
Dietary Polyunsaturated Fatty Acid Deficiency Impairs Renal Lipid Metabolism and Adaptive Response to Proteinuria in Murine Renal Tubules

 , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Methods for Clarifying Research Questions
2.2. Diets, Animals, and Treatments
2.3. Urinary Protein Analysis
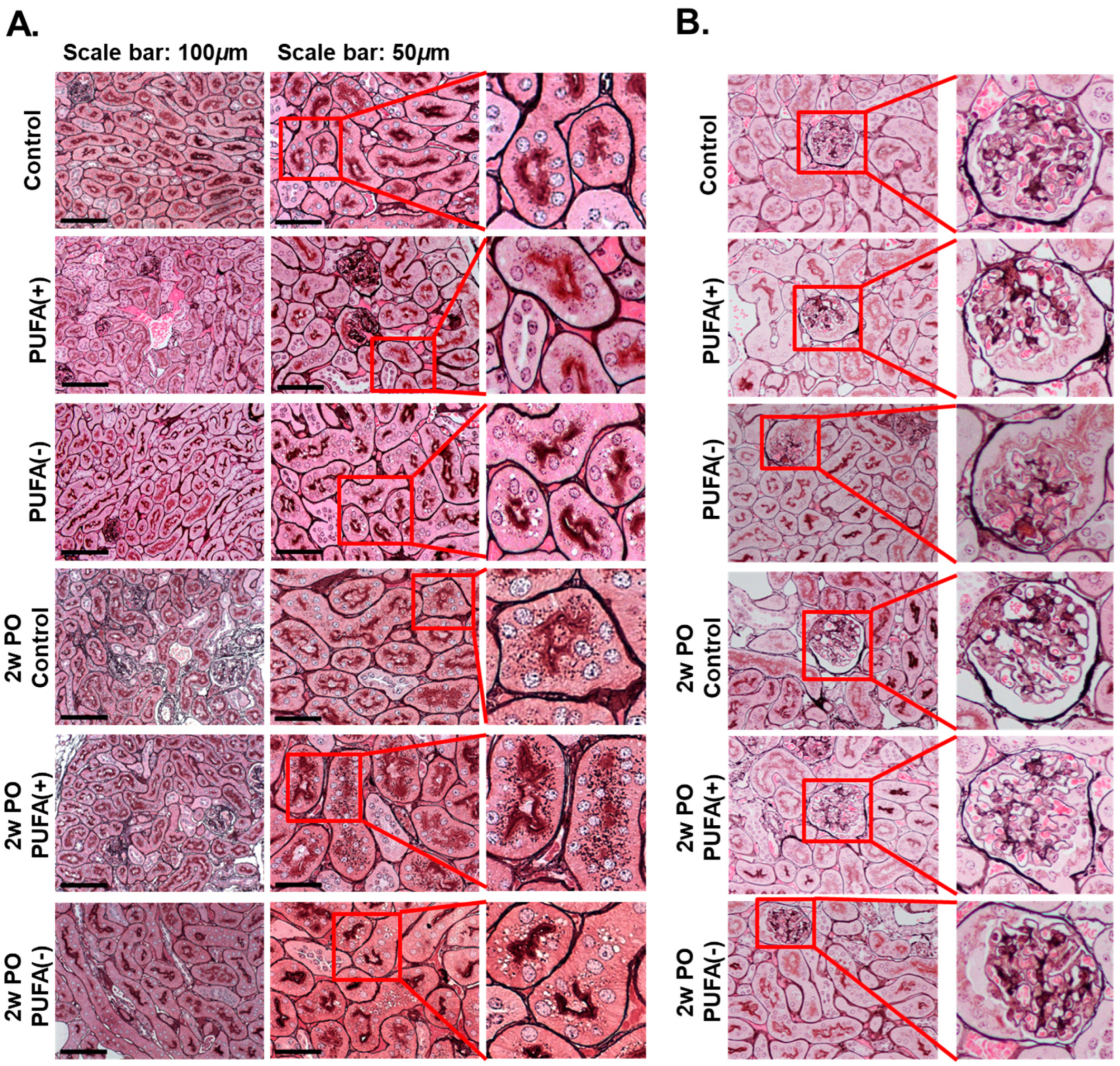
2.4. Pathological Analysis
2.5. mRNA Analysis
2.6. Immunoblot Analysis
2.7. Other Experiments
2.8. Statistical Analysis
3. Results
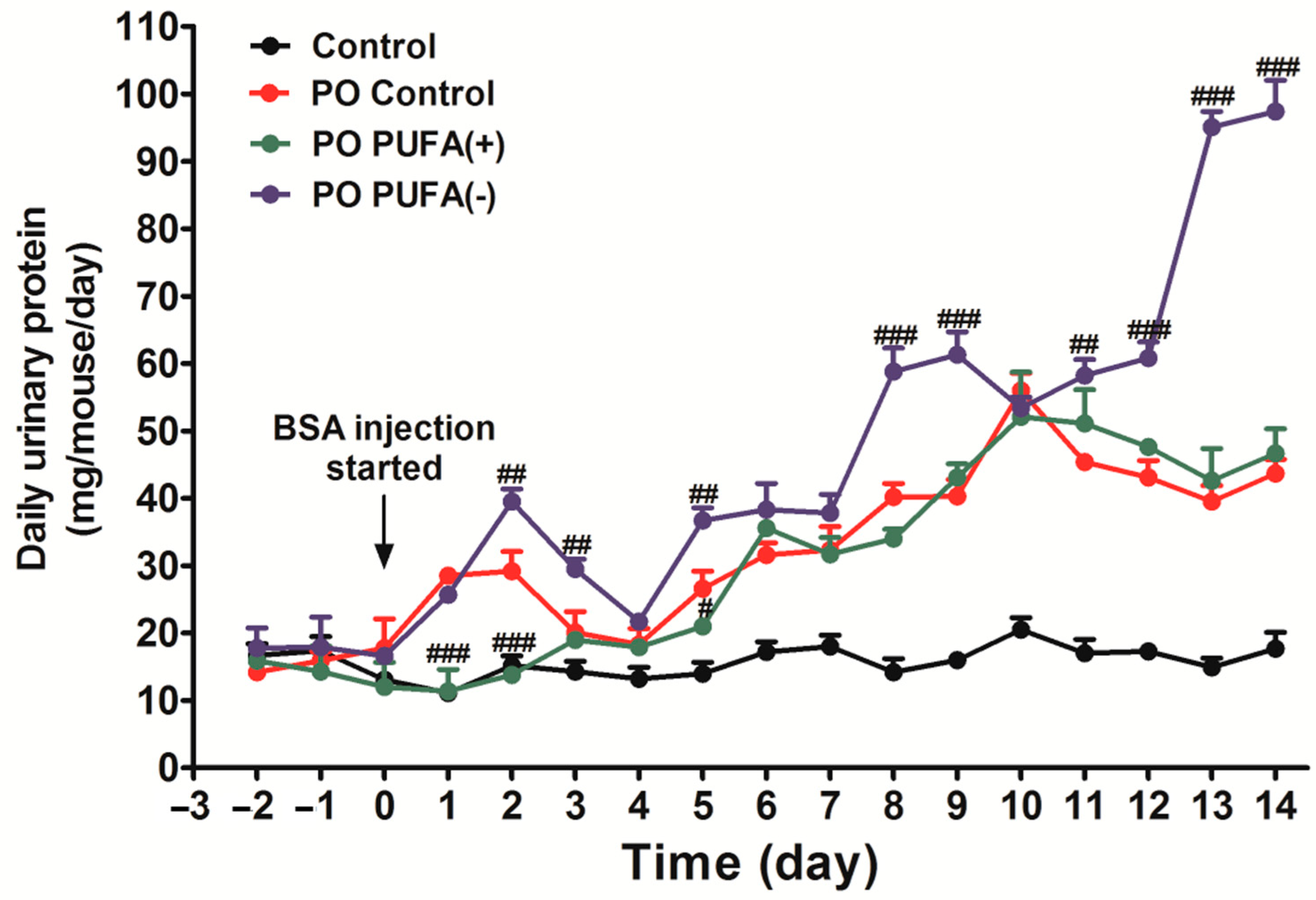
3.1. PUFA-Deficient Diet Significantly Increased Urinary Protein Excretion Following PO
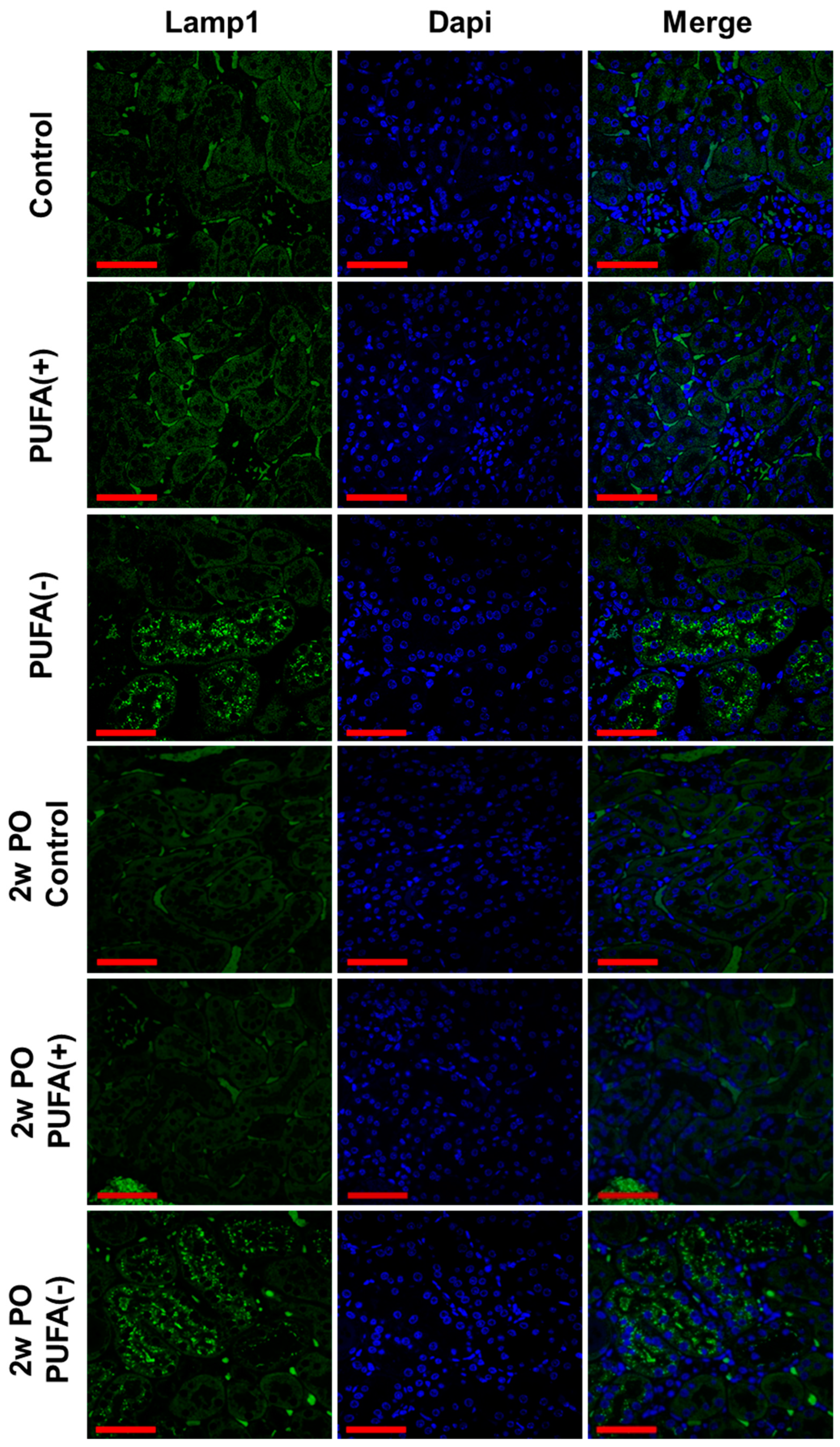
3.2. PUFA-Deficient Diet Increased Lysosomes and Suppressed the PO-Induced Adaptive Endocytosis Activation
3.3. PUFA-Deficient Diet Induced Renal Continuous Autophagy Activation, Possibly Contributing to Lysosomal Increase
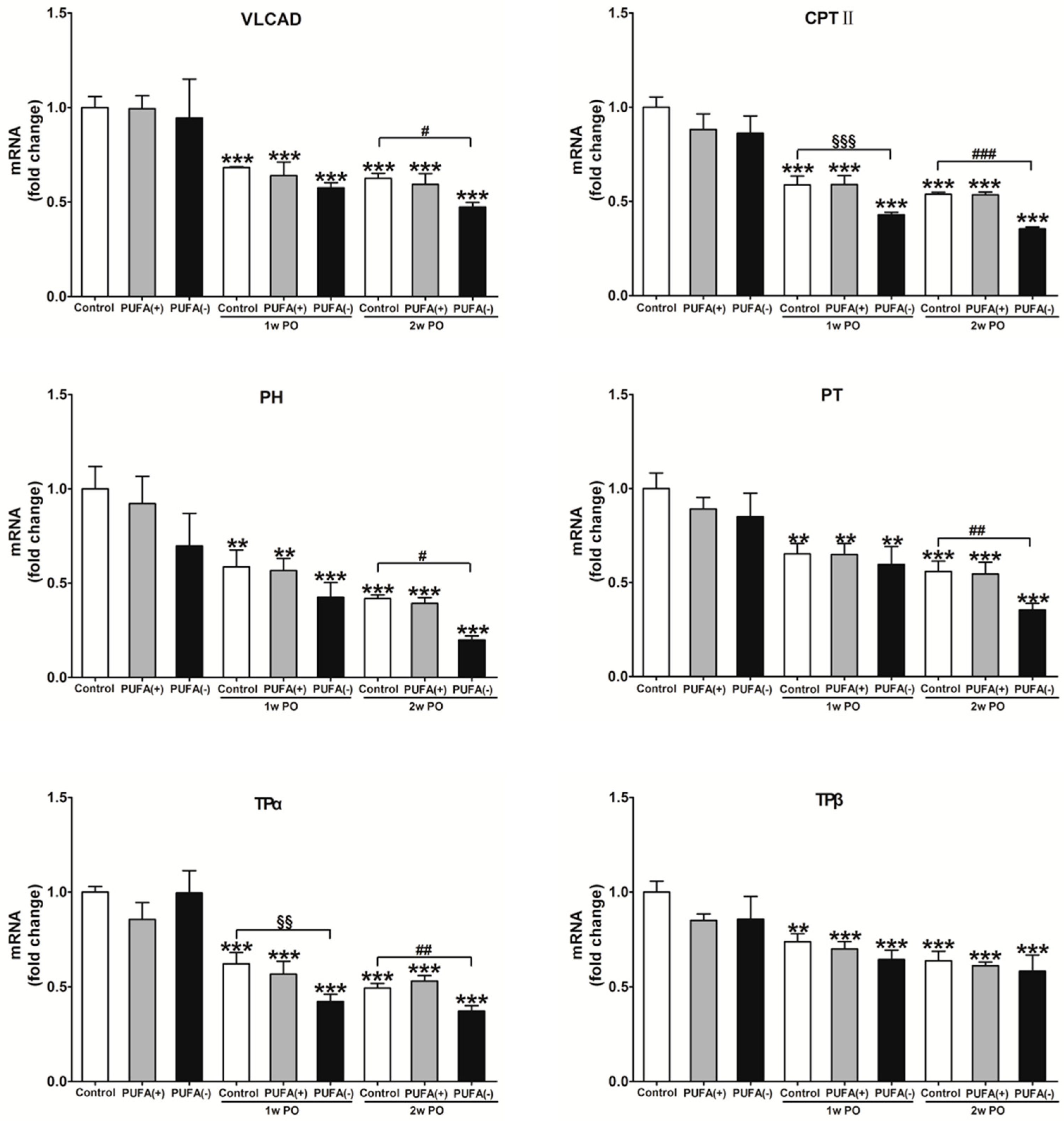
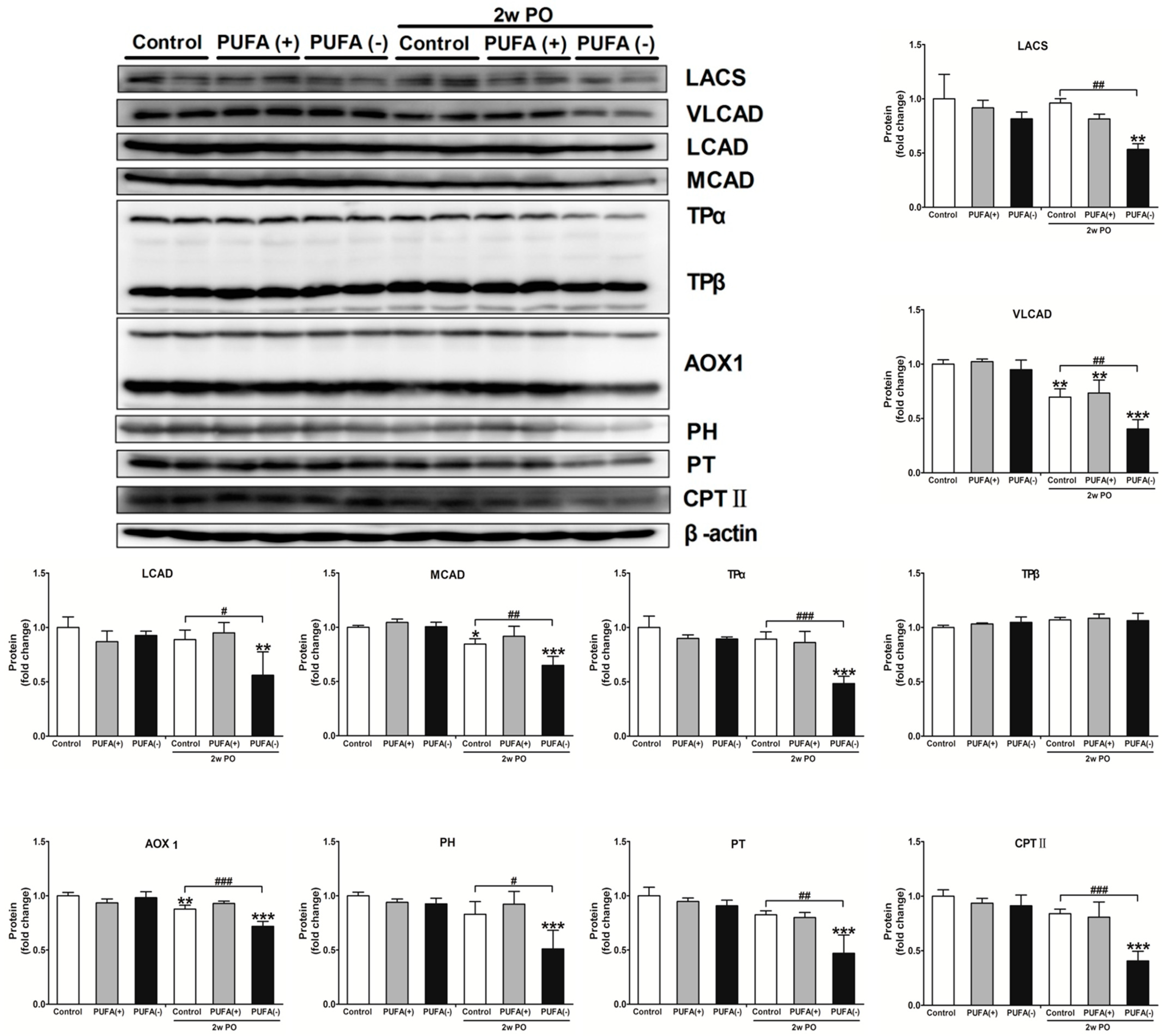
3.4. PUFA-Deficient Diet Significantly Aggravated PO-Induced Impaired FA Metabolism and PPARα Function and Induced the Disruption of Renal Energy Homeostasis
4. Discussion
Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FA | fatty acid |
| ATP | adenosine triphosphate |
| PPARα | peroxisome proliferator-activated receptor α |
| PUFAs | polyunsaturated fatty acids |
| PO | protein overload |
| PTECs | proximal tubular epithelial cells |
| HCO | hydrogenated coconut oil |
| BSA | bovine serum albumin |
| Lamp1 | lysosome-associated membrane protein type 1 |
| LACS | long-chain acyl-CoA synthetase |
| VLCAD | very long-chain acyl-CoA dehydrogenase |
| LCAD | long-chain acyl-CoA dehydrogenase |
| MCAD | medium-chain acyl-CoA dehydrogenase |
| TPα | mitochondrial trifunctional protein α |
| TPβ | mitochondrial trifunctional protein β |
| AOX1 | acyl-CoA oxidase 1 |
| PH | peroxisomal bifunctional protein |
| PT | peroxisomal thiolase |
| CPT II | carnitine palmitoyl-CoA transferase |
| LC3B | microtubule-associated protein 1 light chain 3B |
References
- Shi, M.Y.; Yu, H.C.; Han, C.Y.; Bang, I.H.; Park, H.S.; Jang, K.Y.; Lee, S.; Son, J.B.; Kim, N.D.; Park, B.H.; et al. p21-activated kinase 4 suppresses fatty acid beta-oxidation and ketogenesis by phosphorylating NCoR1. Nat. Commun. 2023, 14, 4987. [Google Scholar] [CrossRef] [PubMed]
- Tahri-Joutey, M.; Andreoletti, P.; Surapureddi, S.; Nasser, B.; Cherkaoui-Malki, M.; Latruffe, N. Mechanisms mediating the regulation of peroxisomal fatty acid beta-oxidation by PPARα. Int. J. Mol. Sci. 2021, 22, 8969. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Xie, X.; Wang, N.; Wang, Y.; Zhao, J.; Chen, F.; Qu, F.; Wen, W.; Miao, J.; Cui, H. Formononetin promotes fatty acid beta-oxidation to treat non-alcoholic steatohepatitis through SIRT1/PGC-1alpha/PPARalpha pathway. Phytomedicine 2024, 124, 155285. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Gao, Y.; Qu, A.; Jiang, Y.; Li, H.; Xie, G.; Yao, X.; Yang, X.; Zhu, S.; Yagai, T.; et al. YAP-TEAD mediates PPAR alpha-induced hepatomegaly and liver regeneration in mice. Hepatology 2022, 75, 74–88. [Google Scholar] [CrossRef]
- Xie, S.Y.; Liu, S.Q.; Zhang, T.; Shi, W.K.; Xing, Y.; Fang, W.X.; Zhang, M.; Chen, M.Y.; Xu, S.C.; Fan, M.Q.; et al. USP28 serves as a key suppressor of mitochondrial morphofunctional defects and cardiac dysfunction in the diabetic heart. Circulation 2024, 149, 684–706. [Google Scholar] [CrossRef]
- Shan, Z.; Zhang, H.; He, C.; An, Y.; Huang, Y.; Fu, W.; Wang, M.; Du, Y.; Xie, J.; Yang, Y.; et al. High-protein mulberry leaves improve glucose and lipid metabolism via activation of the PI3K/Akt/PPARα/CPT-1 pathway. Int. J. Mol. Sci. 2024, 25, 8726. [Google Scholar] [CrossRef]
- Shinkai, Y.; Sasaki, K.; Tamura, R.; Ike, T.; Takahashi, A.; Osaki, Y.; Ishiuchi, N.; Maeoka, Y.; Nakashima, A.; Masaki, T. Selective activation of PPARalpha by pemafibrate mitigates peritoneal inflammation and fibrosis through suppression of NLRP3 inflammasome and modulation of inflammation. Sci. Rep. 2024, 14, 23816. [Google Scholar] [CrossRef]
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 2021, 114, 154338. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, W.; Cheng, N.; Zhu, Y.; Li, H.; Zhang, S.; Guo, W.; Ge, G. Pectolinarigenin ameliorates acetaminophen-induced acute liver injury via attenuating oxidative stress and inflammatory response in Nrf2 and PPARa dependent manners. Phytomedicine 2023, 113, 154726. [Google Scholar] [CrossRef]
- Camoglio, C.; Balla, J.; Fadda, P.; Dedoni, S. Oleoylethanolamide and palmitoylethanolamide enhance IFNbeta-induced apoptosis in human neuroblastoma SH-SY5Y cells. Molecules 2024, 29, 1592. [Google Scholar] [CrossRef]
- Yoo, J.; Jeong, I.K.; Ahn, K.J.; Chung, H.Y.; Hwang, Y.C. Fenofibrate, a PPARalpha agonist, reduces hepatic fat accumulation through the upregulation of TFEB-mediated lipophagy. Metabolism 2021, 120, 154798. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Su, L.Y.; Li, G.; Yang, J.; Liu, Q.; Yang, L.X.; Zhang, D.F.; Zhou, H.; Xu, M.; Fan, Y.; et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 2020, 16, 52–69. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauca, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef]
- Sahara, Y.; Fukui, C.; Kuniyoshi, Y.; Takasato, M. Proximal tubule cell maturation rate and function are controlled by PPARalpha signaling in kidney organoids. Commun. Biol. 2024, 7, 1532. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, Y.; Hora, K.; Tanaka, N.; Usuda, N.; Kiyosawa, K.; Nakajima, T.; Gonzalez, F.J.; Aoyama, T. Identification of functions of peroxisome proliferator-activated receptor alpha in proximal tubules. J. Am. Soc. Nephrol. 2002, 13, 1691–1702. [Google Scholar] [CrossRef]
- Nakajima, T.; Yang, Y.; Lu, Y.; Kamijo, Y.; Yamada, Y.; Nakamura, K.; Koyama, M.; Yamaguchi, S.; Sugiyama, E.; Tanaka, N.; et al. Decreased fatty acid beta-oxidation is the main cause of fatty liver induced by polyunsaturated fatty acid deficiency in mice. Tohoku J. Exp. Med. 2017, 242, 229–239. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Diao, P.; Yamada, Y.; Nakamura, K.; Nakayama, J.; Tanaka, N.; Aoyama, T.; Kamijo, Y. Polyunsaturated fatty acid deficiency affects sulfatides and other sulfated glycans in lysosomes through autophagy-mediated degradation. FASEB J. 2020, 34, 9594–9614. [Google Scholar] [CrossRef]
- Kamijo, Y.; Hora, K.; Nakajima, T.; Kono, K.; Takahashi, K.; Ito, Y.; Higuchi, M.; Kiyosawa, K.; Shigematsu, H.; Gonzalez, F.J.; et al. Peroxisome proliferator-activated receptor alpha protects against glomerulonephritis induced by long-term exposure to the plasticizer di-(2-ethylhexyl)phthalate. J. Am. Soc. Nephrol. 2007, 18, 176–188. [Google Scholar] [CrossRef]
- Yamada, Y.; Harada, M.; Hashimoto, K.; Guo, R.; Nakajima, T.; Kashihara, T.; Yamada, M.; Aoyama, T.; Kamijo, Y. Impact of chronic kidney dysfunction on serum sulfatides and its metabolic pathway in mice. Glycoconj. J. 2019, 36, 1–11. [Google Scholar] [CrossRef]
- Shindo, Y.; Hashimoto, T. Acyl-Coenzyme A synthetase and fatty acid oxidation in rat liver peroxisomes. J. Biochem. 1978, 84, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Izai, K.; Uchida, Y.; Orii, T.; Yamamoto, S.; Hashimoto, T. Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. I. Purification and properties of very-long-chain acyl-coenzyme A dehydrogenase. J. Biol. Chem. 1992, 267, 1027–1033. [Google Scholar] [CrossRef]
- Miyazawa, S.; Furuta, S.; Osumi, T.; Hashimoto, T.; Ui, N. Properties of peroxisomal 3-ketoacyl-coA thiolase from rat liver. J. Biochem. 1981, 90, 511–519. [Google Scholar] [CrossRef]
- Furuta, S.; Miyazawa, S.; Hashimoto, T. Purification and properties of rat liver acyl-CoA dehydrogenases and electron transfer flavoprotein. J. Biochem. 1981, 90, 1739–1750. [Google Scholar] [CrossRef]
- Uchida, Y.; Izai, K.; Orii, T.; Hashimoto, T. Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. II. Purification and properties of enoyl-coenzyme A (CoA) hydratase/3-hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase trifunctional protein. J. Biol. Chem. 1992, 267, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Osumi, T.; Hashimoto, T.; Ui, N. Purification and properties of acyl-CoA oxidase from rat liver. J. Biochem. 1980, 87, 1735–1746. [Google Scholar] [CrossRef]
- Osumi, T.; Hashimoto, T. Purification and properties of mitochondrial and peroxisomal 3-hydroxyacyl-CoA dehydrogenase from rat liver. Arch. Biochem. Biophys. 1980, 203, 372–383. [Google Scholar] [CrossRef]
- Furuta, S.; Miyazawa, S.; Osumi, T.; Hashimoto, T.; Ui, N. Properties of mitochondria and peroxisomal enoyl-CoA hydratases from rat liver. J. Biochem. 1980, 88, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, S.; Ozasa, H.; Osumi, T.; Hashimoto, T. Purification and properties of carnitine octanoyltransferase and carnitine palmitoyltransferase from rat liver. J. Biochem. 1983, 94, 529–542. [Google Scholar] [CrossRef]
- Figueiredo, M.; Daryadel, A.; Sihn, G.; Muller, D.N.; Popova, E.; Rouselle, A.; Nguyen, G.; Bader, M.; Wagner, C.A. The (pro)renin receptor (ATP6ap2) facilitates receptor-mediated endocytosis and lysosomal function in the renal proximal tubule. Pflug. Arch. 2021, 473, 1229–1246. [Google Scholar] [CrossRef]
- Devuyst, O.; Luciani, A. Chloride transporters and receptor-mediated endocytosis in the renal proximal tubule. J. Physiol. 2015, 593, 4151–4164. [Google Scholar] [CrossRef]
- Melk, A.; Baisantry, A.; Schmitt, R. The yin and yang of autophagy in acute kidney injury. Autophagy 2016, 12, 596–597. [Google Scholar] [CrossRef]
- Fukushima, K.; Kitamura, S.; Tsuji, K.; Wada, J. Sodium-glucose cotransporter 2 inhibitors work as a “regulator” of autophagic activity in overnutrition diseases. Front. Pharmacol. 2021, 12, 761842. [Google Scholar] [CrossRef]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Livingston, M.J.; Dong, Z. Autophagy in acute kidney injury and repair. Nephron Clin. Pract. 2014, 127, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Peng, X.; Wang, Y.; Cao, S.; Xiong, L.; Fan, J.; Wang, Y.; Zhuang, S.; Yu, X.; Mao, H. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy 2016, 12, 1472–1486. [Google Scholar] [CrossRef]
- Ashabi, G.; Ahmadiani, A.; Abdi, A.; Abraki, S.B.; Khodagholi, F. Time course study of Abeta formation and neurite outgrowth disruption in differentiated human neuroblastoma cells exposed to H2O2: Protective role of autophagy. Toxicol. In Vitro 2013, 27, 1780–1788. [Google Scholar] [CrossRef]
- Liang, S.; Wu, Y.S.; Li, D.Y.; Tang, J.X.; Liu, H.F. Autophagy and renal fibrosis. Aging Dis. 2022, 13, 712–731. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Takabatake, Y.; Minami, S.; Sakai, S.; Fujimura, R.; Takahashi, A.; Namba-Hamano, T.; Matsuda, J.; Kimura, T.; Matsui, I.; et al. Eicosapentaenoic acid attenuates renal lipotoxicity by restoring autophagic flux. Autophagy 2021, 17, 1700–1713. [Google Scholar] [CrossRef]
- Minami, S.; Sakai, S.; Yamamoto, T.; Takabatake, Y.; Namba-Hamano, T.; Takahashi, A.; Matsuda, J.; Yonishi, H.; Nakamura, J.; Maeda, S.; et al. FGF21 and autophagy coordinately counteract kidney disease progression during aging and obesity. Autophagy 2024, 20, 489–504. [Google Scholar] [CrossRef]
- Park, H.S.; Song, J.W.; Park, J.H.; Lim, B.K.; Moon, O.S.; Son, H.Y.; Lee, J.H.; Gao, B.; Won, Y.S.; Kwon, H.J. TXNIP/VDUP1 attenuates steatohepatitis via autophagy and fatty acid oxidation. Autophagy 2021, 17, 2549–2564. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.S.; Zhang, S.F.; Luo, G.; Cheng, B.C.; Zhang, C.; Wang, Y.W.; Qiu, X.Y.; Zhou, X.H.; Wang, Q.G.; Song, X.L.; et al. Schisandrin B mitigates hepatic steatosis and promotes fatty acid oxidation by inducing autophagy through AMPK/mTOR signaling pathway. Metabolism 2022, 131, 155200. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Kuma, A.; Sugiura, Y.; Ichimura, Y.; Obata, M.; Kitamura, H.; Okuda, S.; Lee, H.C.; Ikeda, K.; Kanegae, Y.; et al. Autophagy regulates lipid metabolism through selective turnover of NCoR1. Nat. Commun. 2019, 10, 1567. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Rajak, S.; Singh, B.K.; Yen, P.M. Hepatic lipid catabolism via PPARalpha-lysosomal crosstalk. Int. J. Mol. Sci. 2020, 21, 2391. [Google Scholar] [CrossRef]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef]
- Liang, J.; Xu, C.; Xu, J.; Yang, C.; Kong, W.; Xiao, Z.; Chen, X.; Liu, Q.; Weng, Z.; Wang, J.; et al. PPARalpha senses bisphenol S to trigger EP300-mediated autophagy blockage and hepatic steatosis. Environ. Sci. Technol. 2023, 57, 21581–21592. [Google Scholar] [CrossRef]
- Molitoris, B.A.; Sandoval, R.M.; Yadav, S.P.S.; Wagner, M.C. Albumin uptake and processing by the proximal tubule: Physiological, pathological, and therapeutic implications. Physiol. Rev. 2022, 102, 1625–1667. [Google Scholar] [CrossRef]
- Hodgkins, K.S.; Schnaper, H.W. Tubulointerstitial injury and the progression of chronic kidney disease. Pediatr. Nephrol. 2012, 27, 901–909. [Google Scholar] [CrossRef]
- Ix, J.H.; Shlipak, M.G. The Promise of Tubule Biomarkers in Kidney Disease: A Review. Am. J. Kidney Dis. 2021, 78, 719–727. [Google Scholar] [CrossRef]
- Koh, E.S.; Kim, G.H.; Chung, S. Intrarenal Mechanisms of Sodium-Glucose Cotransporter-2 Inhibitors on Tubuloglomerular Feedback and Natriuresis. Endocrinol. Metab. 2023, 38, 359–372. [Google Scholar] [CrossRef]
- Koh, H.B.; Kim, H.W.; Joo, Y.S.; Jung, C.Y.; Kim, H.J.; Chang, T.I.; Park, J.T.; Yoo, T.H.; Kang, S.W.; Han, S.H. Plasma levels of polyunsaturated fatty acids and adverse kidney outcomes. Am. J. Kidney Dis. 2024, 84, 179–194.e1. [Google Scholar] [CrossRef] [PubMed]
- Syren, M.L.; Turolo, S.; Marangoni, F.; Milani, G.P.; Edefonti, A.; Montini, G.; Agostoni, C. The polyunsaturated fatty acid balance in kidney health and disease: A review. Clin. Nutr. 2018, 37, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Favor, O.K.; Chauhan, P.S.; Pourmand, E.; Edwards, A.M.; Wagner, J.G.; Lewandowski, R.P.; Heine, L.K.; Harkema, J.R.; Lee, K.S.S.; Pestka, J.J. Lipidome modulation by dietary omega-3 polyunsaturated fatty acid supplementation or selective soluble epoxide hydrolase inhibition suppresses rough LPS-accelerated glomerulonephritis in lupus-prone mice. Front. Immunol. 2023, 14, 1124910. [Google Scholar] [CrossRef]
- Han, S.; Choi, H.; Park, H.; Kim, J.J.; Lee, E.J.; Ham, Y.R.; Na, K.R.; Lee, K.W.; Chang, Y.K.; Choi, D.E. Omega-3 fatty acids attenuate renal fibrosis via AMPK-mediated autophagy flux activation. Biomedicines 2023, 11, 2553. [Google Scholar] [CrossRef]
- Huang, X.; Lindholm, B.; Stenvinkel, P.; Carrero, J.J. Dietary fat modification in patients with chronic kidney disease: N-3 fatty acids and beyond. J. Nephrol. 2013, 26, 960–974. [Google Scholar] [CrossRef]
- Moriyama, T.; Kumon, S.; Kamiyama, T.; Karasawa, K.; Uchida, K.; Nitta, K. The renoprotective effects of docosahexaenoic acid as an add-on therapy in patients receiving eicosapentaenoic acid as treatment for IgA nephropathy: A pilot uncontrolled trial. Intern. Med. 2018, 57, 173–179. [Google Scholar] [CrossRef]
- Roveta, A.; Parodi, E.L.; Brezzi, B.; Tunesi, F.; Zanetti, V.; Merlotti, G.; Francese, A.; Maconi, A.G.; Quaglia, M. Lupus nephritis from pathogenesis to new therapies: An Update. Int. J. Mol. Sci. 2024, 25, 8981. [Google Scholar] [CrossRef] [PubMed]
- Kochan, Z.; Szupryczynska, N.; Malgorzewicz, S.; Karbowska, J. Dietary lipids and dyslipidemia in chronic kidney disease. Nutrients 2021, 13, 3138. [Google Scholar] [CrossRef]
- Schulze, M.B.; Minihane, A.M.; Saleh, R.N.M.; Riserus, U. Intake and metabolism of omega-3 and omega-6 polyunsaturated fatty acids: Nutritional implications for cardiometabolic diseases. Lancet Diabetes Endocrinol. 2020, 8, 915–930. [Google Scholar] [CrossRef]
- Djuricic, I.; Calder, P.C. Pros andcons of long-chain Omega-3 polyunsaturated fatty acids in cardiovascular health. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 383–406. [Google Scholar] [CrossRef]
- Park, S.; Lee, J.J.; Lee, J.; Lee, J.K.; Byun, J.; Kim, I.; Ha, J.H. Lowering n-6/n-3 ratio as an important dietary intervention to prevent LPS-inducible dyslipidemia and hepatic abnormalities in ob/ob mice. Int. J. Mol. Sci. 2022, 23, 6384. [Google Scholar] [CrossRef] [PubMed]
- Omer, E.; Chiodi, C. Fat digestion and absorption: Normal physiology and pathophysiology of malabsorption, including diagnostic testing. Nutr. Clin. Pract. 2024, 39 (Suppl. S1), S6–S16. [Google Scholar] [CrossRef]
- Akombi, B.J.; Agho, K.E.; Merom, D.; Renzaho, A.M.; Hall, J.J. Child malnutrition in sub-Saharan Africa: A meta-analysis of demographic and health surveys (2006–2016). PLoS ONE 2017, 12, e0177338. [Google Scholar] [CrossRef]
- Carroll, G.J.; Lama, S.D.; Martinez-Brockman, J.L.; Perez-Escamilla, R. Evaluation of nutrition interventions in children in conflict zones: A narrative review. Adv. Nutr. 2017, 8, 770–779. [Google Scholar] [CrossRef]
- Mani, I.; Kurpad, A.V. Fats & fatty acids in Indian diets: Time for serious introspection. Indian J. Med. Res. 2016, 144, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Arakawa, H.; Tamai, I. Uric acid in health and disease: From physiological functions to pathogenic mechanisms. Pharmacol. Ther. 2024, 256, 108615. [Google Scholar] [CrossRef]
- Ahmad, A.A.; Draves, S.O.; Rosca, M. Mitochondria in diabetic kidney disease. Cells 2021, 10, 2945. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Calcia, T.B.B.; Silva, C.P.; Guilherme, R.F.; Almeida-Souza, F.; Lemos, F.S.; Calabrese, K.S.; Caruso-Neves, C.; Neves, J.S.; Benjamim, C.F. ATRvD1 attenuates renal tubulointerstitial injury induced by albumin overload in sepsis-surviving mice. Int. J. Mol. Sci. 2021, 22, 11634. [Google Scholar] [CrossRef]
- Wang, Z.; You, L.; Ren, Y.; Zhu, X.; Mao, X.; Liang, X.; Wang, T.; Guo, Y.; Liu, T.; Xue, J. Finasteride alleviates high fat associated protein-overload nephropathy by inhibiting trimethylamine n-oxide synthesis and regulating gut microbiota. Front. Physiol. 2022, 13, 900961. [Google Scholar] [CrossRef]
- Zhang, X.; Nakajima, T.; Kamijo, Y.; Li, G.; Hu, R.; Kannagi, R.; Kyogashima, M.; Aoyama, T.; Hara, A. Acute kidney injury induced by protein-overload nephropathy down-regulates gene expression of hepatic cerebroside sulfotransferase in mice, resulting in reduction of liver and serum sulfatides. Biochem. Biophys. Res. Commun. 2009, 390, 1382–1388. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Primer Sequence (5′ to 3′) |
|---|---|---|
| Acadvl (VLCAD) | NM_017366 | F GCGTGTGCTCCGAGATATTC |
| R CCAGTGAGTTCCTTTCCTTTG | ||
| CPT II | NM_009949 | F ATCGTACCCACCATGCACTAC |
| R CTGTCATTCAAGAGAGGCTTCTG | ||
| Acaa1 (PT) | NM_130864 | F TCTACGGTCAACAGACAGTGTTCA |
| R GGCCATGCCAATGTCATAAGA | ||
| PH | NM_023737 | F CGATACTCTTCCCCCACTACCA |
| R CAGTTACCAACAACGACTCCAATC | ||
| HADHA (TPα) | NM_178878 | F CCTTTATCCTGCCCCTTTG |
| R GCGATTCAGCAAGATAACCA | ||
| HADHB (TPβ) | NM_145558 | F AGCGCCTGTCCTTACTCAGT |
| R CATGGTCTCATTAGTGGAGAACTC | ||
| Lamp1 | NM_010684 | F ACACTGCACACAGGATGGAC |
| R CTCTGGTCACCGTCTTGTTGT | ||
| Gusb (β-glucuronidase) | NM_010368 | F GCAGCCCTTCGGGACTTTAT |
| R CCCATTCACCCACACAACTG | ||
| Becn1 (beclin 1) | NM_019584 | F GGCTAACTCAGGAGAGGAGC |
| R ATCAGATGCCTCCCCGATCA | ||
| GAPDH | M32599 | F TGCACCACCAACTGCTTAG |
| R GGATGCAGGGATGATGTTCTG |
| Body Weight (g) | Body Weight Change (%) | Kidney/Body Weight (%) | Liver/Body Weight (%) | |
|---|---|---|---|---|
| Control | 25.1 ± 0.8 | nd | 1.3 ± 0.1 | 4.0 ± 0.4 |
| PUFA (+) | 27.8 ± 0.3 | 109.7 ± 3.5 | 1.4 ± 0.1 | 4.4 ± 0.6 |
| PUFA (−) | 26.6 ± 1.7 | 105.1 ± 9.2 | 1.3 ± 0.1 | 4.6 ± 0.3 |
| 1w PON Control | 27.5 ± 1.6 | 125.6 ± 10.0 | 1.4 ± 0.1 | 4.6 ± 0.1 |
| 1w PON PUFA (+) | 26.5 ± 1.5 | 121.1 ± 10.0 | 1.5 ± 0.2 | 4.3 ± 0.8 |
| 1w PON PUFA (−) | 28.0 ± 0.7 | 128.0 ± 8.4 | 1.4 ± 0.1 | 4.2 ± 0.3 |
| 2w PON Control | 26.9 ± 1.0 | 123.2 ± 10.7 | 1.3 ± 0.1 | 4.4 ± 0.2 |
| 2w PON PUFA (+) | 27.1 ± 1.0 | 124.0 ± 10.1 | 1.5 ± 0.1 | 4.4 ± 0.5 |
| 2w PON PUFA (−) | 27.0 ± 1.1 | 123.2 ± 5.8 | 1.5 ± 0.2 | 4.5 ± 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Diao, P.; Aomura, D.; Nimura, T.; Harada, M.; Jia, F.; Nakajima, T.; Tanaka, N.; Kamijo, Y. Dietary Polyunsaturated Fatty Acid Deficiency Impairs Renal Lipid Metabolism and Adaptive Response to Proteinuria in Murine Renal Tubules. Nutrients 2025, 17, 961. https://doi.org/10.3390/nu17060961
Wang Y, Diao P, Aomura D, Nimura T, Harada M, Jia F, Nakajima T, Tanaka N, Kamijo Y. Dietary Polyunsaturated Fatty Acid Deficiency Impairs Renal Lipid Metabolism and Adaptive Response to Proteinuria in Murine Renal Tubules. Nutrients. 2025; 17(6):961. https://doi.org/10.3390/nu17060961
Chicago/Turabian StyleWang, Yaping, Pan Diao, Daiki Aomura, Takayuki Nimura, Makoto Harada, Fangping Jia, Takero Nakajima, Naoki Tanaka, and Yuji Kamijo. 2025. "Dietary Polyunsaturated Fatty Acid Deficiency Impairs Renal Lipid Metabolism and Adaptive Response to Proteinuria in Murine Renal Tubules" Nutrients 17, no. 6: 961. https://doi.org/10.3390/nu17060961
APA StyleWang, Y., Diao, P., Aomura, D., Nimura, T., Harada, M., Jia, F., Nakajima, T., Tanaka, N., & Kamijo, Y. (2025). Dietary Polyunsaturated Fatty Acid Deficiency Impairs Renal Lipid Metabolism and Adaptive Response to Proteinuria in Murine Renal Tubules. Nutrients, 17(6), 961. https://doi.org/10.3390/nu17060961