
Mechanisms of Fetal Overgrowth in Gestational Diabetes: The Potential Role of SOCS2

 , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
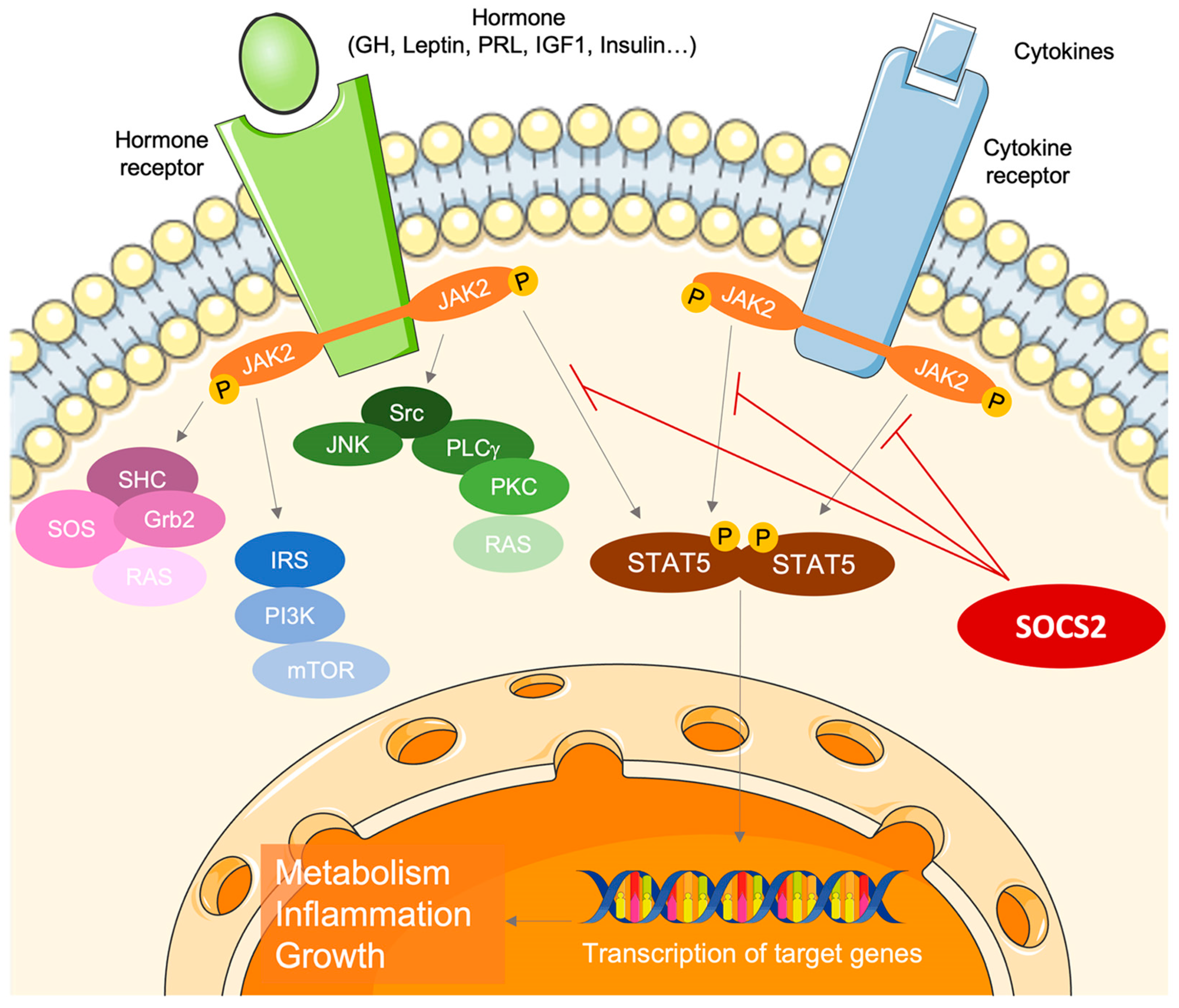
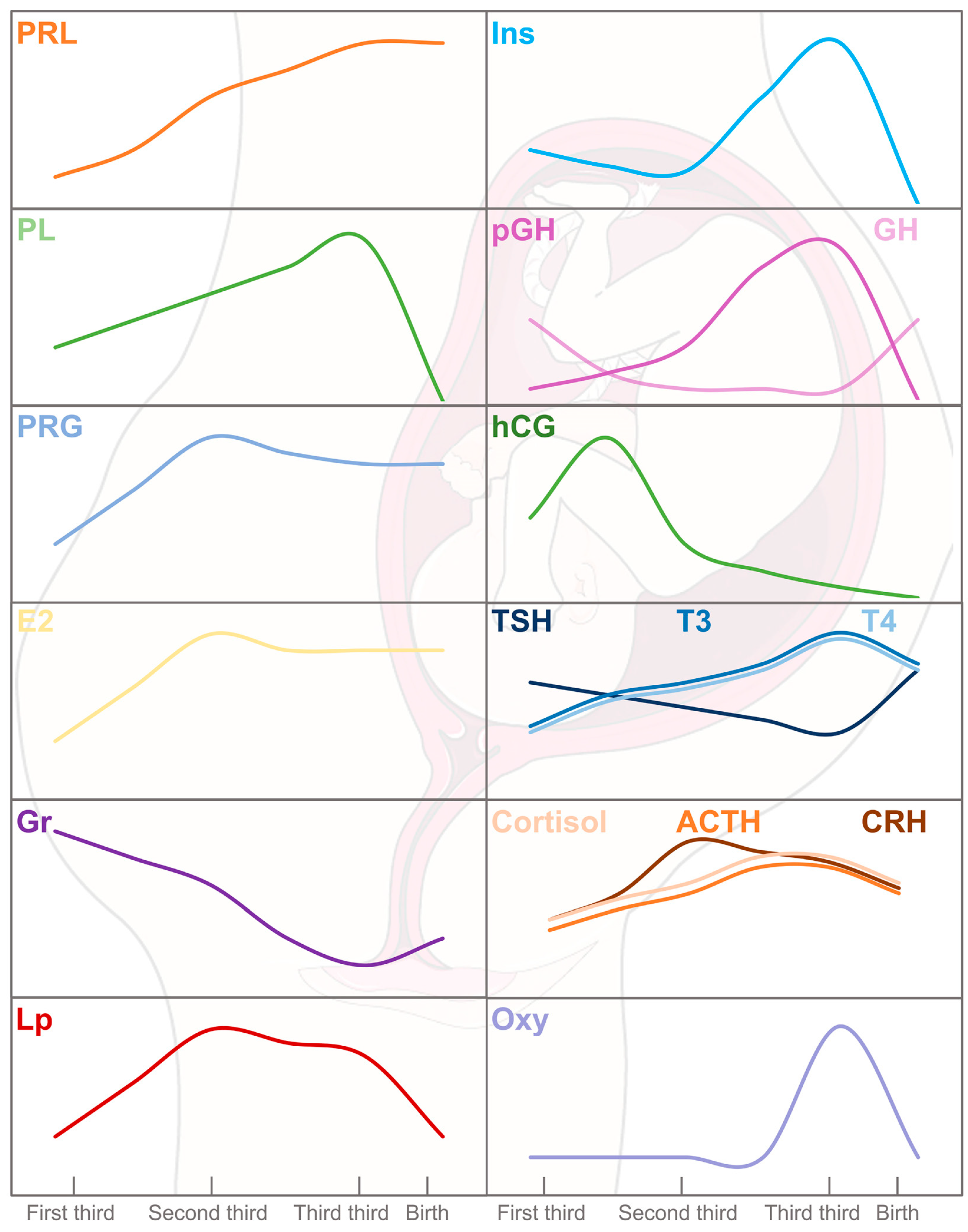
2. Endocrine–Metabolic Sketch of Pregnancy
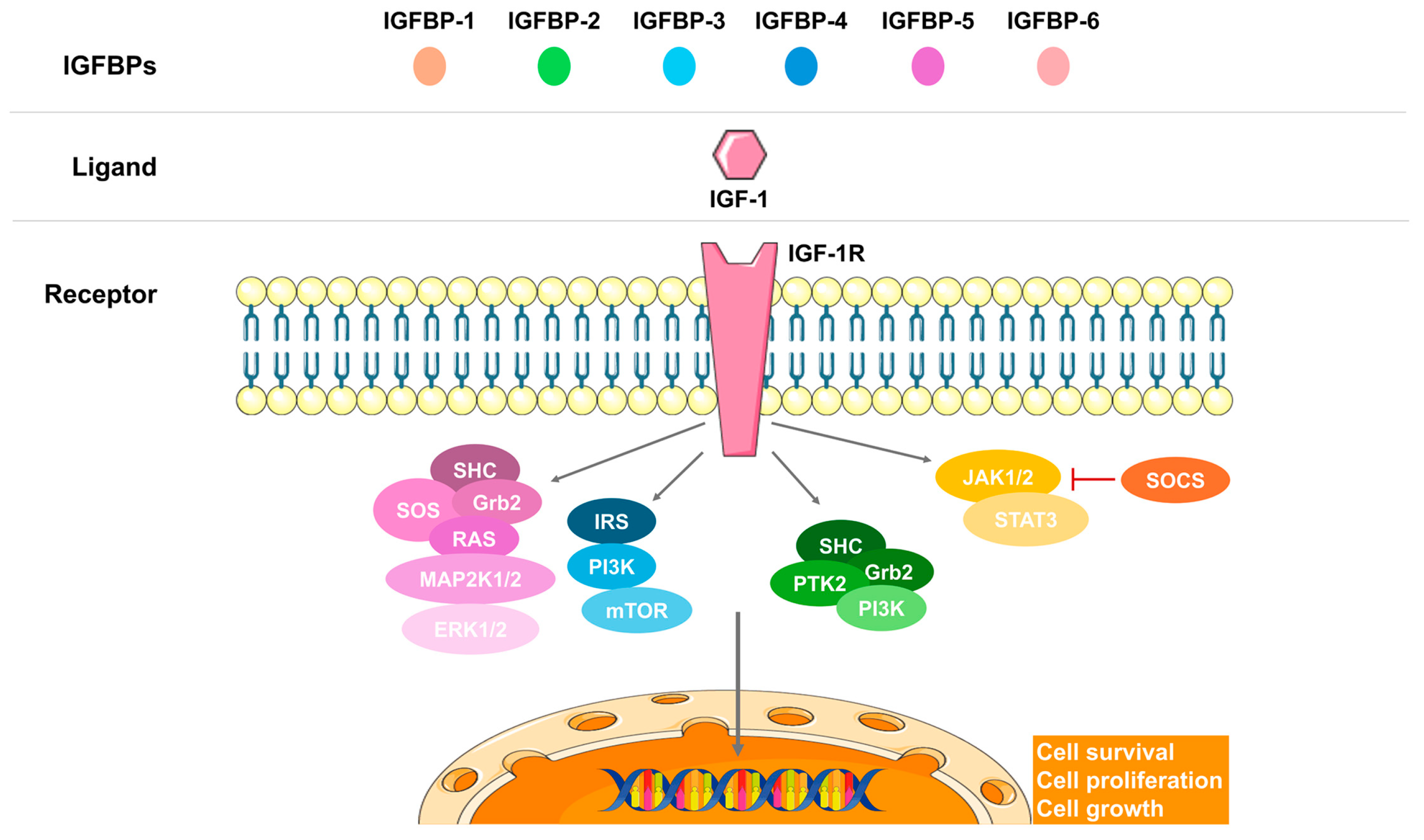
3. Embryonic and Fetal Growth
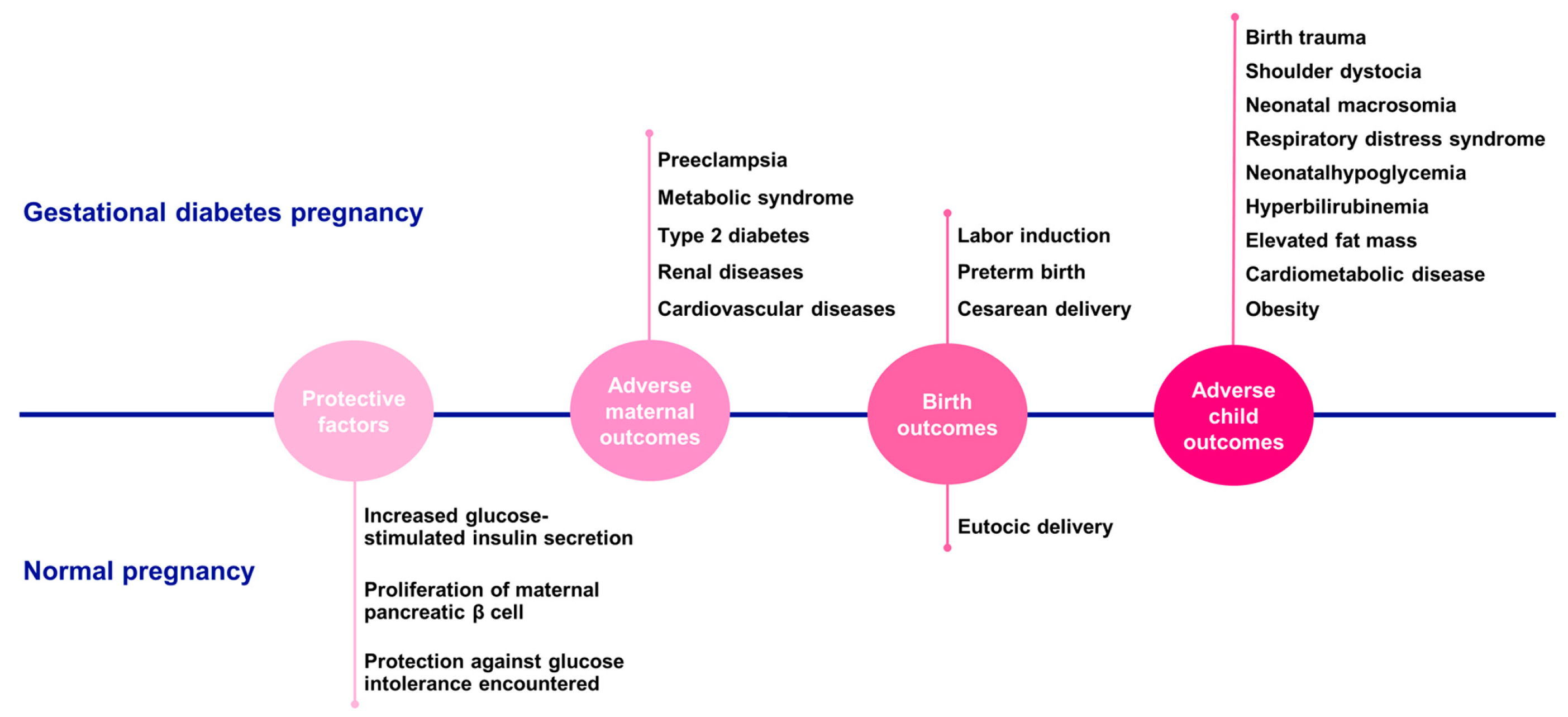
4. Gestational Diabetes
5. Macrosomia
6. Biomarkers of Gestational Diabetes and Macrosomia
6.1. Gestational Diabetes Mellitus Biomarkers
- a.
- Maternal serum biomarkers
- b.
- Placental and cord blood biomarkers
6.2. Macrosomia Biomarkers
- a.
- Maternal serum biomarkers
- b.
- Placental and cord blood biomarkers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarker | Origin | Up (↑)/Down (↓) Regulation | Role/Function | Type of Study | Evidence |
|---|---|---|---|---|---|
| 2-Hydroxybutyric ## 3-Hydroxybutyric ## | Maternal | ↑ | Ketone body derived from fatty acid oxidation | Metabolic plasma analysis (maternal blood samples) | [57,104] |
| Adiponectin #& | Maternal | ↓ | Stimulation of FAO, enhancement glucose metabolism and reduction in insulin resistance throughout | Primary cell culture of human placentas/Human placenta and maternal blood samples | [22,82] |
| AMPK && | Placental | ↓ | Activation of cellular metabolism | Human placenta and maternal blood samples | [44] |
| ANGPL-3-4-8 #& | Placental | ↑ | Increased transplacental nutrition and lipid and glucose metabolism | Maternal and cord blood and human placenta samples | [122] |
| ARHGEF11 ## | Placental | ↑ | Activation of G protein signaling and cellular processes such as insulin secretion, insulin signaling, and lipid metabolism | Human placenta samples | [38] |
| BCAA ## Valine, Isoleucine, Leucine | Maternal | ↑ | Essential amino acids that cannot be produced by the body and must be primarily obtained from the diet. Related with GDM. | Metabolic plasma analysis (maternal blood samples) | [57] |
| C-peptide #& | Maternal | ↑ | Marker of endogenous insulin secretion. Derived from the cleavage of proinsulin in insulin. | Maternal and cord blood samples | [86] |
| E2 ## | Maternal/Cord blood | ↓ | Sex female steroid that maintains β-cell survival and glucose homeostasis | Clinical, anthropometrical data and maternal blood samples/Cord blood samples | [71,108] |
| EL | Placental | ↑ | Mediation of the uptake of fatty acids from high-density lipoproteins | Human placenta samples | [68] |
| ER-α $$ | Placental | ↑ | Enhancement glucose uptake capacity | Human placenta samples | [75] |
| FAO #& | Placental | ↓ | Breakdown of fatty acids into CoA for use as an energy resource | Human placenta and maternal blood samples | [85,95] |
| FFA #& | Cord Blood/Maternal | ↑/= | Source of energy for the body | Maternal blood samples/Maternal and cord blood samples | [97,98] |
| FGF21 #& | Maternal | ↑ | Enhancement carbohydrate and lipid metabolism | Blood samples/Meta-analysis | [101,102] |
| GHR $$ | Placental | ↑ | Regulation of GH function in growth, metabolism, and other physiological functions | In vitro analysis with Sprague Dawley rats embryo samples | [47] |
| Gluconeogenic AA ## (Glutamine and Glutamate) | Maternal | ↑ | Aminoacids related to glucose metabolism | Metabolomics (maternal blood and urine samples) | [57,103] |
| GLUT4 ## | Placental | ↓ | Insulin-regulated glucose uptake transporter | Human placenta samples | [38] |
| I-CAM-1 && | Maternal | ↑ | Cell adhesion molecule related with a proinflammatory state | Prospective case–control study | [58] |
| IGF-1 #$ | Placental/ Maternal | ↑/↓ | Increased fetal growth | Human placenta samples/Human placenta and maternal blood samples | [38,44,46] |
| IGFBP-1 #$ | Placental | ↓ | Modulation of biological functions of IGFs | Human placenta | [116] |
| Insulin ## | Maternal | ↑ | Reduction in blood glucose levels and induction of glucose storage in the liver | In vivo study with rat pancreas | [61] |
| IR ## | Placental | ↑ | Promotion of cellular metabolism | Human placenta and maternal blood samples | [44] |
| Irisin ## | Maternal | ↑ | Transformation of white adipose tissue to brown adipose tissue, inducing energy expenditure | Maternal blood, umbilical cord and placenta samples | [127] |
| IRS-1 (ph) ## | Placental | ↑ | Docking protein for insulin signal transmission | Human placenta samples | [75] |
| Kisspeptin $$ | Maternal | ↓ | Neuroactive hormone that adapts maternal physiology to pregnancy | Maternal and cord blood and human placenta samples/Review | [107,110] |
| Leptin #& | Maternal | ↑ | Suppression of appetite and control of the energy expenditure in white adipose tissue. In pregnancy, maintains increased energy intake for the fetal growth | Maternal blood samples/Human placenta and maternal blood samples | [22,121] |
| miR-21 $$ | ↑ | micro-RNA in JAK/STAT pathway linked to macrosomic babies. | Human placenta samples/Maternal blood samples/Review | [53,124,125] | |
| miR-675 $$ | Maternal | micro-RNA increases placental growth | In vitro analysis (cell line culture) and in vivo analysis (C57/BL6 and H19Δ3 transgenic line)/Review | [53,126] | |
| NEFA #& | Maternal | ↑ | Source of energy for skeletal muscle | Primary cell culture of human placentas | [95] |
| p-S307 ## | Placental | ↓ | Phosphorylated Ser307 of IRS-1, blocks insulin action | Human placenta samples | [38] |
| p-Y612 ## | Placental | ↓ | Phosphorylated site of IRS-1 intermediates with PI3K activity | Human placenta samples | [38] |
| p58a $$ | Placental | ↑ | An ER molecular chaperone | Human placenta samples | [75] |
| pAKT ## | Placental | ↓/↑ | Phosphorylation and regulation of multiple intracellular signaling pathways (metabolism, apoptosis, and proliferation) | Human placenta samples | [38] |
| pGH $$ | Maternal | ↑ | Activation of maternal and fetal metabolism and growth | Maternal blood samples/Review | [54,115] |
| PI3K ## | Placental | ↓ | Activation of multiple intracellular signaling pathways (growth, proliferation, migration, secretion, differentiation, transcription, and translation) | Human placenta samples | [38] |
| Proinflamatory Cytokines && (TNF-a, IL-6, IL-1, MCP-1) | Maternal | ↑ | Activation of multiple cellular-mediated responses | A prospective case–control study | [58] |
| Resistin ## | Placental | ↑ | Inflammation and impaired glucose tolerance | Human placenta and maternal blood samples | [22] |
| SNAT1 #$ | Placental | ↑ | Amino acid transporter in the placenta | Human placenta and maternal blood samples | [44] |
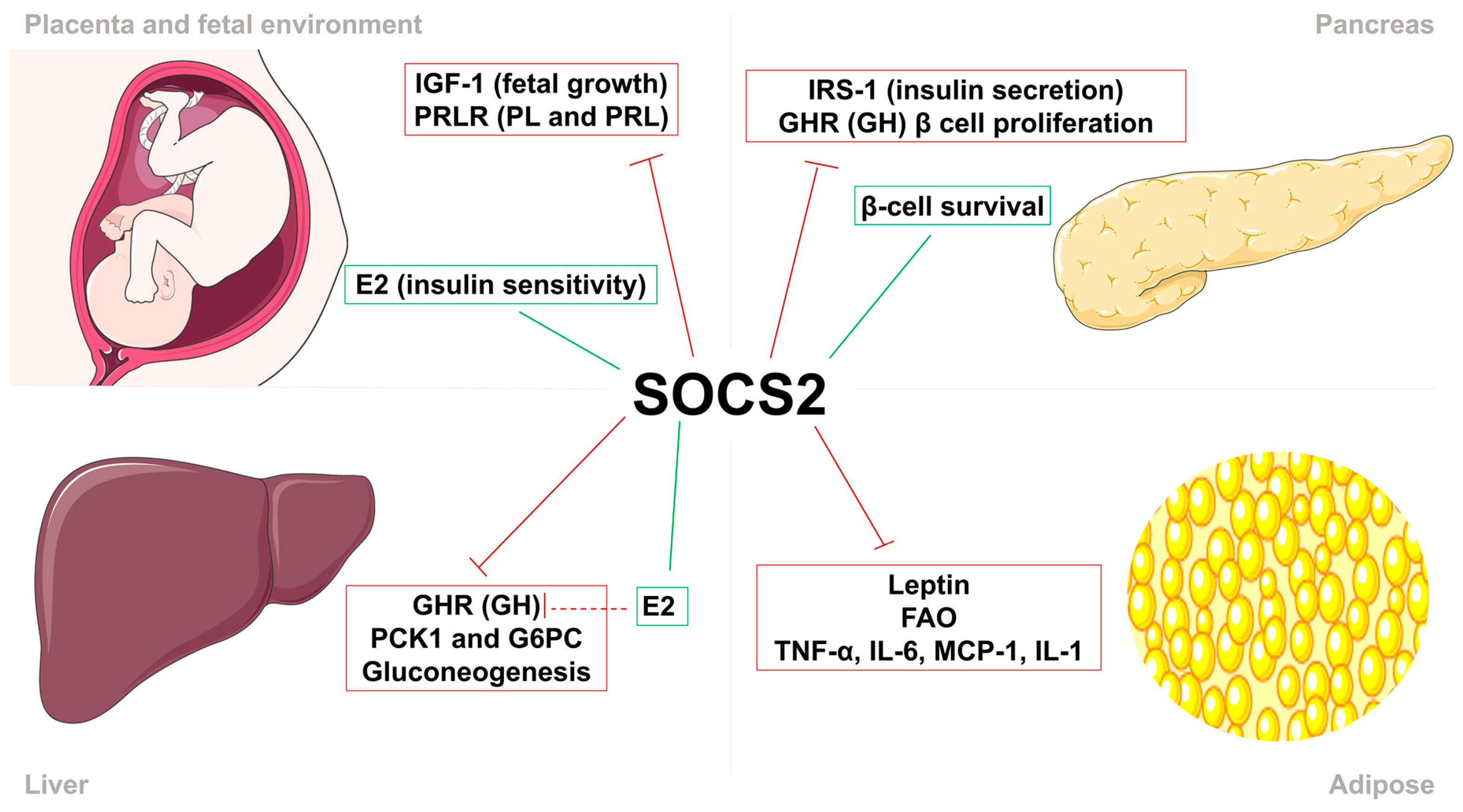
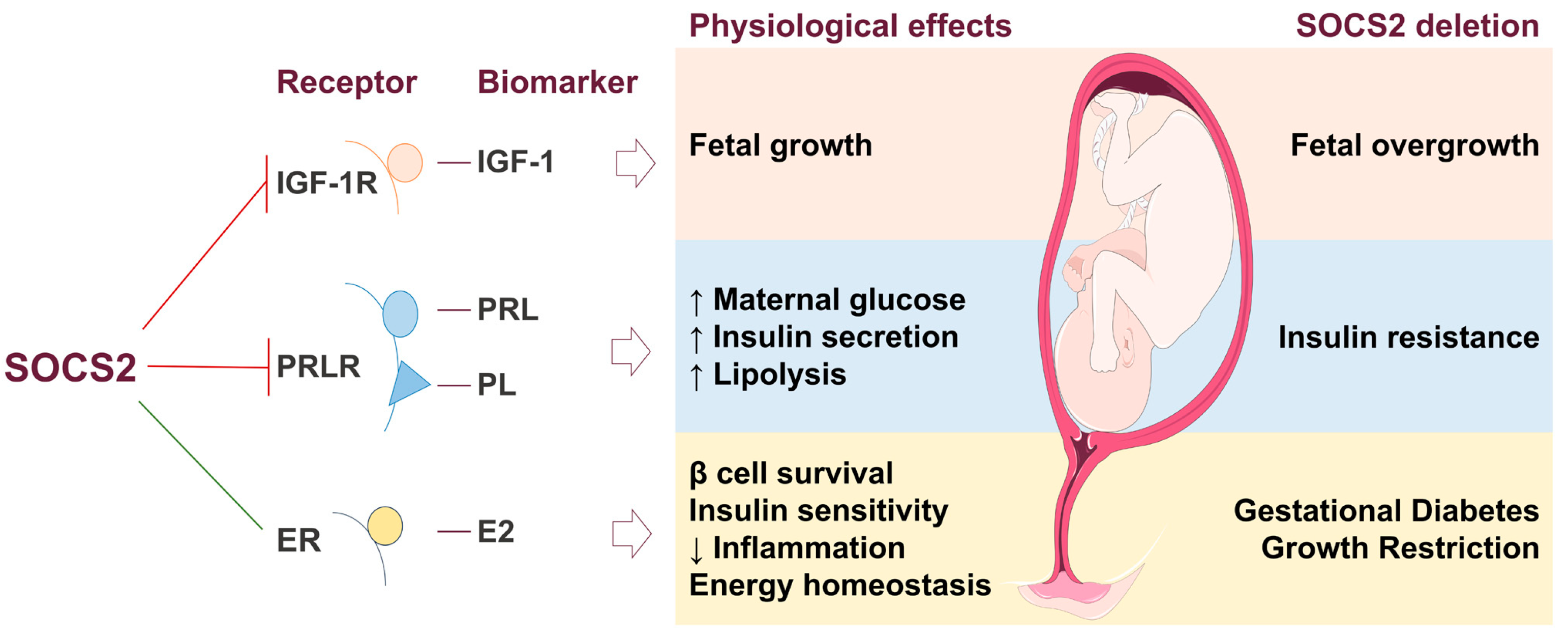
| SOCS2 #$& | Maternal | ↓ | Inhibition of GH and IGF-1 signaling, thus regulates body growth and development, immune response, lipid and glucose metabolism, and β-cell physiology | Human placenta samples/In vivo evaluation of Socs2−/− mice | [35,128,129] |
| Succinic acid ## | Maternal | ↑ | Intermediate product of the Krebs cycle. To produce energy in Krebs cycle | Metabolic plasma analysis (maternal blood samples) | [57,104] |
| TG #& | Placental/Cord Blood/Maternal | ↑/= | Source of energy for the body | Human placenta and maternal blood samples/Maternal and cord blood samples | [85,86,97] |
| V-CAM-1 && | Maternal | ↑ | Cell adhesion molecule related to a proinflammatory state | A prospective case–control study | [58] |
7. SOCS2 Mechanism in Gestational Diabetes and Macrosomia
8. Limitations, Directions for Future Research, and General Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTH | Adrenocorticotropic hormone |
| ADA | American Diabetes Association |
| AKT | Protein kinase B |
| AMPK | AMP-activated protein kinase |
| ARHGEF11 | Rho guanine nucleotide exchange factor 11 |
| ANGPTL | Angiopoietin-like protein |
| BCAAs | Branched-chain amino acids |
| BMI | Body mass index |
| CBG | Corticosteroid-binding globulin |
| CISH | Cytokine-inducible SH2 protein |
| CRH | Corticotropin |
| DOC | Deoxycorticosterone |
| E2 | Estrogen Estradiol |
| EL | Endothelial lipase |
| ER | Estrogen receptor |
| ER-stress | Endoplasmic reticulum stress |
| FAO | Fatty acid oxidation |
| FDA | United States Food and Drug Administration |
| FFA | Free fatty acids |
| G6PC | Glucose-6-phosphatase catalytic subunit |
| GDM | Gestational diabetes mellitus |
| GH | Growth hormone |
| GHR | Growth hormone receptor |
| GLUT | Glucose transporter |
| HbAc1 | Glycated hemoglobin |
| HFD | High fat diet |
| HOMA-IR | Homeostasis model assessment-estimated insulin resistance |
| HPA | Hypothalamus–pituitary–adrenal |
| ICAM | Intracellular adhesion molecule |
| IGF | Insulin growth factor |
| IGFR | Insulin growth factor receptor |
| IL | Interleukin |
| IR | Insulin receptor |
| IRS | Insulin receptor substrate |
| IUGR | Intrauterine growth restriction |
| JAK | Janus kinase |
| LGA | Large for gestational age |
| MCP | Monocyte chemoattractant protein |
| mTOR | Mammalian target of rapamycin |
| NEFA | Nonesterified fatty acids |
| NF-kB | Nuclear factor kappa light chain enhancer of activated B cells |
| PCK1 | Phosphoenolpyruvate carboxykinase 1 |
| PI3K | Phosphatidyllinositol-3 kinase |
| p58α | Phosphatidyllinositol-3 kinase subunit |
| PL | Placental lactogen |
| PRL | Prolactin |
| PRLR | Prolactin receptor |
| SNAT1 | Sodium-coupled neutral amino acid transporter |
| SNPs | Single nucleotide polymorphisms SNPs |
| SOCS | Suppressor of cytokine signaling |
| STAT | Signal transducers and activator or transcription |
| T2D | Type 2 diabetes |
| T3 | Triiodothyronine |
| T4 | Thyroxine |
| TBG | Thyroxine-binding globulin |
| TNF-α | Tumor necrosis factor α |
| TSH | Thyroid-stimulating hormone |
| VCAM | Vascular cell adhesion molecule |
References
- Letellier, E.; Haan, S. SOCS2: Physiological and Pathological Functions. Front. Biosci.—Elite 2016, 8, 189–204. [Google Scholar] [CrossRef]
- Dodington, D.W.; Desai, H.R.; Woo, M. JAK/STAT—Emerging Players in Metabolism. Trends Endocrinol. Metab. 2018, 29, 55–65. [Google Scholar] [CrossRef]
- Rico-Bautista, E.; Flores-Morales, A.; Fernández-Pérez, L. Suppressor of Cytokine Signaling (SOCS) 2, a Protein with Multiple Functions. Cytokine Growth Factor Rev. 2006, 17, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef]
- Wang, X.; Athayde, N.; Trudinger, B. A Proinflammatory Cytokine Response Is Present in the Fetal Placental Vasculature in Placental Insufficiency. Am. J. Obstet. Gynecol. 2003, 189, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Nomura, K.; Osabe, D.; Shinohara, S.; Mizumori, O.; Katashima, R.; Iwasaki, S.; Nishimura, K.; Yoshino, M.; Kobori, M.; et al. Association of Single-Nucleotide Polymorphisms in the Suppressor of Cytokine Signaling 2 (SOCS2) Gene with Type 2 Diabetes in the Japanese. Genomics 2006, 87, 446–458. [Google Scholar] [CrossRef]
- Metcalf, D.; Greenhalgh, C.J.; Viney, E.; Wilison, T.A.; Starr, R.; Nicola, N.A.; Hilton, D.J.; Alexander, W.S. Gigantism in Mice Lacking Suppressor of Cytokine Signalling-2. Nature 2000, 405, 1069–1073. [Google Scholar] [CrossRef]
- Pan, J.; Tong, R.; Deng, Q.; Tian, Y.; Wang, N.; Peng, Y.; Fei, S.; Zhang, W.; Cui, J.; Guo, C.; et al. The Effect of SOCS2 Polymorphisms on Type 2 Diabetes Mellitus Susceptibility and Diabetic Complications in the Chinese Han Population. Pharmgenomics Pers. Med. 2022, 15, 65–79. [Google Scholar] [CrossRef]
- Metzger, B.E.; Buchanan, T.A. Gestational Diabetes. In Diabetes in America; Cowie, C., Casagrande, S., Menke, A., Cissell, M., Eberhardt, M., Meigs, J., Gregg, E., Knowler, W., Barrett-Connor, E., Becker, D., et al., Eds.; National Institute of Diabetes and Digestive and Kidney Diseases (US): Bethesda, MD, USA, 2018. [Google Scholar]
- American Diabetes Association. Gestational Diabetes Mellitus. Diabetes Care 2003, 26, s103–s105. [Google Scholar] [CrossRef]
- McIntyre, H.D.; Catalano, P.; Zhang, C.; Desoye, G.; Mathiesen, E.R.; Damm, P. Gestational Diabetes Mellitus. Nat. Rev. Dis. Primers 2019, 5, 47. [Google Scholar] [CrossRef]
- Dumolt, J.H.; Powell, T.L.; Jansson, T. Placental Function and the Development of Fetal Overgrowth and Fetal Growth Restriction. Obstet. Gynecol. Clin. N. Am. 2021, 48, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Armistead, B.; Johnson, E.; Vanderkamp, R.; Kula-Eversole, E.; Kadam, L.; Drewlo, S.; Kohan-Ghadr, H.-R. Placental Regulation of Energy Homeostasis During Human Pregnancy. Endocrinology 2020, 161, bqaa076. [Google Scholar] [CrossRef] [PubMed]
- Di Cianni, G.; Miccoli, R.; Volpe, L.; Lencioni, C.; Del Prato, S. Intermediate Metabolism in Normal Pregnancy and in Gestational Diabetes. Diabetes Metab. Res. Rev. 2003, 19, 259–270. [Google Scholar] [CrossRef]
- Gicquel, C.; Le Bouc, Y. Hormonal Regulation of Fetal Growth. Horm. Res. Paediatr. 2006, 65, 28–33. [Google Scholar] [CrossRef]
- Jiao, Y.; Rieck, S.; Le Lay, J.; Kaestner, K.H. CISH Has No Non-Redundant Functions in Glucose Homeostasis or Beta Cell Proliferation during Pregnancy in Mice. Diabetologia 2013, 56, 2435–2445. [Google Scholar] [CrossRef] [PubMed]
- Haddad-Tóvolli, R.; Claret, M. Metabolic and Feeding Adjustments during Pregnancy. Nat. Rev. Endocrinol. 2023, 19, 564–580. [Google Scholar] [CrossRef]
- Tal, R.; Taylor, H.S. Endocrinology of Pregnancy. In Endotext [Internet]; Feingold, K.R., Ahmed, S.F., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2021; p. 476. ISBN 9780128151457. [Google Scholar]
- Anderson, S.T.; Isa, N.N.M.; Barclay, J.L.; Waters, M.J.; Curlewis, J.D. Maximal Expression of Suppressors of Cytokine Signaling in the Rat Ovary Occurs in Late Pregnancy. Reproduction 2009, 138, 537–544. [Google Scholar] [CrossRef]
- Sutherland, K.D.; Lindeman, G.J.; Visvader, J.E. Knocking off SOCS Genes in the Mammary Gland. Cell Cycle 2007, 6, 799–803. [Google Scholar] [CrossRef]
- Hufnagel, A.; Dearden, L.; Fernandez-Twinn, D.S.; Ozanne, S.E. Programming of Cardiometabolic Health: The Role of Maternal and Fetal Hyperinsulinaemia. J. Endocrinol. 2022, 253, R47–R63. [Google Scholar] [CrossRef]
- Lappas, M.; Yee, K.; Permezel, M.; Rice, G.E. Release and Regulation of Leptin, Resistin and Adiponectin from Human Placenta, Fetal Membranes, and Maternal Adipose Tissue and Skeletal Muscle from Normal and Gestational Diabetes Mellitus-Complicated Pregnancies. J. Endocrinol. 2005, 186, 457–465. [Google Scholar] [CrossRef]
- Masuzaki, H.; Ogawa, Y.; Sagawa, N.; Hosoda, K.; Matsumoto, T.; Mise, H.; Nishimura, H.; Yoshimasa, Y.; Tanaka, I.; Mori, T.; et al. Nonadipose Tissue Production of Leptin: Leptin as a Novel Placenta-Derived Hormone in Humans. Nat. Med. 1997, 3, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Zeck, W.; Widberg, C.; Maylin, E.; Desoye, G.; Lang, U.; Mcintyre, D.; Prins, J.; Russell, A. Regulation of Placental Growth Hormone Secretion in a Human Trophoblast Model—The Effects of Hormones and Adipokines. Pediatr. Res. 2008, 63, 353–357. [Google Scholar] [CrossRef]
- Kim, J.J.; Accili, D. Signalling through IGF-I and Insulin Receptors: Where Is the Specificity? Growth Horm. IGF Res. 2002, 12, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Cianfarani, S.; Germani, D.; Rossi, P.; Rossi, L.; Germani, A.; Ossicini, C.; Zuppa, A.; Argirò, G.; Holly, J.M.P.; Branca, F. Intrauterine Growth Retardation: Evidence for the Activation of the Insulin-Like Growth Factor (IGF)-Related Growth-Promoting Machinery and the Presence of a Cation-Independent IGF Binding Protein-3 Proteolytic Activity by Two Months of Life. Pediatr. Res. 1998, 44, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Dominici, F.P.; Argentino, D.P.; Muñoz, M.C.; Miquet, J.G.; Sotelo, A.I.; Turyn, D. Influence of the Crosstalk between Growth Hormone and Insulin Signalling on the Modulation of Insulin Sensitivity. Growth Horm. IGF Res. 2005, 15, 324–336. [Google Scholar] [CrossRef]
- Velegrakis, A.; Sfakiotaki, M.; Sifakis, S. Human Placental Growth Hormone in Normal and Abnormal Fetal Growth. Biomed. Rep. 2017, 7, 115. [Google Scholar] [CrossRef]
- Aplin, J.D.; Myers, J.E.; Timms, K.; Westwood, M. Tracking Placental Development in Health and Disease. Nat. Rev. Endocrinol. 2020, 16, 479–494. [Google Scholar] [CrossRef]
- Glinoer, D. The Regulation of Thyroid Function in Pregnancy: Pathways of Endocrine Adaptation from Physiology to Pathology. Endocr. Rev. 1997, 18, 404–433. [Google Scholar] [CrossRef]
- Duthie, L.; Reynolds, R.M. Changes in the Maternal Hypothalamic-Pituitary-Adrenal Axis in Pregnancy and Postpartum: Influences on Maternal and Fetal Outcomes. Neuroendocrinology 2013, 98, 106–115. [Google Scholar] [CrossRef]
- Dôrr, H.G.; Heller, A.; Versmold, H.T.; Sippell, W.G.; Herrmann, M.; Bidlingmaier, F.; Knorr, D. Longitudinal Study of Progestins, Mineralocorticoids, and Glucocorticoids throughout Human Pregnancy. J. Clin. Endocrinol. Metab. 1989, 68, 863–868. [Google Scholar] [CrossRef]
- Fernández-Pérez, L.; Flores-Morales, A.; Guerra, B.; Díaz-Chico, J.C.; Iglesias-Gato, D. Growth Hormone Receptor Signaling Pathways and Its Negative Regulation by SOCS2. In Restricted Growth—Clinical, Genetic and Molecular Aspects; InTechOpen: London, UK, 2016; pp. 125–145. [Google Scholar]
- Fowden, A.L. The Insulin-like Growth Factors and Feto-Placental Growth. Placenta 2003, 24, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Street, M.E.; Viani, I.; Ziveri, M.A.; Volta, C.; Smerieri, A.; Bernasconi, S. Impairment of Insulin Receptor Signal Transduction in Placentas of Intra-Uterine Growth-Restricted Newborns and Its Relationship with Fetal Growth. Eur. J. Endocrinol. 2011, 164, 45–52. [Google Scholar] [CrossRef]
- Jansson, N.; Rosario, F.J.; Gaccioli, F.; Lager, S.; Jones, H.N.; Roos, S.; Jansson, T.; Powell, T.L. Activation of Placental MTOR Signaling and Amino Acid Transporters in Obese Women Giving Birth to Large Babies. J. Clin. Endocrinol. Metab. 2013, 98, 105–113. [Google Scholar] [CrossRef]
- Verhaeghe, J.; Pintiaux, A.; Van Herck, E.; Hennen, G.; Foidart, J.M.; Igout, A. Placental GH, IGF-I, IGF-Binding Protein-1, and Leptin during a Glucose Challenge Test in Pregnant Women: Relation with Maternal Body Weight, Glucose Tolerance, and Birth Weight. J. Clin. Endocrinol. Metab. 2002, 87, 2875–2882. [Google Scholar] [CrossRef]
- Zhang, W.; Su, R.; Lin, L.; Yang, H. ARHGEF11 Affecting the Placental Insulin Signaling Pathway in Fetal Macrosomia of Normal Glucose Tolerance Pregnant Women. Placenta 2018, 63, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Deegan, D.F.; Engel, N. Sexual Dimorphism in the Age of Genomics: How, When, Where. Front. Cell Dev. Biol. 2019, 7, 186. [Google Scholar] [CrossRef] [PubMed]
- Christians, J.K. The Placenta’s Role in Sexually Dimorphic Fetal Growth Strategies. Reprod. Sci. 2021, 29, 1895–1907. [Google Scholar] [CrossRef]
- Martin, E.; Smeester, L.; Bommarito, P.A.; Grace, M.R.; Boggess, K.; Kuban, K.; Karagas, M.R.; Marsit, C.J.; O’Shea, T.M.; Fry, R.C. Sexual Epigenetic Dimorphism in the Human Placenta: Implications for Susceptibility during the Prenatal Period. Epigenomics 2017, 9, 267–278. [Google Scholar] [CrossRef]
- Sun, T.; Gonzalez, T.L.; Deng, N.; Dipentino, R.; Clark, E.L.; Lee, B.; Tang, J.; Wang, Y.; Stripp, B.R.; Yao, C.; et al. Sexually Dimorphic Crosstalk at the Maternal-Fetal Interface. J. Clin. Endocrinol. Metab. 2020, 105, 4831–4847. [Google Scholar] [CrossRef]
- Vatten, L.J.; Nilsen, S.T.; Ødegård, R.A.; Romundstad, P.R.; Austgulen, R. Insulin-like Growth Factor I and Leptin in Umbilical Cord Plasma and Infant Birth Size at Term. Pediatrics 2002, 109, 1131–1135. [Google Scholar] [CrossRef]
- Shang, M.; Wen, Z. Increased Placental IGF-1/MTOR Activity in Macrosomia Born to Women with Gestational Diabetes. Diabetes Res. Clin. Pract. 2018, 146, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Kajantie, E.; Dunkel, L.; Rutanen, E.M.; Seppälä, M.; Koistinen, R.; Sarnesto, A.; Andersson, S. IGF-I, IGF Binding Protein (IGFBP)-3, Phosphoisoforms of IGFBP-1, and Postnatal Growth in Very Low Birth Weight Infants. J. Clin. Endocrinol. Metab. 2002, 87, 2171–2179. [Google Scholar] [CrossRef]
- Iñiguez, G.; Castro, J.J.; Garcia, M.; Kakarieka, E.; Johnson, M.C.; Cassorla, F.; Mericq, V. IGF-IR Signal Transduction Protein Content and Its Activation by IGF-I in Human Placentas: Relationship with Gestational Age and Birth Weight. PLoS ONE 2014, 9, e102252. [Google Scholar] [CrossRef]
- Garcia-Aragon, J.; Lobie, P.E.; Muscat, G.E.O.; Gobius, K.S.; Norstedt, G.; Waters, M.J. Prenatal Expression of the Growth Hormone (GH) Receptor/Binding Protein in the Rat: A Role for GH in Embryonic and Fetal Development? Development 1992, 114, 869–876. [Google Scholar] [CrossRef]
- Trarbach, E.B.; Jorge, A.A.; Duarte, F.H.; Bronstein, M.D.; Jallad, R.S. SOCS2 Polymorphisms Are Not Associated with Clinical and Biochemical Phenotypes in Acromegalic Patients. Pituitary 2017, 20, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Turnley, A.M. Role of SOCS2 in Growth Hormone Actions. Trends Endocrinol. Metab. 2005, 16, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.C.; Nuyt, A.M.; Delvin, E.; Audibert, F.; Girard, I.; Shatenstein, B.; Cloutier, A.; Cousineau, J.; Djemli, A.; Deal, C.; et al. Maternal and Fetal IGF-I and IGF-II Levels, Fetal Growth, and Gestational Diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Nawathe, A.R.; Christian, M.; Kim, S.H.; Johnson, M.; Savvidou, M.D.; Terzidou, V. Insulin-like Growth Factor Axis in Pregnancies Affected by Fetal Growth Disorders. Clin. Epigenetics 2016, 8, 1–13. [Google Scholar] [CrossRef]
- Murphy, V.E.; Smith, R.; Giles, W.B.; Clifton, V.L. Endocrine Regulation of Human Fetal Growth: The Role of the Mother, Placenta, and Fetus. Endocr. Rev. 2006, 27, 141–169. [Google Scholar] [CrossRef]
- Ibarra, A.; Vega-Guedes, B.; Brito-Casillas, Y.; Wägner, A.M. Diabetes in Pregnancy and MicroRNAs: Promises and Limitations in Their Clinical Application. Noncoding RNA 2018, 4, 32. [Google Scholar] [CrossRef]
- Leung, K.C.; Johannsson, G.; Leong, G.M.; Ho, K.K.Y. Estrogen Regulation of Growth Hormone Action. Endocr. Rev. 2004, 25, 693–721. [Google Scholar] [CrossRef]
- Martinez, C.S.; Piazza, V.G.; Díaz, M.E.; Boparai, R.K.; Arum, O.; Ramírez, M.C.; González, L.; Becú-Villalobos, D.; Bartke, A.; Turyn, D.; et al. GH/STAT5 Signaling during the Growth Period in Livers of Mice Overexpressing GH. J. Mol. Endocrinol. 2015, 54, 171–184. [Google Scholar] [CrossRef]
- Metzger, B.; Coustan, D. Proceedings of the Fourth International Work-Shop-Conference on Gestational Diabetes Mellitus. Diabetes Care 1998, 21, B1–B167. [Google Scholar]
- Liu, L.; Liu, L.; Wang, J.; Zheng, Q.; Jin, B.; Sun, L. Differentiation of Gestational Diabetes Mellitus by Nuclear Magnetic Resonance-Based Metabolic Plasma Analysis. J. Biomed. Res. 2021, 35, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Roca-Rodríguez, M.d.M.; López-Tinoco, C.; Fernández-Deudero, Á.; Murri, M.; García-Palacios, M.V.; García-Valero, M.D.A.; Tinahones, F.J.; Aguilar-Diosdado, M. Unfavorable Cytokine and Adhesion Molecule Profiles during and after Pregnancy, in Women with Gestational Diabetes Mellitus. Endocrinol. Diabetes Nutr. 2017, 64, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Wicklow, B.; Retnakaran, R. Gestational Diabetes Mellitus and Its Implications across the Life Span. Diabetes Metab. J. 2023, 47, 333. [Google Scholar] [CrossRef] [PubMed]
- Sweeting, A.; Wong, J.; Murphy, H.R.; Ross, G.P.; Hons, M.; Hl, G. A Clinical Update on Gestational Diabetes Mellitus. Endocr. Rev. 2022, 43, 763–793. [Google Scholar] [CrossRef]
- Rieck, S.; White, P.; Schug, J.; Fox, A.J.; Smirnova, O.; Gao, N.; Gupta, R.K.; Wang, Z.V.; Scherer, P.E.; Keller, M.P.; et al. The Transcriptional Response of the Islet to Pregnancy in Mice. Mol. Endocrinol. 2009, 23, 1702–1712. [Google Scholar] [CrossRef]
- Salazar-Petres, E.R.; Sferruzzi-Perri, A.N. Pregnancy-Induced Changes in β-Cell Function: What Are the Key Players? J. Physiol. 2022, 600, 1089–1117. [Google Scholar] [CrossRef]
- Andersen, M.D.; Alstrup, A.K.O.; Duvald, C.S.; Mikkelsen, E.F.R.; Vendelbo, M.H.; Ovesen, P.G.; Pedersen, M.; Andersen, M.D.; Alstrup, A.K.O.; Duvald, C.S.; et al. Animal Models of Fetal Medicine and Obstetrics. In Experimental Animal Models of Human Diseases—An Effective Therapeutic Strategy; InTech: London, UK, 2018. [Google Scholar] [CrossRef]
- Desoye, G.; Wells, J.C.K. Pregnancies in Diabetes and Obesity: The Capacity-Load Model of Placental Adaptation. Diabetes 2021, 70, 823. [Google Scholar] [CrossRef]
- Pantham, P.; Aye, I.L.M.H.; Powell, T.L. Inflammation in Maternal Obesity and Gestational Diabetes Mellitus. Placenta 2015, 36, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M. Obesity and Pregnancy—The Propagation of a Viscous Cycle? J. Clin. Endocrinol. Metab. 2003, 88, 3505–3506. [Google Scholar] [CrossRef]
- Catalano, P.M. Trying to Understand Gestational Diabetes. Diabet. Med. 2014, 31, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Gauster, M.; Hiden, U.; Van Poppel, M.; Frank, S.; Wadsack, C.; Hauguel-de Mouzon, S.; Desoye, G. Dysregulation of Placental Endothelial Lipase in Obese Women With Gestational Diabetes Mellitus. Diabetes 2011, 60, 2457. [Google Scholar] [CrossRef]
- Roberts, K.A.; Riley, S.C.; Reynolds, R.M.; Barr, S.; Evans, M.; Statham, A.; Hor, K.; Jabbour, H.N.; Norman, J.E.; Denison, F.C. Placental Structure and Inflammation in Pregnancies Associated with Obesity. Placenta 2011, 32, 247–254. [Google Scholar] [CrossRef]
- Challis, J.R.; Lockwood, C.J.; Myatt, L.; Norman, J.E.; Strauss, J.F.; Petraglia, F. Inflammation and Pregnancy. Reprod. Sci. 2009, 16, 206–215. [Google Scholar] [CrossRef]
- Montelongo, A.; Lasuncion, M.A.; Pallardo, L.F.; Herrera, E. Longitudinal Study of Plasma Lipoproteins and Hormones During Pregnancy in Normal and Diabetic Women. Diabetes 1992, 41, 1651–1659. [Google Scholar] [CrossRef]
- Van Lieshout, R.J.; Savoy, C.D.; Ferro, M.A.; Krzeczkowski, J.E.; Colman, I. Macrosomia and Psychiatric Risk in Adolescence. Eur. Child Adolesc. Psychiatry 2020, 29, 1537–1545. [Google Scholar] [CrossRef]
- Garmash, O.V.; Ryabokon, E.N. The Effect of Fetal Macrosomia on the Neonate and Infant Dental Health. Int. J. Clin. Dent. 2017, 10, 199–210. [Google Scholar]
- Opati, P.; Zheng, R.; Wang, J.; Xin, Y.; Zhao, H.; Bi, D. Comparison of Neonatal Outcomes in Macrosomic Infants of Diabetic and Non-Diabetic Mothers. J. Neonatal. Perinatal. Med. 2015, 8, 9–13. [Google Scholar] [CrossRef]
- Alonso, A.; Del Rey, C.G.; Navarro, A.; Tolivia, J.; González, C.G. Effects of Gestational Diabetes Mellitus on Proteins Implicated in Insulin Signaling in Human Placenta. Gynecol. Endocrinol. 2006, 22, 526–535. [Google Scholar] [CrossRef]
- Pedersen, J. The Pregnant Diabetic and Her Newborn: Problems and Management; Williams & Wilkins Company: Philadelphia, PE, USA, 1977. [Google Scholar]
- She, P.; Van Horn, C.; Reid, T.; Hutson, S.M.; Cooney, R.N.; Lynch, C.J. Obesity-Related Elevations in Plasma Leucine Are Associated with Alterations in Enzymes Involved in Branched Chain Amino Acid (BCAA) Metabolism. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1552. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Park, J.Y.; Lee, J.H.; Kim, S.H. Plasma Levels of Lysine, Tyrosine, and Valine during Pregnancy Are Independent Risk Factors of Insulin Resistance and Gestational Diabetes. Metab. Syndr. Relat. Disord 2015, 13, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Owei, I.; Umekwe, N.; Stentz, F.; Wan, J.; Dagogo-Jack, S. Amino Acid Signature Predictive of Incident Prediabetes: A Case-Control Study Nested Within the Longitudinal Pathobiology of Prediabetes in a Biracial Cohort. Metabolism 2019, 98, 76. [Google Scholar] [CrossRef]
- Knopp, R.H.; Magee, M.S.; Walden, C.E.; Bonet, B.; Benedetti, T.J. Prediction of Infant Birth Weight by GDM Screening Tests. Importance of Plasma Triglyceride. Diabetes Care 1992, 15, 1605–1613. [Google Scholar] [CrossRef]
- Aye, I.L.M.H.; Powell, T.L.; Jansson, T. Review: Adiponectin--the Missing Link between Maternal Adiposity, Placental Transport and Fetal Growth? Placenta 2013, 34, S40–S45. [Google Scholar] [CrossRef]
- Aye, I.L.M.H.; Gao, X.; Weintraub, S.T.; Jansson, T.; Powell, T.L. Adiponectin Inhibits Insulin Function in Primary Trophoblasts by PPARα-Mediated Ceramide Synthesis. Mol. Endocrinol. 2014, 28, 512–524. [Google Scholar] [CrossRef]
- Rosario, F.J.; Powell, T.L.; Jansson, T. Activation of Placental Insulin and MTOR Signaling in a Mouse Model of Maternal Obesity Associated with Fetal Overgrowth. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R87–R93. [Google Scholar] [CrossRef]
- Lilao-Garzón, J.; Brito-Casillas, Y.; Quesada-Canales, O.; Wägner, A.M.; Muñoz-Descalzo, S. Maternal Age, Obesity and Hyperglycaemia Are Associated with a Delay in Preimplantation Embryo Development in Mouse. Reproduction 2023, 166, 235–245. [Google Scholar] [CrossRef]
- Visiedo, F.; Bugatto, F.; Sánchez, V.; Cózar-Castellano, I.; Bartha, J.L.; Perdomo, G. High Glucose Levels Reduce Fatty Acid Oxidation and Increase Triglyceride Accumulation in Human Placenta. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E205–E212. [Google Scholar] [CrossRef]
- Helen, L.; Kathryn, L.; Candice, M.; Sci, B.M.E.D.; Blasio, D. Maternal and Neonatal Circulating Markers of Metabolic and Cardiovascular Risk in the Metformin in Gestational Diabetes (MiG) Trial. Diabetes Care 2013, 36, 529. [Google Scholar]
- Lepercq, J.; Cauzac, M.; Lahlou, N.; Timsit, J.; Girard, J.; Auwerx, J.; Mouzon, S.H. De Overexpression of Placental Leptin in Diabetic Pregnancy: A Critical Role for Insulin. Diabetes 1998, 47, 847–850. [Google Scholar] [CrossRef]
- Gonzalez, C.; Alonso, A.; Alvarez, N.; Diaz, F.; Martinez, M.; Fernandez, S.; Patterson, A.M. Role of 17beta-Estradiol and/or Progesterone on Insulin Sensitivity in the Rat: Implications during Pregnancy. J. Endocrinol. 2000, 166, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Barbour, L.A.; McCurdy, C.E.; Hernandez, T.L.; Kirwan, J.P.; Catalano, P.M.; Friedman, J.E. Cellular Mechanisms for Insulin Resistance in Normal Pregnancy and Gestational Diabetes. Diabetes Care 2007, 30, S112–S119. [Google Scholar] [CrossRef]
- Scioscia, M.; Gumaa, K.; Kunjara, S.; Paine, M.A.; Selvaggi, L.E.; Rodeck, C.H.; Rademacher, T.W. Insulin Resistance in Human Preeclamptic Placenta Is Mediated by Serine Phosphorylation of Insulin Receptor Substrate-1 and -2. J. Clin. Endocrinol. Metab. 2006, 91, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Gallo, L.A.; Barrett, H.L.; Dekker Nitert, M. Placental Transport and Metabolism of Energy Substrates in Maternal Obesity and Diabetes. Placenta 2017, 54, 59–67. [Google Scholar] [CrossRef]
- Zhu, Z.; Cao, F.; Li, X. Epigenetic Programming and Fetal Metabolic Programming. Front. Endocrinol. 2019, 10, 422860. [Google Scholar] [CrossRef]
- Ott, R.; Stupin, J.H.; Melchior, K.; Schellong, K.; Ziska, T.; Dudenhausen, J.W.; Henrich, W.; Rancourt, R.C.; Plagemann, A. Alterations of Adiponectin Gene Expression and DNA Methylation in Adipose Tissues and Blood Cells Are Associated with Gestational Diabetes and Neonatal Outcome. Clin. Epigenetics 2018, 10, 131. [Google Scholar] [CrossRef]
- Food and Drug Administration (US); National Institutes of Health (US). BEST (Biomarkers, EndpointS, and Other Tools) Resource; Food and Drug Administration (US): Silver Spring, MD, USA, 2016. [Google Scholar]
- Visiedo, F.; Bugatto, F.; Quintero-Prado, R.; Cózar-Castellano, I.; Bartha, J.L.; Perdomo, G. Glucose and Fatty Acid Metabolism in Placental Explants from Pregnancies Complicated with Gestational Diabetes Mellitus. Reprod. Sci. 2015, 22, 798–801. [Google Scholar] [CrossRef]
- Schaefer-Graf, U.M.; Graf, K.; Kulbacka, I.; Kjos, S.L.; Dudenhausen, J.; Vetter, K.; Herrera, E. Maternal Lipids as Strong Determinants of Fetal Environment and Growth in Pregnancies with Gestational Diabetes Mellitus. Diabetes Care 2008, 31, 1858–1863. [Google Scholar] [CrossRef]
- Schaefer-Graf, U.M.; Meitzner, K.; Ortega-Senovilla, H.; Graf, K.; Vetter, K.; Abou-Dakn, M.; Herrera, E. Differences in the Implications of Maternal Lipids on Fetal Metabolism and Growth between Gestational Diabetes Mellitus and Control Pregnancies. Diabetes Med. 2011, 28, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Guo, J.; Lu, W.; Fang, X.; Zhang, R.; Tang, M.; Luo, Q.; Liang, W.; Yu, X.; Hu, C. The Mediating Role of Maternal Metabolites between Lipids and Adverse Pregnancy Outcomes of Gestational Diabetes Mellitus. Front. Med. 2022, 9, 925602. [Google Scholar] [CrossRef]
- Radaelli, T.; Lepercq, J.; Varastehpour, A.; Basu, S.; Catalano, P.M.; Hauguel-De Mouzon, S. Differential Regulation of Genes for Feto-Placental Lipid Pathways in Pregnancy with Gestational and Type 1 Diabetes. Am. J. Obstet. Gynecol. 2009, 201, 209.e1. [Google Scholar] [CrossRef] [PubMed]
- Shang, M.; Zhao, J.; Yang, L.; Lin, L. Oxidative Stress and Antioxidant Status in Women with Gestational Diabetes Mellitus Diagnosed by IADPSG Criteria. Diabetes Res. Clin. Pract. 2015, 109, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhu, W.; Li, J.; An, C.; Wang, Z. Serum Concentrations of Fibroblast Growth Factors 19 and 21 in Women with Gestational Diabetes Mellitus: Association with Insulin Resistance, Adiponectin, and Polycystic Ovary Syndrome History. PLoS ONE 2013, 8, e81190. [Google Scholar] [CrossRef]
- Jia, J.; Wei, W.; Yu, F.; Liu, R.; Shen, Y.; Zhang, R.; Yuan, G.; Zhou, H. Circulating Levels of Fibroblast Growth Factor 21 in Gestational Diabetes Mellitus: A Meta-Analysis. Endrocrine J. 2021, 68, 345–352. [Google Scholar] [CrossRef]
- Dudzik, D.; Zorawski, M.; Skotnicki, M.; Zarzycki, W.; Kozlowska, G.; Bibik-Malinowska, K.; Vallejo, M.; García, A.; Barbas, C.; Ramos, M.P. Metabolic Fingerprint of Gestational Diabetes Mellitus. J. Proteom. 2014, 103, 57–71. [Google Scholar] [CrossRef]
- Dudzik, D.; Zorawski, M.; Skotnicki, M.; Zarzycki, W.; García, A.; Angulo, S.; Lorenzo, M.P.; Barbas, C.; Ramos, M.P. GC-MS Based Gestational Diabetes Mellitus Longitudinal Study: Identification of 2-and 3-Hydroxybutyrate as Potential Prognostic Biomarkers. J. Pharm. Biomed. Anal. 2017, 144, 90–98. [Google Scholar] [CrossRef]
- Banerjee, R.R.; Cyphert, H.A.; Walker, E.M.; Chakravarthy, H.; Peiris, H.; Gu, X.; Liu, Y.; Conrad, E.; Goodrich, L.; Stein, R.W.; et al. Gestational Diabetes Mellitus From Inactivation of Prolactin Receptor and MafB in Islet β-Cells. Diabetes 2016, 65, 2331–2341. [Google Scholar] [CrossRef]
- Le, T.N.; Elsea, S.H.; Romero, R.; Chaiworapongsa, T.; Francis, G.L. Prolactin Receptor Gene Polymorphisms Are Associated with Gestational Diabetes. Genet. Test. Mol. Biomark. 2013, 17, 567–571. [Google Scholar] [CrossRef]
- Napso, T.; Yong, H.E.J.; Lopez-Tello, J.; Sferruzzi-Perri, A.N. The Role of Placental Hormones in Mediating Maternal Adaptations to Support Pregnancy and Lactation. Front. Physiol. 2018, 9, 1091. [Google Scholar] [CrossRef]
- Qi, X.; Gong, B.; Yu, J.; Shen, L.; Jin, W.; Wu, Z.; Wang, J.; Wang, J.; Li, Z. Decreased Cord Blood Estradiol Levels in Related to Mothers with Gestational Diabetes. Medicine 2017, 96, e6962. [Google Scholar] [CrossRef] [PubMed]
- Hiden, U.; Bilban, M.; Knöfler, M.; Desoye, G. Kisspeptins and the Placenta: Regulation of Trophoblast Invasion. Rev. Endocr. Metab. Disord. 2007, 8, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Matjila, M.; Millar, R.; Van Der Spuy, Z.; Katz, A. Elevated Placental Expression at the Maternal–Fetal Interface but Diminished Maternal Circulatory Kisspeptin in Preeclamptic Pregnancies. Pregnancy Hypertens. Int. J. Women’s Cardiovasc. Health 2016, 6, 79–87. [Google Scholar] [CrossRef]
- Colomiere, M.; Permezel, M.; Riley, C.; Desoye, G.; Lappas, M. Defective Insulin Signaling in Placenta from Pregnancies Complicated by Gestational Diabetes Mellitus. Eur. J. Endocrinol. 2009, 160, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Muralimanoharan, S.; Maloyan, A.; Myatt, L. Mitochondrial Function and Glucose Metabolism in the Placenta with Gestational Diabetes Mellitus: Role of MiR-143. Clin. Sci. 2016, 130, 931–941. [Google Scholar] [CrossRef]
- Leong, G.M.; Moverare, S.; Brce, J.; Doyle, N.; Sjögren, K.; Dahlman-Wright, K.; Gustafsson, J.A.; Ho, K.K.Y.; Ohlsson, C.; Leung, K.C. Estrogen Up-Regulates Hepatic Expression of Suppressors of Cytokine Signaling-2 and -3 in Vivo and in Vitro. Endocrinology 2004, 145, 5525–5531. [Google Scholar] [CrossRef]
- Barbour, L.A.; Shao, J.; Qiao, L.; Leitner, W.; Anderson, M.; Friedman, J.E.; Draznin, B. Human Placental Growth Hormone Increases Expression of the P85 Regulatory Unit of Phosphatidylinositol 3-Kinase and Triggers Severe Insulin Resistance in Skeletal Muscle. Endocrinology 2004, 145, 1144–1150. [Google Scholar] [CrossRef]
- Caufriez, A.; Frankenne, F.; Englert, Y.; Golstein, J.; Cantraine, F.; Hennen, G.; Copinschi, G. Placental Growth Hormone as a Potential Regulator of Maternal IGF-I during Human Pregnancy. Am. J. Physiol. 1990, 258, E1014–E1019. [Google Scholar] [CrossRef]
- Hivert, M.-F.; White, F.; Allard, C.; James, K.; Majid, S.; Aguet, F.; Ardlie, K.G.; Florez, J.C.; Edlow, A.G.; Bouchard, L.; et al. Placental IGFBP1 Levels during Early Pregnancy and the Risk of Insulin Resistance and Gestational Diabetes. Nat. Med. 2024, 30, 1689–1695. [Google Scholar] [CrossRef]
- Osmond, D.T.D.; Nolan, C.J.; King, R.G.; Brennecke, S.P.; Gude, N.M. Effects of Gestational Diabetes on Human Placental Glucose Uptake, Transfer, and Utilisation. Diabetologia 2000, 43, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Oken, E.; Morton-Eggleston, E.; Rifas-Shiman, S.L.; Switkowski, K.M.; Hivert, M.F.; Fleisch, A.F.; Mantzoros, C.; Gillman, M.W. Sex-Specific Associations of Maternal Gestational Glycemia with Hormones in Umbilical Cord Blood at Delivery. Am. J. Perinatol. 2016, 33, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Le Moullec, N.; Fianu, A.; Maillard, O.; Chazelle, E.; Naty, N.; Schneebeli, C.; Gérardin, P.; Huiart, L.; Charles, M.A.; Favier, F. Sexual Dimorphism in the Association between Gestational Diabetes Mellitus and Overweight in Offspring at 5-7 Years: The OBEGEST Cohort Study. PLoS ONE 2018, 13, e0195531. [Google Scholar] [CrossRef]
- Yang, M.-N.; Chiu, H.-C.; Wang, W.-J.; Fang, F.; Zhang, G.-H.; Zhu, H.; Zhang, L.; Zhang, D.-L.; Du, Q.; He, H.; et al. Sex Dimorphism in the Associations of Gestational Diabetes with Cord Blood Adiponectin and Retinol-Binding Protein 4. BMJ Open Diab. Res. Care 2020, 8, 1310. [Google Scholar] [CrossRef] [PubMed]
- Stock, S.M.; Bremme, K.A. Elevation of Plasma Leptin Levels during Pregnancy in Normal and Diabetic Women. Metabolism 1998, 47, 840–843. [Google Scholar] [CrossRef]
- Klid, S.; Maymó-Masip, E.; Algaba-Chueca, F.; Ballesteros, M.; Inglès-Puig, M.; Guarque, A.; Madeira, A.; Jareño, C.; Vendrell, J.; Fernández-Veledo, S.; et al. The ANGPTL3-4-8 Axis in Normal Gestation and in Gestational Diabetes, and Its Potential Involvement in Fetal Growth. Int. J. Mol. Sci. 2023, 24, 2486. [Google Scholar] [CrossRef]
- Orbak, Z.; Darcan, Ş.; Çoker, M.; Gökşen, D. Maternal and Fetal Serum Insulin-like Growth Factor-I (IGF-I), IGF Binding Protein-3 (IGFBP-3), Leptin Levels and Early Postnatal Growth in Infants Born Asymmetrically Small for Gestational Age. J. Pediatr. Endocrinol. Metab. 2001, 14, 1119–1127. [Google Scholar] [CrossRef]
- Jiang, H.; Wen, Y.; Hu, L.; Miao, T.; Zhang, M.; Dong, J. Serum MicroRNAs as Diagnostic Biomarkers for Macrosomia. Reprod. Sci. 2015, 22, 664–671. [Google Scholar] [CrossRef]
- Maccani, M.A.; Padbury, J.F.; Marsit, C.J. MiR-16 and MiR-21 Expression in the Placenta Is Associated with Fetal Growth. PLoS ONE 2011, 6, e21210. [Google Scholar] [CrossRef]
- Keniry, A.; Oxley, D.; Monnier, P.; Kyba, M.; Dandolo, L.; Smits, G.; Reik, W. The H19 LincRNA Is a Developmental Reservoir of MiR-675 That Suppresses Growth and Igf1r. Nat. Cell Biol. 2012, 14, 659–665. [Google Scholar] [CrossRef]
- Ebert, T.; Stepan, H.; Schrey, S.; Kralisch, S.; Hindricks, J.; Hopf, L.; Platz, M.; Lossner, U.; Jessnitzer, B.; Drewlo, S.; et al. Serum Levels of Irisin in Gestational Diabetes Mellitus during Pregnancy and after Delivery. Cytokine 2014, 65, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Brito-Casillas, Y.; Aranda-Tavío, H.; Rodrigo-González, L.; Expósito-Montesdeoca, A.B.; Martín-Rodríguez, P.; Guerra, B.; Wägner, A.M.; Fernández-Pérez, L. Socs2-/- Mouse as a Potential Model of Macrosomia and Gestational Diabetes. Diabetologia 2017, 60, S43. [Google Scholar]
- Hernandez-Baraza, L.; Valverde-Tercedor, C.; Díaz, M.; Wägner, A.M.; Fernández-Pérez, L.; Guerra, B.; Brito-Castillas, Y. Body Composition and Effect of Insulin Treatment during Pregnancy in Socs2-/- Mice with Gestational Diabetes and Macrosomia. Diabetologia 2022, 65, S223. [Google Scholar]
- Rønn, S.G.; Billestrup, N.; Mandrup-Poulsen, T. Diabetes and Suppressors of Cytokine Signaling Proteins. Diabetes 2007, 56, 541–548. [Google Scholar] [CrossRef]
- Accili, D. Insulin Receptor Knock-out Mice. Trends Endocrinol. Metab. 1997, 8, 101–104. [Google Scholar] [CrossRef]
- Zadjali, F.; Santana-Farre, R.; Vesterlund, M.; Carow, B.; Mirecki-Garrido, M.; Hernandez-Hernandez, I.; Flodström-Tullberg, M.; Parini, P.; Rottenberg, M.; Norstedt, G.; et al. SOCS2 Deletion Protects against Hepatic Steatosis but Worsens Insulin Resistance in High-fat-diet-fed Mice. FASEB J. 2012, 26, 3282–3291. [Google Scholar] [CrossRef]
- Zhang, X.; Zhuang, Y.; Qin, T.; Chang, M.; Ji, X.; Wang, N.; Zhang, Z.; Zhou, H.; Wang, Q.; Li, J.Z. Suppressor of Cytokine Signalling-2 Controls Hepatic Gluconeogenesis and Hyperglycemia by Modulating JAK2/STAT5 Signalling Pathway. Metabolism 2021, 122, 154823. [Google Scholar] [CrossRef]
- Lebrun, P.; Cognard, E.; Gontard, P.; Bellon-Paul, R.; Filloux, C.; Berthault, M.F.; Magnan, C.; Ruberte, J.; Luppo, M.; Pujol, A.; et al. The Suppressor of Cytokine Signalling 2 (SOCS2) Is a Key Repressor of Insulin Secretion. Diabetologia 2010, 53, 1935–1946. [Google Scholar] [CrossRef]
- Chowdhury, A.I.; Bergsten, P. GLP-1 Analogue Recovers Impaired Insulin Secretion from Human Islets Treated with Palmitate via down-Regulation of SOCS2. Mol. Cell Endocrinol. 2017, 439, 194–202. [Google Scholar] [CrossRef]
- Suba, Z. Low Estrogen Exposure and/or Defective Estrogen Signaling Induces Disturbances in Glucose Uptake and Energy Expenditure. J. Diabetes Metab. 2013, 04, 1–10. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef] [PubMed]
- Linossi, E.M.; Babon, J.J.; Hilton, D.J.; Nicholson, S.E. Suppression of Cytokine Signaling: The SOCS Perspective. Cytokine Growth Factor Rev. 2013, 24, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Satyajit, K.K.; Hainan, C.; Graeme, M.W.; Jeremy, H.J.; Xueying, G.; Zhang, A.Y.; Magali, F.; Michael, Y.H.; Seung, K.K. Menin Controls Growth of Pancreatic B-Cells in Pregnant Mice and Promotes Gestational Diabetes Mellitus. Proc. Natl. Acad. Sci. USA 2005, 102, 181. [Google Scholar] [CrossRef]
- Hernández Baraza, L.; Alemán Cabrera, I.; Valverde Tercedor, C.; Wägner, A.M.; Fernández Pérez, L.; Guerra, B.; Brito Casillas, Y. Insulin Reverts Macrosomia in a Mouse Model of Gestational Diabetes. Diabetologia 2023, 66, S261–S262. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Baraza, L.; Brito-Casillas, Y.; Valverde-Tercedor, C.; Recio, C.; Fernández-Pérez, L.; Guerra, B.; Wägner, A.M. Mechanisms of Fetal Overgrowth in Gestational Diabetes: The Potential Role of SOCS2. Nutrients 2025, 17, 1519. https://doi.org/10.3390/nu17091519
Hernández-Baraza L, Brito-Casillas Y, Valverde-Tercedor C, Recio C, Fernández-Pérez L, Guerra B, Wägner AM. Mechanisms of Fetal Overgrowth in Gestational Diabetes: The Potential Role of SOCS2. Nutrients. 2025; 17(9):1519. https://doi.org/10.3390/nu17091519
Chicago/Turabian StyleHernández-Baraza, Luisa, Yeray Brito-Casillas, Carmen Valverde-Tercedor, Carlota Recio, Leandro Fernández-Pérez, Borja Guerra, and Ana M. Wägner. 2025. "Mechanisms of Fetal Overgrowth in Gestational Diabetes: The Potential Role of SOCS2" Nutrients 17, no. 9: 1519. https://doi.org/10.3390/nu17091519
APA StyleHernández-Baraza, L., Brito-Casillas, Y., Valverde-Tercedor, C., Recio, C., Fernández-Pérez, L., Guerra, B., & Wägner, A. M. (2025). Mechanisms of Fetal Overgrowth in Gestational Diabetes: The Potential Role of SOCS2. Nutrients, 17(9), 1519. https://doi.org/10.3390/nu17091519