell line. Cancer Lett. 2002, 184, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Fabbri, A.; Del Corno, M.; Conti, L. Dietary Polyphenols: Promising Adjuvants for Colorectal Cancer Therapies. Cancers 2021, 13, 4499. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.I.; Ke, Y.F.; Ko, Y.C.; Lin, J.K. Curcumin inhibits SK-Hep-1 hepatocellular carcinoma cell invasion in vitro and suppresses matrix metalloproteinase-9 secretion. Oncology 1998, 55, 349–353. [Google Scholar] [CrossRef]
- Cao, J.; Liu, Y.; Jia, L.; Zhou, H.M.; Kong, Y.; Yang, G.; Jiang, L.P.; Li, Q.J.; Zhong, L.F. Curcumin induces apoptosis through mitochondrial hyperpolarization and mtDNA damage in human hepatoma G2 cells. Free Radic. Biol. Med. 2007, 43, 968–975. [Google Scholar] [CrossRef]
- Ma, K.; Shen, Y.; Hu, J.; Li, J.; Zhang, X. Curcumin as a therapeutic agent in liver cancer: A systematic review of preclinical models and mechanisms. Eur. J. Med. Res. 2025, 30, 640. [Google Scholar] [CrossRef]
- Schwarz, K.; Dobiasch, S.; Nguyen, L.; Schilling, D.; Combs, S.E. Modification of radiosensitivity by Curcumin in human pancreatic cancer cell lines. Sci. Rep. 2020, 10, 3815. [Google Scholar] [CrossRef] [PubMed]
- de Matos, R.P.A.; Calmon, M.F.; Amantino, C.F.; Villa, L.L.; Primo, F.L.; Tedesco, A.C.; Rahal, P. Effect of Curcumin-Nanoemulsion Associated with Photodynamic Therapy in Cervical Carcinoma Cell Lines. BioMed Res. Int. 2018, 2018, 4057959. [Google Scholar] [CrossRef] [PubMed]
- Abdull Rahim, U.; Mustapa, M.; Mohamed Shakrin, N.N.S.; Nurdin, A.; Mohamad Taridi, N.; Yusof, Y.A.M.; Mad Nordin, M.F.; Che Roos, N.A. Current evidence and future direction on evaluating the anticancer effects of curcumin, gingerols, and shogaols in cervical cancer: A systematic review. PLoS ONE 2024, 19, e0314280. [Google Scholar] [CrossRef]
- Zong, H.; Wang, F.; Fan, Q.X.; Wang, L.X. Curcumin inhibits metastatic progression of breast cancer cell through suppression of urokinase-type plasminogen activator by NF-kappa B signaling pathways. Mol. Biol. Rep. 2012, 39, 4803–4808. [Google Scholar] [CrossRef]
- Desai, S.J.; Prickril, B.; Rasooly, A. Mechanisms of Phytonutrient Modulation of Cyclooxygenase-2 (COX-2) and Inflammation Related to Cancer. Nutr. Cancer 2018, 70, 350–375. [Google Scholar] [CrossRef]
- Goel, A.; Boland, C.R.; Chauhan, D.P. Specific inhibition of cyclooxygenase-2 (COX-2) expression by dietary curcumin in HT-29 human colon cancer cells. Cancer Lett. 2001, 172, 111–118. [Google Scholar] [CrossRef]
- Waghela, B.N.; Sharma, A.; Dhumale, S.; Pandey, S.M.; Pathak, C. Curcumin conjugated with PLGA potentiates sustainability, anti-proliferative activity and apoptosis in human colon carcinoma cells. PLoS ONE 2015, 10, e0117526. [Google Scholar] [CrossRef]
- Li, L.; Braiteh, F.S.; Kurzrock, R. Liposome-encapsulated curcumin: In vitro and in vivo effects on proliferation, apoptosis, signaling, and angiogenesis. Cancer 2005, 104, 1322–1331. [Google Scholar] [CrossRef]
- Aggarwal, S.; Ndinguri, M.W.; Solipuram, R.; Wakamatsu, N.; Hammer, R.P.; Ingram, D.; Hansel, W. [DLys(6)]-luteinizing hormone releasing hormone-curcumin conjugate inhibits pancreatic cancer cell growth in vitro and in vivo. Int. J. Cancer 2011, 129, 1611–1623. [Google Scholar] [CrossRef]
- Weissenberger, J.; Priester, M.; Bernreuther, C.; Rakel, S.; Glatzel, M.; Seifert, V.; Kogel, D. Dietary curcumin attenuates glioma growth in a syngeneic mouse model by inhibition of the JAK1,2/STAT3 signaling pathway. Clin. Cancer Res. 2010, 16, 5781–5795. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Zhang, B.; Duan, D.; Wu, J.; Fang, J. Curcumin targeting the thioredoxin system elevates oxidative stress in HeLa cells. Toxicol. Appl. Pharmacol. 2012, 262, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Xu, B.; Beevers, C.S.; Odaka, Y.; Chen, L.; Liu, L.; Luo, Y.; Zhou, H.; Chen, W.; Shen, T.; et al. Curcumin inhibits protein phosphatases 2A and 5, leading to activation of mitogen-activated protein kinases and death in tumor cells. Carcinogenesis 2012, 33, 868–875. [Google Scholar] [CrossRef]
- Bose, S.; Panda, A.K.; Mukherjee, S.; Sa, G. Curcumin and tumor immune-editing: Resurrecting the immune system. Cell Div. 2015, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Varalakshmi, C.; Ali, A.M.; Pardhasaradhi, B.V.; Srivastava, R.M.; Singh, S.; Khar, A. Immunomodulatory effects of curcumin: In-vivo. Int. Immunopharmacol. 2008, 8, 688–700. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Mandal, D.; Saha, B.; Sen, G.S.; Das, T.; Sa, G. Curcumin prevents tumor-induced T cell apoptosis through Stat-5a-mediated Bcl-2 induction. J. Biol. Chem. 2007, 282, 15954–15964. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Mandal, D.; Sen, G.S.; Pal, S.; Banerjee, S.; Lahiry, L.; Finke, J.H.; Tannenbaum, C.S.; Das, T.; Sa, G. Tumor-induced oxidative stress perturbs nuclear factor-kappaB activity-augmenting tumor necrosis factor-alpha-mediated T-cell death: Protection by curcumin. Cancer Res. 2007, 67, 362–370. [Google Scholar] [CrossRef]
- Pratheeshkumar, P.; Sreekala, C.; Zhang, Z.; Budhraja, A.; Ding, S.; Son, Y.O.; Wang, X.; Hitron, A.; Hyun-Jung, K.; Wang, L.; et al. Cancer prevention with promising natural products: Mechanisms of action and molecular targets. Anticancer Agents Med. Chem. 2012, 12, 1159–1184. [Google Scholar] [CrossRef] [PubMed]
- Afshari, H.; Noori, S.; Nourbakhsh, M.; Daraei, A.; Azami Movahed, M.; Zarghi, A. A novel imidazo[1,2-a]pyridine derivative and its co-administration with curcumin exert anti-inflammatory effects by modulating the STAT3/NF-kappaB/iNOS/COX-2 signaling pathway in breast and ovarian cancer cell lines. Bioimpacts 2024, 14, 27618. [Google Scholar] [CrossRef]
- Cheng, A.L.; Hsu, C.H.; Lin, J.K.; Hsu, M.M.; Ho, Y.F.; Shen, T.S.; Ko, J.Y.; Lin, J.T.; Lin, B.R.; Ming-Shiang, W.; et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001, 21, 2895–2900. [Google Scholar]
- Khosravi, M.A.; Seifert, R. Clinical trials on curcumin in relation to its bioavailability and effect on malignant diseases: Critical analysis. Naunyn Schmiedebergs Arch. Pharmacol. 2024, 397, 3477–3491. [Google Scholar] [CrossRef]
- Wong, S.C.; Kamarudin, M.N.A.; Naidu, R. Anticancer Mechanism of Curcumin on Human Glioblastoma. Nutrients 2021, 13, 950. [Google Scholar] [CrossRef] [PubMed]
- Panahi, Y.; Saadat, A.; Beiraghdar, F.; Sahebkar, A. Adjuvant therapy with bioavailability-boosted curcuminoids suppresses systemic inflammation and improves quality of life in patients with solid tumors: A randomized double-blind placebo-controlled trial. Phytother. Res. 2014, 28, 1461–1467. [Google Scholar] [CrossRef]
- Basak, S.K.; Bera, A.; Yoon, A.J.; Morselli, M.; Jeong, C.; Tosevska, A.; Dong, T.S.; Eklund, M.; Russ, E.; Nasser, H.; et al. A randomized, phase 1, placebo-controlled trial of APG-157 in oral cancer demonstrates systemic absorption and an inhibitory effect on cytokines and tumor-associated microbes. Cancer 2020, 126, 1668–1682. [Google Scholar] [CrossRef]
- Man, S.; Zhang, L.; Cui, J.; Yang, L.; Ma, L.; Gao, W. Curcumin enhances the anti-cancer effects of Paris Saponin II in lung cancer cells. Cell Prolif. 2018, 51, e12458. [Google Scholar] [CrossRef]
- Zou, J.; Zhu, L.; Jiang, X.; Wang, Y.; Wang, Y.; Wang, X.; Chen, B. Curcumin increases breast cancer cell sensitivity to cisplatin by decreasing FEN1 expression. Oncotarget 2018, 9, 11268–11278. [Google Scholar] [CrossRef]
- Bojko, A.; Cierniak, A.; Adamczyk, A.; Ligeza, J. Modulatory Effects of Curcumin and Tyrphostins (AG494 and AG1478) on Growth Regulation and Viability of LN229 Human Brain Cancer Cells. Nutr. Cancer 2015, 67, 1170–1182. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Xiong, X.; Lu, M.; Huang, S.; Luo, Y.; Wang, Y.; Ying, Y. Curcumin suppresses colorectal tumorigenesis through restoring the gut microbiota and metabolites. BMC Cancer 2024, 24, 1141. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Niu, S.; Wang, H.; Li, J. Microbiome Modulation with Lactobacillus rhamnosus GG Potentiates Curcumin’s Efficacy in Reversing Gemcitabine Resistance of Gallbladder Cancer through Gut Microbiota-PI3K/AKT Axis. J. Microbiol. Biotechnol. 2026, 36, e2601007. [Google Scholar] [CrossRef]
- Mahmud, M.; Piwoni, A.; Filipczak, N.; Janicka, M.; Gubernator, J. Long-Circulating Curcumin-Loaded Liposome Formulations with High Incorporation Efficiency, Stability and Anticancer Activity towards Pancreatic Adenocarcinoma Cell Lines In Vitro. PLoS ONE 2016, 11, e0167787. [Google Scholar] [CrossRef]
- Qasim Almajidi, Y.; Jawad, A.Q.; Abdulameer Albadri, A. Biocompatible PAMAM-PLGA-PCL Nanocarrier for Efficient Curcumin Delivery to Lung Cancer Cells: In Vitro Studies. Chem. Biodivers. 2024, 21, e202401106. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, P.; Hou, X.; Yan, F.; Jiang, Z.; Shi, J.; Xie, X.; Shen, J.; Fan, Q.; Wang, Z.; et al. Hybrid curcumin-phospholipid complex-near-infrared dye oral drug delivery system to inhibit lung metastasis of breast cancer. Int. J. Nanomed. 2019, 14, 3311–3330. [Google Scholar] [CrossRef]
- Atlan, M.; Neman, J. Targeted Transdermal Delivery of Curcumin for Breast Cancer Prevention. Int. J. Environ. Res. Public. Health 2019, 16, 4949. [Google Scholar] [CrossRef] [PubMed]
- Busquets, S.; Carbo, N.; Almendro, V.; Quiles, M.T.; Lopez-Soriano, F.J.; Argiles, J.M. Curcumin, a natural product present in turmeric, decreases tumor growth but does not behave as an anticachectic compound in a rat model. Cancer Lett. 2001, 167, 33–38. [Google Scholar] [CrossRef]
- Heidari, H.; Bagherniya, M.; Majeed, M.; Sathyapalan, T.; Jamialahmadi, T.; Sahebkar, A. Curcumin-piperine co-supplementation and human health: A comprehensive review of preclinical and clinical studies. Phytother. Res. 2023, 37, 1462–1487. [Google Scholar] [CrossRef]
- Shoba, G.; Joy, D.; Joseph, T.; Majeed, M.; Rajendran, R.; Srinivas, P.S. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998, 64, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Boroughani, M.; Moaveni, A.K.; Hatami, P.; Mansoob Abasi, N.; Seyedoshohadaei, S.A.; Pooladi, A.; Moradi, Y.; Rahimi Darehbagh, R. Nanocurcumin in cancer treatment: A comprehensive systematic review. Discov. Oncol. 2024, 15, 515. [Google Scholar] [CrossRef] [PubMed]
- Janjua, K.A.; Shehzad, A.; Shahzad, R.; Islam, S.U.; Islam, M.U. Nanocurcumin: A Double-Edged Sword for Microcancers. Curr. Pharm. Des. 2020, 26, 5783–5792. [Google Scholar] [CrossRef]
- Karthikeyan, A.; Senthil, N.; Min, T. Nanocurcumin: A Promising Candidate for Therapeutic Applications. Front. Pharmacol. 2020, 11, 487. [Google Scholar] [CrossRef]
- Gagliardi, S.; Morasso, C.; Stivaktakis, P.; Pandini, C.; Tinelli, V.; Tsatsakis, A.; Prosperi, D.; Hickey, M.; Corsi, F.; Cereda, C. Curcumin Formulations and Trials: What’s New in Neurological Diseases. Molecules 2020, 25, 5389. [Google Scholar] [CrossRef]
- Maiti, K.; Mukherjee, K.; Gantait, A.; Saha, B.P.; Mukherjee, P.K. Curcumin-phospholipid complex: Preparation, therapeutic evaluation and pharmacokinetic study in rats. Int. J. Pharm. 2007, 330, 155–163. [Google Scholar] [CrossRef]
- Jiao, Y.; Wilkinson, J.t.; Di, X.; Wang, W.; Hatcher, H.; Kock, N.D.; D’Agostino, R., Jr.; Knovich, M.A.; Torti, F.M.; Torti, S.V. Curcumin, a cancer chemopreventive and chemotherapeutic agent, is a biologically active iron chelator. Blood 2009, 113, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Pavithra, B.H.; Prakash, N.; Jayakumar, K. Modification of pharmacokinetics of norfloxacin following oral administration of curcumin in rabbits. J. Vet. Sci. 2009, 10, 293–297. [Google Scholar] [CrossRef]
- Liu, A.C.; Zhao, L.X.; Lou, H.X. Curcumin alters the pharmacokinetics of warfarin and clopidogrel in Wistar rats but has no effect on anticoagulation or antiplatelet aggregation. Planta Med. 2013, 79, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Lamb, S.R.; Wilkinson, S.M. Contact allergy to tetrahydrocurcumin. Contact Dermat. 2003, 48, 227. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Curcumin Formulation | Cancer Cell Line(s) | Study (Year) | Key Findings |
|---|---|---|---|
| Free curcumin | SK-Hep-1 and Huh-7 (liver) | Lin et al., 1998 [89] | Inhibited invasion and migration of HCC cells via MMP-9 suppression |
| Free curcumin | HepG2 (liver) | Cao et al., 2007 [90] | Induced apoptosis through a mitochondrial pathway; promoted cytochrome c release |
| Free curcumin | MCF-7 and MDA-MB-231 (breast) | Goksu Guneydas and Topcul, 2022 [84] | Dose-dependent anti-proliferative effects in luminal A and triple-negative breast cancer cells |
| Free curcumin | Panc-1 and MIAPaCa-2 (pancreatic) | Schwarz et al., 2020 [92] | Radiosensitized pancreatic cancer cells; enhanced radiation-induced apoptosis and G2/M arrest |
| Curcumin + piperine (nanoparticles) | HCT-116 (colon) | Puri and Arora, 2025 [12] | Synergistic anticancer effect; enhanced cellular uptake; IC50 lower than free curcumin |
| Liposomal curcumin | Pancreatic carcinoma (in vivo) | Li et al., 2005 [99] | Activity comparable to or greater than free curcumin; inhibited tumor growth and suppressed angiogenesis in murine xenografts |
| Liposomal curcumin | AsPC-1 and BxPC-3 (pancreatic) | Mahmud et al., 2016 [120] | Potent anticancer activity on pancreatic adenocarcinoma cells; less toxic to normal cells than free curcumin |
| Nanocurcumin (PLGA) | A549 (lung) | Almajidi et al., 2024 [121] | IC50 of 50 nM at 24 h; 6–8-fold upregulation of apoptotic genes (caspase-9 and Bax); no toxicity in normal cells |
| Nanocurcumin (PLGA) | HCT-116 (colon) | Waghela et al., 2015 [98] | Inhibited colony formation and cell migration; enhanced anti-cancer activity vs. free curcumin |
| Nanocurcumin (PLGA) | MDA-MB-231 (breast) and A549 (lung) | Khan et al., 2018 [85] | Reduced HIF-1a and nuclear p65 expression; enhanced anti-cancer activity under hypoxic conditions |
| Curcumin-phospholipid complex | HC11 and BME-UV (mammary epithelial) | Cucuzza et al., 2008 [86] | Induced apoptosis in mammary epithelial cells via the STAT-3 signaling pathway |
| CUR/IR780@SMEDDS | 4T1 (breast and metastatic) | Liu et al., 2019 [122] | Enhanced oral bioavailability; inhibited lung metastasis of breast cancer in vivo |
| [DLys6]-LHRH-Curcumin | MIAPaCa-2, Panc-1, and BxPC-3 (pancreatic) | Aggarwal et al., 2011 [100] | Water-soluble conjugate selectively targeted and inhibited pancreatic cancer cell growth; enabled i.v. administration |
| Curcumin-infused bio-textiles | Transdermal delivery (proof of concept) | Atlan et al., 2019 [123] | Delivered targeted transdermal curcumin therapy through simple skin contact |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
LeBlanc, K.; Brewer, E.; Aggarwal, S. Curcumin and Cancer-Related Inflammation. Nutrients 2026, 18, 1636. https://doi.org/10.3390/nu18101636
LeBlanc K, Brewer E, Aggarwal S. Curcumin and Cancer-Related Inflammation. Nutrients. 2026; 18(10):1636. https://doi.org/10.3390/nu18101636
Chicago/Turabian StyleLeBlanc, Kaitlyn, Emilee Brewer, and Sita Aggarwal. 2026. "Curcumin and Cancer-Related Inflammation" Nutrients 18, no. 10: 1636. https://doi.org/10.3390/nu18101636
APA StyleLeBlanc, K., Brewer, E., & Aggarwal, S. (2026). Curcumin and Cancer-Related Inflammation. Nutrients, 18(10), 1636. https://doi.org/10.3390/nu18101636