The Effect of Fatty Acid Desaturase on Cardiovascular Lipid Biomarkers Depends on Circulating ω-3 and ω-6 Polyunsaturated Fatty Acids in the UK Biobank


Abstract
1. Introduction
2. Methods
2.1. UK Biobank Dataset
2.2. Variables
2.3. Statistical Analyses
3. Results
3.1. Sample Characteristics
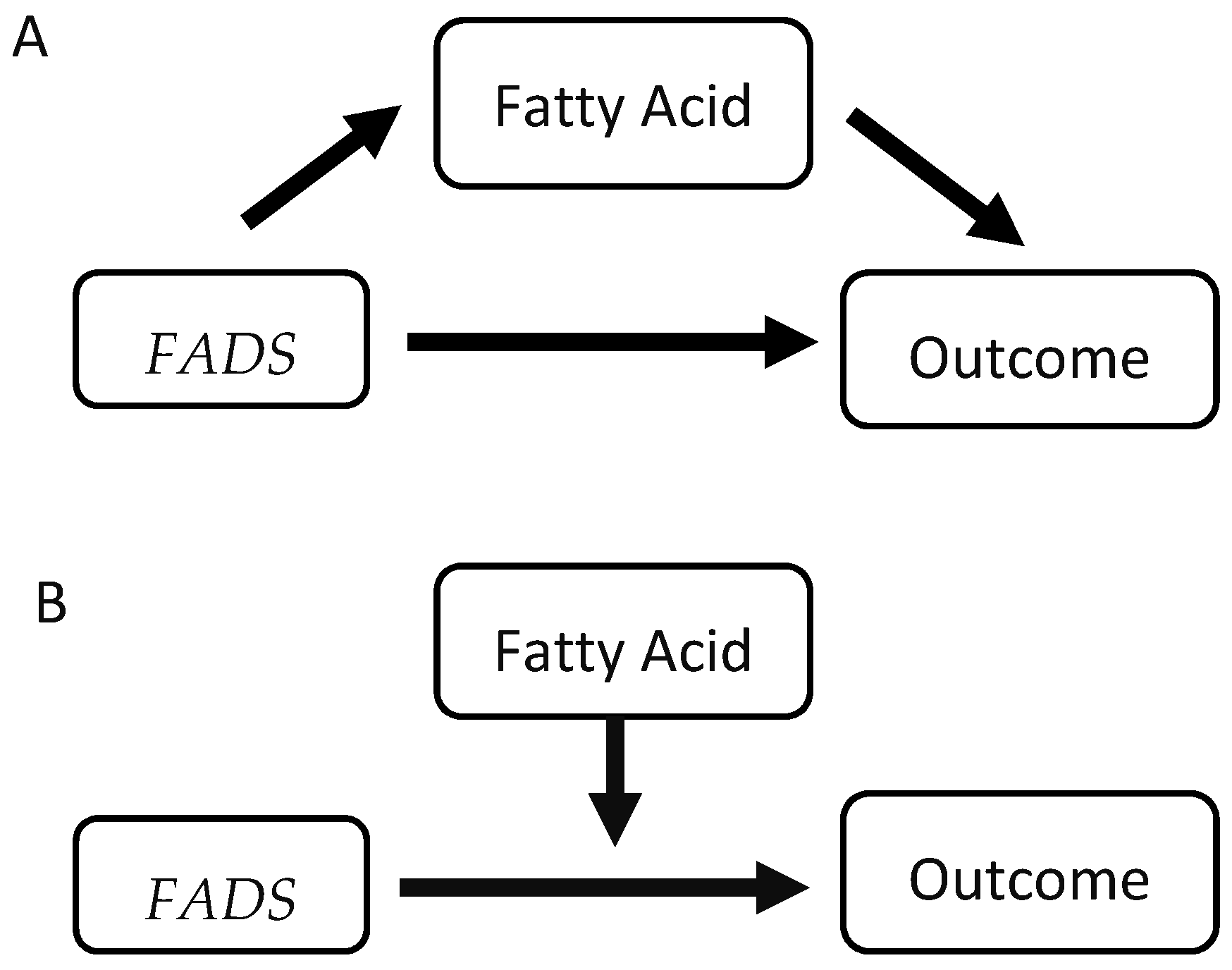
3.2. Components of Mediation and Moderation Models
3.3. Moderation
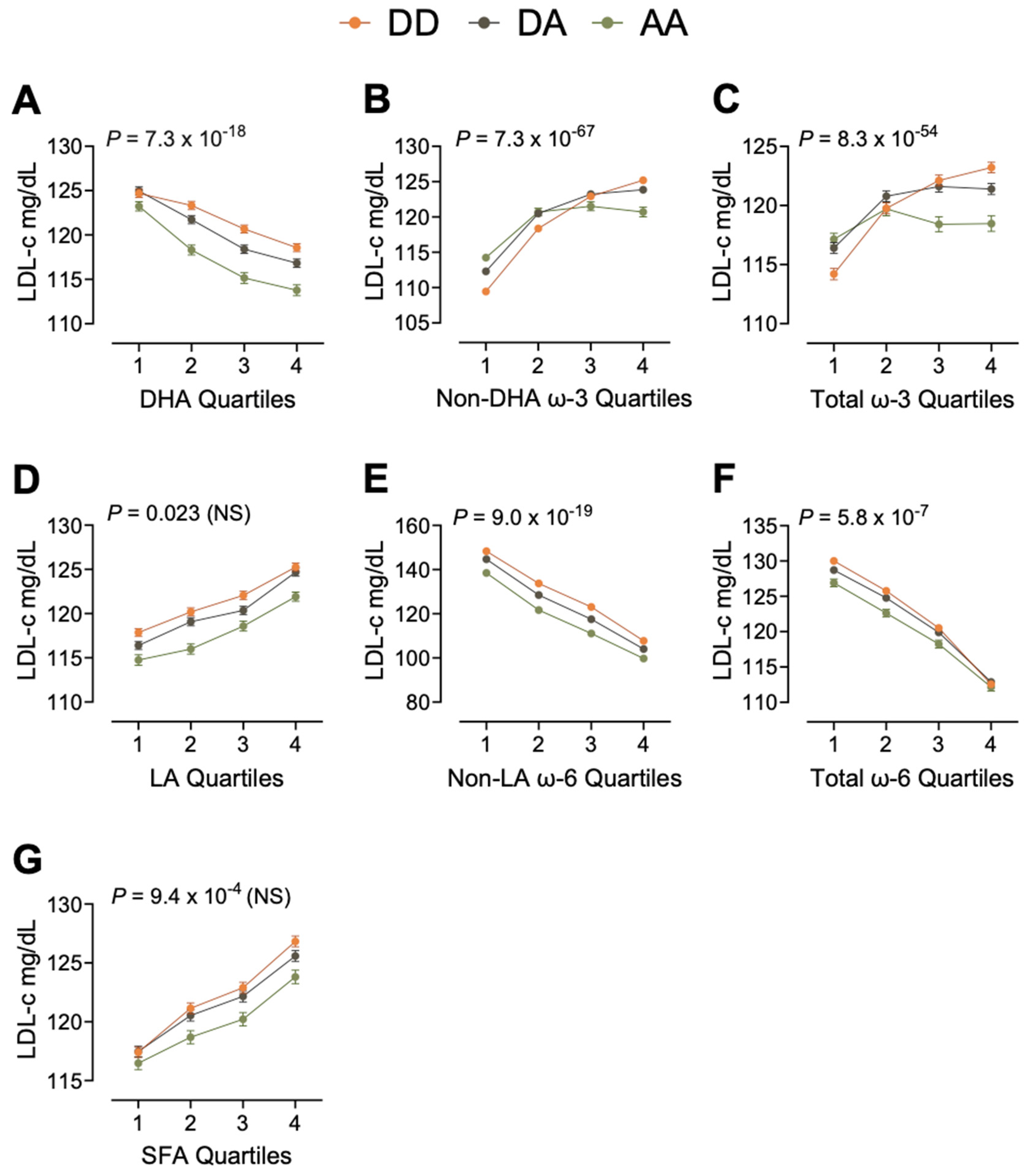
3.4. Example 1: Moderation of LDL-c
3.5. Mediation/Suppression
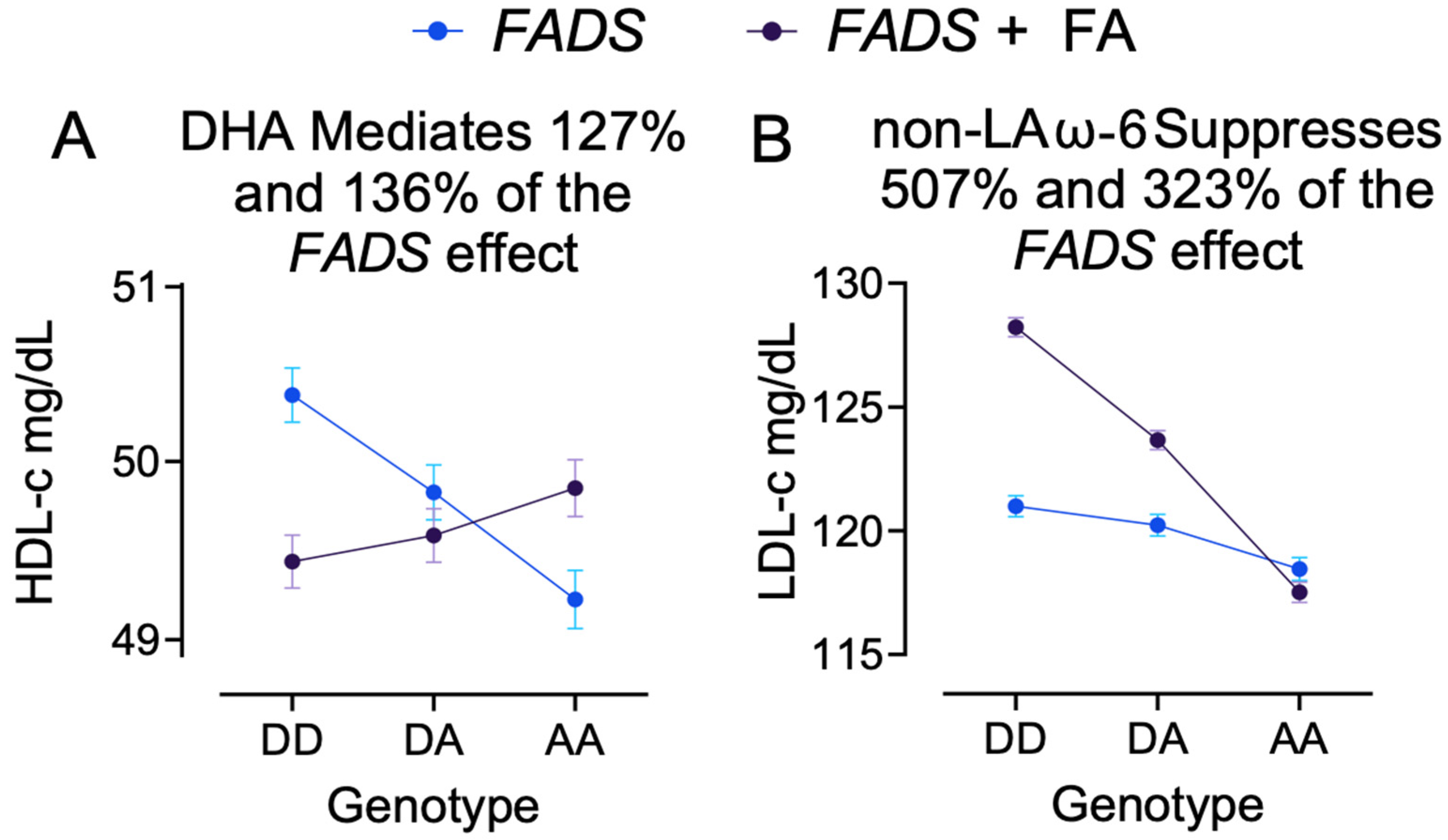
3.6. Example 2: DHA Mediates FADS Effect on HDL-c
3.7. Example 3: Non-LA ω-6 PUFA Suppresses FADS Effect on LDL-c
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AA | arachidonic acid |
| ALA | alpha-linolenic acid |
| ANOVA | analysis of variance |
| BMI | body mass index |
| CI | confidence interval |
| CVD | cardiovascular disease |
| DHA | docosahexaenoic acid |
| DPA | docosapentaenoic acid |
| EDTA | ethylenediaminetetraacetic acid |
| EPA | eicosapentaenoic acid |
| FA | fatty acid |
| FADS | fatty acid desaturase |
| GISSI | Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico |
| HDL-c | high-density lipoprotein cholesterol |
| JELIS | Japan EPA Lipid Intervention Study |
| LA | linoleic acid |
| LD | linkage disequilibrium |
| LDL-c | low-density lipoprotein cholesterol |
| NMR | nuclear magnetic resonance |
| ω-3 | omega−3 |
| ω-6 | omega−6 |
| PSU | Pennsylvania State University |
| PUFA | polyunsaturated fatty acid |
| SFA | saturated fatty acid |
| SNP | single-nucleotide polymorphism |
| TC | total cholesterol |
| TDI | Townsend’s Deprivation Index |
| TG | total triglycerides |
| UKB | United Kingdom Biobank |
References
- Papackova, Z.; Cahova, M. Fatty acid signaling: The new function of intracellular lipases. Int. J. Mol. Sci. 2015, 16, 3831–3855. [Google Scholar] [CrossRef] [PubMed]
- Eyster, K.M. The membrane and lipids as integral participants in signal transduction: Lipid signal transduction for the non-lipid biochemist. Adv. Physiol. Educ. 2007, 31, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Stillwell, W.; Jenski, L.J.; Crump, F.T.; Ehringer, W. Effect of docosahexaenoic acid on mouse mitochondrial membrane properties. Lipids 1997, 32, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in Our Understanding of Oxylipins Derived from Dietary PUFAs. Adv. Nutr. 2015, 6, 513–540. [Google Scholar] [CrossRef]
- O’Keefe, J.H., Jr.; Harris, W.S. From Inuit to implementation: Omega-3 fatty acids come of age. Mayo Clin. Proc. 2000, 75, 607–614. [Google Scholar] [CrossRef]
- Papanikolaou, Y.; Brooks, J.; Reider, C.; Fulgoni, V.L., 3rd. U.S. adults are not meeting recommended levels for fish and omega-3 fatty acid intake: Results of an analysis using observational data from NHANES 2003–2008. Nutr. J. 2014, 13, 31. [Google Scholar] [CrossRef]
- Djousse, L.; Akinkuolie, A.O.; Wu, J.H.; Ding, E.L.; Gaziano, J.M. Fish consumption, omega-3 fatty acids and risk of heart failure: A meta-analysis. Clin. Nutr. 2012, 31, 846–853. [Google Scholar] [CrossRef]
- Harris, W.S.; Isley, W.L. Clinical trial evidence for the cardioprotective effects of omega-3 fatty acids. Curr. Atheroscler. Rep. 2001, 3, 174–179. [Google Scholar] [CrossRef]
- Kris-Etherton, P.M.; Richter, C.K.; Bowen, K.J.; Skulas-Ray, A.C.; Jackson, K.H.; Petersen, K.S.; Harris, W.S. Recent Clinical Trials Shed New Light on the Cardiovascular Benefits of Omega-3 Fatty Acids. Methodist. Debakey Cardiovasc. J. 2019, 15, 171–178. [Google Scholar] [CrossRef]
- Rizos, E.C.; Elisaf, M.S. Does Supplementation with Omega-3 PUFAs Add to the Prevention of Cardiovascular Disease? Curr. Cardiol. Rep. 2017, 19, 47. [Google Scholar] [CrossRef]
- Maki, K.C.; Palacios, O.M.; Bell, M.; Toth, P.P. Use of supplemental long-chain omega-3 fatty acids and risk for cardiac death: An updated meta-analysis and review of research gaps. J. Clin. Lipidol. 2017, 11, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Qi, S.; Ji, Y.; Wang, X.; Fang, S.; Cao, R. Effects of omega-3 fatty acid on major cardiovascular outcomes: A systematic review and meta-analysis. Medicine 2022, 101, e29556. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Calviello, G. Omega-3 PUFA Responders and Non-Responders and the Prevention of Lipid Dysmetabolism and Related Diseases. Nutrients 2020, 12, 1363. [Google Scholar] [CrossRef] [PubMed]
- Minihane, A.M. Impact of Genotype on EPA and DHA Status and Responsiveness to Increased Intakes. Nutrients 2016, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Brayner, B.; Kaur, G.; Keske, M.A.; Livingstone, K.M. FADS Polymorphism, Omega-3 Fatty Acids and Diabetes Risk: A Systematic Review. Nutrients 2018, 10, 758. [Google Scholar] [CrossRef]
- Tanaka, T.; Shen, J.; Abecasis, G.R.; Kisialiou, A.; Ordovas, J.M.; Guralnik, J.M.; Singleton, A.; Bandinelli, S.; Cherubini, A.; Arnett, D.; et al. Genome-wide association study of plasma polyunsaturated fatty acids in the InCHIANTI Study. PLoS Genet. 2009, 5, e1000338. [Google Scholar] [CrossRef]
- Westra, J.; Annevelink, C.; Orchard, T.; Hou, L.; Harris, W.S.; O’Connell, T.D.; Shearer, G.; Tintle, N. Genome-wide association study of Red Blood Cell fatty acids in the Women’s Health Initiative Memory Study. Prostaglandins Leukot. Essent. Fatty Acids 2023, 194, 102577. [Google Scholar] [CrossRef]
- Tintle, N.L.; Pottala, J.V.; Lacey, S.; Ramachandran, V.; Westra, J.; Rogers, A.; Clark, J.; Olthoff, B.; Larson, M.; Harris, W.; et al. A genome-wide association study of saturated, mono- and polyunsaturated red blood cell fatty acids in the Framingham Heart Offspring Study. Prostaglandins Leukot. Essent. Fatty Acids 2015, 94, 65–72. [Google Scholar] [CrossRef]
- Francis, M.S.Y.; Xu, H.; Brenna, J.T.; Ye, K. Fifty-one novel and replicated GWAS loci for polyunsaturated and monounsaturated fatty acids in 124,024 Europeans. medRxiv 2022. [Google Scholar] [CrossRef]
- Lemaitre, R.N.; Tanaka, T.; Tang, W.; Manichaikul, A.; Foy, M.; Kabagambe, E.K.; Nettleton, J.A.; King, I.B.; Weng, L.C.; Bhattacharya, S.; et al. Genetic loci associated with plasma phospholipid n-3 fatty acids: A meta-analysis of genome-wide association studies from the CHARGE Consortium. PLoS Genet. 2011, 7, e1002193. [Google Scholar] [CrossRef]
- Hu, Y.; Li, H.; Lu, L.; Manichaikul, A.; Zhu, J.; Chen, Y.D.; Sun, L.; Liang, S.; Siscovick, D.S.; Steffen, L.M.; et al. Genome-wide meta-analyses identify novel loci associated with n-3 and n-6 polyunsaturated fatty acid levels in Chinese and European-ancestry populations. Hum. Mol. Genet. 2016, 25, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Veenstra, J.; Bartz, T.M.; Pahl, M.C.; Hallmark, B.; Chen, Y.I.; Westra, J.; Steffen, L.M.; Brown, C.D.; Siscovick, D.; et al. Genome-wide association studies and fine-mapping identify genomic loci for n-3 and n-6 polyunsaturated fatty acids in Hispanic American and African American cohorts. Commun. Biol. 2023, 6, 852. [Google Scholar] [CrossRef] [PubMed]
- Coltell, O.; Sorli, J.V.; Asensio, E.M.; Barragan, R.; Gonzalez, J.I.; Gimenez-Alba, I.M.; Zanon-Moreno, V.; Estruch, R.; Ramirez-Sabio, J.B.; Pascual, E.C.; et al. Genome-Wide Association Study for Serum Omega-3 and Omega-6 Polyunsaturated Fatty Acids: Exploratory Analysis of the Sex-Specific Effects and Dietary Modulation in Mediterranean Subjects with Metabolic Syndrome. Nutrients 2020, 12, 310. [Google Scholar] [CrossRef]
- Veenstra, J.; Kalsbeek, A.; Westra, J.; Disselkoen, C.; Smith, C.; Tintle, N. Genome-Wide Interaction Study of Omega-3 PUFAs and Other Fatty Acids on Inflammatory Biomarkers of Cardiovascular Health in the Framingham Heart Study. Nutrients 2017, 9, 900. [Google Scholar] [CrossRef]
- Hellstrand, S.; Ericson, U.; Gullberg, B.; Hedblad, B.; Orho-Melander, M.; Sonestedt, E. Genetic variation in FADS1 has little effect on the association between dietary PUFA intake and cardiovascular disease. J. Nutr. 2014, 144, 1356–1363. [Google Scholar] [CrossRef]
- Chen, H.Y.; Cairns, B.J.; Small, A.M.; Burr, H.A.; Ambikkumar, A.; Martinsson, A.; Theriault, S.; Munter, H.M.; Steffen, B.; Zhang, R.; et al. Association of FADS1/2 Locus Variants and Polyunsaturated Fatty Acids With Aortic Stenosis. JAMA Cardiol. 2020, 5, 694–702. [Google Scholar] [CrossRef]
- Chaaba, R.; Bouaziz, A.; Ben Amor, A.; Mnif, W.; Hammami, M.; Mehri, S. Fatty Acid Profile and Genetic Variants of Proteins Involved in Fatty Acid Metabolism Could Be Considered as Disease Predictor. Diagnostics 2023, 13, 979. [Google Scholar] [CrossRef]
- Krus, D.J.; Wilkinson, S.M. Demonstration of properties of a suppressor variable. Behav. Res. Methods Instrum. Comput. 1986, 18, 21–24. [Google Scholar] [CrossRef]
- Ameur, A.; Enroth, S.; Johansson, A.; Zaboli, G.; Igl, W.; Johansson, A.C.; Rivas, M.A.; Daly, M.J.; Schmitz, G.; Hicks, A.A.; et al. Genetic adaptation of fatty-acid metabolism: A human-specific haplotype increasing the biosynthesis of long-chain omega-3 and omega-6 fatty acids. Am. J. Hum. Genet. 2012, 90, 809–820. [Google Scholar] [CrossRef]
- Zak, A.; Jachymova, M.; Burda, M.; Stankova, B.; Zeman, M.; Slaby, A.; Vecka, M.; Seda, O. FADS Polymorphisms Affect the Clinical and Biochemical Phenotypes of Metabolic Syndrome. Metabolites 2022, 12, 568. [Google Scholar] [CrossRef]
- Schaeffer, L.; Gohlke, H.; Muller, M.; Heid, I.M.; Palmer, L.J.; Kompauer, I.; Demmelmair, H.; Illig, T.; Koletzko, B.; Heinrich, J. Common genetic variants of the FADS1 FADS2 gene cluster and their reconstructed haplotypes are associated with the fatty acid composition in phospholipids. Hum. Mol. Genet. 2006, 15, 1745–1756. [Google Scholar] [CrossRef] [PubMed]
- Chilton, F.H.; Manichaikul, A.; Yang, C.; O’Connor, T.D.; Johnstone, L.M.; Blomquist, S.; Schembre, S.M.; Sergeant, S.; Zec, M.; Tsai, M.Y.; et al. Interpreting Clinical Trials With Omega-3 Supplements in the Context of Ancestry and FADS Genetic Variation. Front. Nutr. 2021, 8, 808054. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.N.; Ruczinski, I.; Yanek, L.R.; Becker, L.C.; Becker, D.M.; Guio, H.; Cui, T.; Chilton, F.H.; Mathias, R.A.; O’Connor, T.D. Evolution of Hominin Polyunsaturated Fatty Acid Metabolism: From Africa to the New World. Genome Biol. Evol. 2019, 11, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.M.; Minihane, A.M. The impact of fatty acid desaturase genotype on fatty acid status and cardiovascular health in adults. Proc. Nutr. Soc. 2017, 76, 64–75. [Google Scholar] [CrossRef]
- Dumitrescu, L.; Carty, C.L.; Franceschini, N.; Hindorff, L.A.; Cole, S.A.; Buzkova, P.; Schumacher, F.R.; Eaton, C.B.; Goodloe, R.J.; Duggan, D.J.; et al. Post-genome-wide association study challenges for lipid traits: Describing age as a modifier of gene-lipid associations in the Population Architecture using Genomics and Epidemiology (PAGE) study. Ann. Hum. Genet. 2013, 77, 416–425. [Google Scholar] [CrossRef]
- Lv, X.; Zhang, Y.; Rao, S.; Qiu, J.; Wang, M.; Luo, X.; Zuo, X.; Su, D.; Feng, X.; Yang, Y.; et al. Joint effects of genetic variants in multiple loci on the risk of coronary artery disease in Chinese Han subjects. Circ. J. 2012, 76, 1987–1992. [Google Scholar] [CrossRef]
- Muzsik, A.; Bajerska, J.; Jelen, H.H.; Gaca, A.; Chmurzynska, A. Associations between Fatty Acid Intake and Status, Desaturase Activities, and FADS Gene Polymorphism in Centrally Obese Postmenopausal Polish Women. Nutrients 2018, 10, 1068. [Google Scholar] [CrossRef]
- Hovsepian, S.; Javanmard, S.H.; Mansourian, M.; Hashemipour, M.; Tajadini, M.; Kelishadi, R. Lipid regulatory genes polymorphism in children with and without obesity and cardiometabolic risk factors: The CASPIAN-III study. J. Res. Med. Sci. 2018, 23, 11. [Google Scholar] [CrossRef]
- Vaittinen, M.; Lankinen, M.A.; Kakela, P.; Agren, J.; Wheelock, C.E.; Laakso, M.; Schwab, U.; Pihlajamaki, J. The FADS1 genotypes modify the effect of linoleic acid-enriched diet on adipose tissue inflammation via pro-inflammatory eicosanoid metabolism. Eur. J. Nutr. 2022, 61, 3707–3718. [Google Scholar] [CrossRef]
- Zhuang, P.; Liu, X.; Li, Y.; Wu, Y.; Li, H.; Wan, X.; Zhang, L.; Xu, C.; Jiao, J.; Zhang, Y. Circulating fatty acids, genetic risk, and incident coronary artery disease: A prospective, longitudinal cohort study. Sci. Adv. 2023, 9, eadf9037. [Google Scholar] [CrossRef]
- Lu, Y.; Vaarhorst, A.; Merry, A.H.; Dolle, M.E.; Hovenier, R.; Imholz, S.; Schouten, L.J.; Heijmans, B.T.; Muller, M.; Slagboom, P.E.; et al. Markers of endogenous desaturase activity and risk of coronary heart disease in the CAREMA cohort study. PLoS ONE 2012, 7, e41681. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Back, M.; Bruzelius, M.; Mason, A.M.; Burgess, S.; Larsson, S. Plasma Phospholipid Fatty Acids, FADS1 and Risk of 15 Cardiovascular Diseases: A Mendelian Randomisation Study. Nutrients 2019, 11, 3001. [Google Scholar] [CrossRef]
- Park, S.; Kim, D.S.; Kang, S. Carrying minor allele of FADS1 and haplotype of FADS1 and FADS2 increased the risk of metabolic syndrome and moderate but not low fat diets lowered the risk in two Korean cohorts. Eur. J. Nutr. 2019, 58, 831–842. [Google Scholar] [CrossRef]
- Hellstrand, S.; Sonestedt, E.; Ericson, U.; Gullberg, B.; Wirfalt, E.; Hedblad, B.; Orho-Melander, M. Intake levels of dietary long-chain PUFAs modify the association between genetic variation in FADS and LDL-C. J. Lipid Res. 2012, 53, 1183–1189. [Google Scholar] [CrossRef]
- Hermant, X.; Delay, C.; Flaig, A.; Luque-Bedregal, J.; Briand, G.; Bout, M.A.; Cottel, D.; Wagner, A.; Arveiler, D.; Simon, C.; et al. Identification of a functional FADS1 3’UTR variant associated with erythrocyte n-6 polyunsaturated fatty acids levels. J. Clin. Lipidol. 2018, 12, 1280–1289. [Google Scholar] [CrossRef]
- Hovsepian, S.; Javanmard, S.H.; Mansourian, M.; Tajadini, M.; Hashemipour, M.; Kelishadi, R. Relationship of lipid regulatory gene polymorphisms and dyslipidemia in a pediatric population: The CASPIAN III study. Hormones 2018, 17, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Ching, Y.K.; Chin, Y.S.; Appukutty, M.; Chan, Y.M.; Lim, P.Y.; Nasir, K.H. Interactions of genetic and macronutrient intake with abdominal obesity among middle-aged vegetarians in Malaysia. Asia Pac. J. Clin. Nutr. 2023, 32, 168–182. [Google Scholar] [CrossRef]
- Liu, S.J.; Zhi, H.; Chen, P.Z.; Chen, W.; Lu, F.; Ma, G.S.; Dai, J.C.; Shen, C.; Liu, N.F.; Hu, Z.B.; et al. Fatty acid desaturase 1 polymorphisms are associated with coronary heart disease in a Chinese population. Chin. Med. J. 2012, 125, 801–806. [Google Scholar]
- Huang, M.C.; Chang, W.T.; Chang, H.Y.; Chung, H.F.; Chen, F.P.; Huang, Y.F.; Hsu, C.C.; Hwang, S.J. FADS Gene Polymorphisms, Fatty Acid Desaturase Activities, and HDL-C in Type 2 Diabetes. Int. J. Environ. Res. Public Health 2017, 14, 572. [Google Scholar] [CrossRef]
- Murphy, C.; Johnson, A.P.; Koenekoop, R.K.; Seiple, W.; Overbury, O. The Relationship Between Cognitive Status and Known Single Nucleotide Polymorphisms in Age-Related Macular Degeneration. Front. Aging Neurosci. 2020, 12, 586691. [Google Scholar] [CrossRef]
- Plunde, O.; Larsson, S.C.; Artiach, G.; Thanassoulis, G.; Carracedo, M.; Franco-Cereceda, A.; Eriksson, P.; Back, M. FADS1 (Fatty Acid Desaturase 1) Genotype Associates With Aortic Valve FADS mRNA Expression, Fatty Acid Content and Calcification. Circ. Genom. Precis. Med. 2020, 13, e002710. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, Z.; Lv, X.; Ma, J. Dietary n-3 polyunsaturated fatty acid intakes modify the effect of genetic variation in fatty acid desaturase 1 on coronary artery disease. PLoS ONE 2015, 10, e0121255. [Google Scholar] [CrossRef] [PubMed]
- Ching, Y.K.; Chin, Y.S.; Appukutty, M.; Ramanchadran, V.; Yu, C.Y.; Ang, G.Y.; Gan, W.Y.; Chan, Y.M.; Teh, L.K.; Salleh, M.Z. Interaction of Dietary Linoleic Acid and alpha-Linolenic Acids with rs174547 in FADS1 Gene on Metabolic Syndrome Components among Vegetarians. Nutrients 2019, 11, 1686. [Google Scholar] [CrossRef]
- Dumont, J.; Goumidi, L.; Grenier-Boley, B.; Cottel, D.; Marecaux, N.; Montaye, M.; Wagner, A.; Arveiler, D.; Simon, C.; Ferrieres, J.; et al. Dietary linoleic acid interacts with FADS1 genetic variability to modulate HDL-cholesterol and obesity-related traits. Clin. Nutr. 2018, 37, 1683–1689. [Google Scholar] [CrossRef]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M.; et al. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef]
- Baron, R.M.; Kenny, D.A. The moderator-mediator variable distinction in social psychological research: Conceptual, strategic, and statistical considerations. J. Pers. Soc. Psychol. 1986, 51, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.; Matute, H. Positive and negative mediation as a function of whether the absent cue was previously associated with the outcome. Q. J. Exp. Psychol. 2010, 63, 2359–2375. [Google Scholar] [CrossRef]
- Mensink, R.P.; Temme, E.H.; Hornstra, G. Dietary saturated and trans fatty acids and lipoprotein metabolism. Ann. Med. 1994, 26, 461–464. [Google Scholar] [CrossRef]
- Rosenberg, I.H.; Schaefer, E.J. Dietary saturated fatty acids and blood cholesterol. N. Engl. J. Med. 1988, 318, 1270–1271. [Google Scholar] [CrossRef]
- Ren, X.L.; Liu, Y.; Chu, W.J.; Li, Z.W.; Zhang, S.S.; Zhou, Z.L.; Tang, J.; Yang, B. Blood levels of omega-6 fatty acids and coronary heart disease: A systematic review and metaanalysis of observational epidemiology. Crit. Rev. Food Sci. Nutr. 2022, 63, 7983–7995. [Google Scholar] [CrossRef]
- Mensink, R.P.; Zock, P.L.; Kester, A.D.; Katan, M.B. Effects of dietary fatty acids and carbohydrates on the ratio of serum total to HDL cholesterol and on serum lipids and apolipoproteins: A meta-analysis of 60 controlled trials. Am. J. Clin. Nutr. 2003, 77, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Innes, J.K.; Calder, P.C. The Differential Effects of Eicosapentaenoic Acid and Docosahexaenoic Acid on Cardiometabolic Risk Factors: A Systematic Review. Int. J. Mol. Sci. 2018, 19, 532. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, A.M.; Ding, E.L.; Willett, W.C.; Rimm, E.B. A meta-analysis shows that docosahexaenoic acid from algal oil reduces serum triglycerides and increases HDL-cholesterol and LDL-cholesterol in persons without coronary heart disease. J. Nutr. 2012, 142, 99–104. [Google Scholar] [CrossRef]
- Dumont, J.; Huybrechts, I.; Spinneker, A.; Gottrand, F.; Grammatikaki, E.; Bevilacqua, N.; Vyncke, K.; Widhalm, K.; Kafatos, A.; Molnar, D.; et al. FADS1 genetic variability interacts with dietary alpha-linolenic acid intake to affect serum non-HDL-cholesterol concentrations in European adolescents. J. Nutr. 2011, 141, 1247–1253. [Google Scholar] [CrossRef]
- Lankinen, M.A.; Fauland, A.; Shimizu, B.I.; Agren, J.; Wheelock, C.E.; Laakso, M.; Schwab, U.; Pihlajamaki, J. Inflammatory response to dietary linoleic acid depends on FADS1 genotype. Am. J. Clin. Nutr. 2019, 109, 165–175. [Google Scholar] [CrossRef]
- Lu, Y.; Feskens, E.J.; Dolle, M.E.; Imholz, S.; Verschuren, W.M.; Muller, M.; Boer, J.M. Dietary n-3 and n-6 polyunsaturated fatty acid intake interacts with FADS1 genetic variation to affect total and HDL-cholesterol concentrations in the Doetinchem Cohort Study. Am. J. Clin. Nutr. 2010, 92, 258–265. [Google Scholar] [CrossRef]
- Malerba, G.; Schaeffer, L.; Xumerle, L.; Klopp, N.; Trabetti, E.; Biscuola, M.; Cavallari, U.; Galavotti, R.; Martinelli, N.; Guarini, P.; et al. SNPs of the FADS gene cluster are associated with polyunsaturated fatty acids in a cohort of patients with cardiovascular disease. Lipids 2008, 43, 289–299. [Google Scholar] [CrossRef]
- Wu, W.C.; Wu, P.Y.; Chan, C.Y.; Lee, M.F.; Huang, C.Y. Effect of FADS1 rs174556 Genotype on Polyunsaturated Fatty Acid Status: A Systematic Review and Meta-Analysis. Adv. Nutr. 2023, 14, 352–362. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar] [CrossRef]
- MacKinnon, D.P.; Fairchild, A.J.; Fritz, M.S. Mediation analysis. Annu. Rev. Psychol. 2007, 58, 593–614. [Google Scholar] [CrossRef]
- Micha, R.; Khatibzadeh, S.; Shi, P.; Fahimi, S.; Lim, S.; Andrews, K.G.; Engell, R.E.; Powles, J.; Ezzati, M.; Mozaffarian, D.; et al. Global, regional, and national consumption levels of dietary fats and oils in 1990 and 2010: A systematic analysis including 266 country-specific nutrition surveys. BMJ 2014, 348, g2272. [Google Scholar] [CrossRef] [PubMed]
- Buzina, R.; Suboticanec, K.; Saric, M. Diet patterns and health problems: Diet in southern Europe. Ann. Nutr. Metab. 1991, 35 (Suppl. S1), 32–40. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Variables | Total Sample (N = 229,859) | FADS-DD Sample (N = 107,680) | FADS-DA Sample (N = 96,308) | FADS-AA Sample (N = 25,871) | p-Value 1 |
|---|---|---|---|---|---|
| Women—n (%) | 124,037 (54) | 58,108 (54) | 51,948 (53.9) | 13,981 (54) | 0.96 |
| Age, years—Mean (SD) | 57.1 (8.1) | 57.0 (8.1) | 57.2 (8.1) | 57.3 (8.0) | 2.2 × 10−7 |
| Race/Ethnicity—n (%) | <2.0 × 10−16 | ||||
| White | 218,314 (95.0) | 99,735 (92.6) | 93,549 (97.1) | 25,030 (96.7) | |
| Mixed | 1261 (0.5) | 713 (0.7) | 459 (0.5) | 89 (0.3) | |
| Asian | 4374 (1.9) | 2742 (2.5) | 1214 (1.3) | 418 (1.6) | |
| Black | 3057 (1.3) | 2855 (2.7) | 195 (0.2) | 7 (0) | |
| Not Reported | 2853 (1.2) | 1635 (1.5) | 891 (0.9) | 327 (1.3) | |
| BMI, kg/m2—Mean (SD) | 27.5 (4.8) | 27.5 (4.8) | 27.5 (4.8) | 27.4 (4.7) | 4.6 × 10−4 |
| TDI—Mean (SD) | −1.4 (3.1) | −1.3 (3.1) | −1.5 (3) | −1.5 (3) | <2.0 × 10−16 |
| Education—n (%) | 1.7 × 10−5 | ||||
| College degree | 84,542 (36.8) | 39,664 (36.8) | 35,268 (36.6) | 9610 (37.1) | |
| Associates degree | 25,180 (11) | 11,642 (10.8) | 10,607 (11) | 2931 (11.3) | |
| High school degree | 77,360 (33.7) | 36,212 (33.6) | 32,623 (33.9) | 8525 (33) | |
| No high school degree | 40,144 (17.5) | 18,817 (17.5) | 16,827 (17.5) | 4500 (17.4) | |
| Unknown | 2633 (1.1) | 1345 (1.2) | 983 (1) | 305 (1.2) | |
| Smoking—n (%) | 5.5 × 10−6 | ||||
| Never | 125,108 (53.8) | 59,046 (54.2) | 51,995 (53.4) | 14,067 (53.7) | |
| Previous | 79,670 (34.3) | 36,882 (33.8) | 33,754 (34.7) | 9034 (34.5) | |
| Current | 24,000 (10.3) | 11,181 (10.3) | 10,148 (10.4) | 2671 (10.2) | |
| Not Reported | 1081 (0.5) | 571 (0.5) | 411 (0.4) | 99 (0.4) | |
| Alcohol Intake—n (%) | <2.0 × 10−16 | ||||
| Never | 9677 (4.2) | 5098 (4.7) | 3627 (3.8) | 952 (3.7) | |
| Previous | 7913 (3.4) | 3793 (3.5) | 3294 (3.4) | 826 (3.2) | |
| Current | 211,780 (92.1) | 98,496 (91.5) | 89,237 (92.7) | 24,047 (92.9) | |
| Not Reported | 489 (0.2) | 293 (0.3) | 150 (0.2) | 46 (0.2) | |
| Fish Oil Supplement—n (%) | 72,270 (31.4) | 34,001 (31.6) | 30,197 (31.4) | 8072 (31.2) | 0.38 |
| Cholesterol Med Use—n (%) | 40,357 (17.6) | 19,099 (17.7) | 16,777 (17.4) | 4481 (17.3) | 0.062 |
| TC, mg/dL—Mean (SD) | 220 (42.7) | 220.6 (42.9) | 220 (42.6) | 217.3 (41.8) | <2.0 × 10−16 |
| LDL-c, mg/dL—Mean (SD) | 137.7 (32.6) | 138 (32.8) | 137.8 (32.6) | 136 (32) | <2.0 × 10−16 |
| HDL-c, mg/dL—Mean (SD) | 56 (14.2) | 56.3 (14.3) | 55.8 (14.3) | 55.2 (13.8) | <2.0 × 10−16 |
| Total TG, mg/dL—Mean (SD) | 152.6 (83.4) | 149.7 (81.8) | 154.5 (84.1) | 157.4 (86.7) | <2.0 × 10−16 |
| ω-3 PUFA, %—Mean (SD) | 4.4 (1.5) | 4.7 (1.4) | 4.2 (1.4) | 3.7 (1.5) | <2.0 × 10−16 |
| DHA | 2 (0.6) | 2.1 (0.6) | 1.9 (0.6) | 1.8 (0.7) | <2.0 × 10−16 |
| Non-DHA | 2.4 (1) | 2.6 (0.9) | 2.3 (0.9) | 1.9 (0.9) | <2.0 × 10−16 |
| ω-6 PUFA, %—Mean (SD) | 37.9 (3.4) | 37.9 (3.5) | 37.9 (3.4) | 38 (3.4) | 8.8 × 10−8 |
| LA | 28.9 (3.3) | 28.6 (3.2) | 29.1 (3.3) | 29.8 (3.3) | <2.0 × 10−16 |
| Non-LA | 9 (1.9) | 9.3 (1.9) | 8.8 (1.8) | 8.2 (1.7) | <2.0 × 10−16 |
| Total SFA, %—Mean (SD) | 34 (1.9) | 34 (1.9) | 34 (1.9) | 33.9 (1.9) | <2.0 × 10−16 |
| Outcome | FA | FADS-Only Model | Evidence of Moderation FA × FADS | Evidence of Mediation FA + FADS |
|---|---|---|---|---|
| p-Value 1 | p-Value 2 | p-Value 3 | ||
| TC | DHA | <2.0 × 10−16 | 6.1 × 10−12 | <2.0 × 10−16 |
| Non-DHA ω-3 | <2.0 × 10−16 | 3.73 × 10−15 | ||
| Total ω-3 PUFA | <2.0 × 10−16 | 3.3 × 10−3 | ||
| LA | 0.014 | <2.0 × 10−16 | ||
| Non-LA ω-6 | 4.0 × 10−14 | <2.0 × 10−16 | ||
| Total ω-6 PUFA | 0.034 | <2.0 × 10−16 | ||
| Total SFA | 2.1 × 10−3 | <2.0 × 10−16 | ||
| LDL-c | DHA | <2.0 × 10−16 | <2.0 × 10−16 | <2.0 × 10−16 |
| Non-DHA ω-3 | <2.0 × 10−16 | 3.7 × 10−13 | ||
| Total ω-3 PUFA | <2.0 × 10−16 | 6.2 × 10−3 | ||
| LA | 0.023 | <2.0 × 10−16 | ||
| Non-LA ω-6 | <2.0 × 10−16 | <2.0 × 10−16 | ||
| Total ω-6 PUFA | 5.8 × 10−7 | <2.0 × 10−16 | ||
| Total SFA | 9.4 × 10−4 | <2.0 × 10−16 | ||
| logHDL-c | DHA | <2.0 × 10−16 | 4.3 × 10−6 | 1.1 × 10−8 |
| Non-DHA ω-3 | 3.9 × 10−15 | <2.0 × 10−16 | ||
| Total ω-3 PUFA | 1.3 × 10−8 | 2.2 × 10−10 | ||
| LA | 2.0 × 10−4 | <2.0 × 10−16 | ||
| Non-LA ω-6 | <2.0 × 10−16 | 1.3 × 10−8 | ||
| Total ω-6 PUFA | 0.037 | <2.0 × 10−16 | ||
| Total SFA | 0.24 | <2.0 × 10−16 | ||
| logTG | DHA | <2.0 × 10−16 | 1.7 × 10−13 | <2.0 × 10−16 |
| Non-DHA ω-3 | <2.0 × 10−16 | <2.0 × 10−16 | ||
| Total ω-3 PUFA | <2.0 × 10−16 | <2.0 × 10−16 | ||
| LA | <2.0 × 10−16 | <2.0 × 10−16 | ||
| Non-LA ω-6 | <2.0 × 10−16 | <2.0 × 10−16 | ||
| Total ω-6 PUFA | 0.74 | <2.0 × 10−16 | ||
| Total SFA | 0.57 | <2.0 × 10−16 |
| Outcome | FA | FADS Group | FA Quartile 1 Mean Difference (95% CI) | FA Quartile 2 Mean Difference (95% CI) | FA Quartile 3 Mean Difference (95% CI) | FA Quartile 4 Mean Difference (95% CI) |
|---|---|---|---|---|---|---|
| TC | DHA | DD | − | − | − | − |
| DA | −0.1 (−1.4, 1.1) | −2.3 (−3.4, −1.1) | −2.9 (−4, −1.8) | −2.2 (−3.3, −1.1) | ||
| AA | −2.8 (−4.4, −1.2) | −7.1 (−8.9, −5.3) | −7.3 (−9.2, −5.3) | −6.5 (−8.5, −4.5) | ||
| Non-DHA | DD | − | − | − | − | |
| DA | 3.2 (2.1, 4.4) | 2.9 (1.8, 4) | 0.9 (−0.2, 2) | −1.4 (−2.5, −0.3) | ||
| AA | 5.2 (3.8, 6.7) | 3.5 (1.7, 5.2) | −0.8 (−2.9, 1.3) | −5.7 (−8.1, −3.3) | ||
| Total ω-3 | DD | − | − | − | − | |
| DA | 2.7 (1.5, 4) | 1.6 (0.5, 2.7) | −0.5 (−1.7, 0.6) | −2.2 (−3.3, −1.1) | ||
| AA | 3.5 (2.1, 5) | 0.5 (−1.4, 2.3) | −4.9 (−7, −2.8) | −6.3 (−8.5, −4) | ||
| Non-LA | DD | − | − | − | − | |
| DA | −5.2 (−6.3, −4.1) | −6.7 (−7.7, −5.7) | −7.5 (−8.5, −6.6) | −5.7 (−6.6, −4.7) | ||
| AA | −13.8 (−15.3, −12.3) | −16.3 (−17.8, −14.8) | −16.9 (−18.5, −15.2) | −12.5 (−14.4, −10.7) | ||
| LDL-c | DHA | DD | − | − | − | − |
| DA | 0.2 (−0.7, 1.1) | −1.6 (−2.5, −0.8) | −2.3 (−3.1, −1.4) | −1.6 (−2.5, −0.8) | ||
| AA | −1.6 (−2.8, −0.4) | −5.1 (−6.5, −3.7) | −5.6 (−7.1, −4.1) | −4.6 (−6.1, −3.1) | ||
| Non-DHA | DD | − | − | − | − | |
| DA | 2.8 (1.9, 3.7) | 2.2 (1.3, 3) | 0.3 (−0.5, 1.2) | −1.3 (−2.2, −0.4) | ||
| AA | 4.6 (3.5, 5.7) | 2.3 (1, 3.7) | −1.3 (−2.9, 0.3) | −4.4 (−6.2, −2.6) | ||
| Total ω-3 | DD | − | − | − | − | |
| DA | 2.2 (1.3, 3.2) | 1.1 (0.2, 1.9) | −0.5 (−1.4, 0.3) | −1.8 (−2.6, −0.9) | ||
| AA | 2.9 (1.7, 4.0) | 0 (−1.4, 1.4) | −3.7 (−5.4, −2.1) | −4.7 (−6.5, −3) | ||
| Non-LA | DD | − | − | − | − | |
| DA | −3.7 (−4.6, −2.8) | −5.2 (−6, −4.4) | −5.4 (−6.2, −4.7) | −3.7 (−4.4, −3) | ||
| AA | −10 (−11.2, −8.9) | −12 (−13.2, −10.8) | −11.9 (−13.2, −10.6) | −8.2 (−9.5, −6.8) | ||
| Total ω-6 | DD | − | − | − | − | |
| DA | −1.1 (−2.0, −0.3) | −1 (−1.9, −0.2) | −0.5 (−1.4, 0.3) | 0.3 (−0.5, 1.1) | ||
| AA | −3.1 (−4.4, −1.7) | −3.2 (−4.5, −1.8) | −2.3 (−3.6, −0.9) | −0.5 (−1.7, 0.7) | ||
| logHDL-c | DHA | DD | − | − | − | − |
| DA | 0.005 (−0.001, 0.011) | 0.009 (0.003, 0.015) | 0.002 (−0.004, 0.008) | −0.003 (−0.009, 0.004) | ||
| AA | 0.013 (0.005, 0.021) | 0.017 (0.007, 0.026) | 0.01 (−0.001, 0.02) | −0.007 (−0.018, 0.004) | ||
| Non-DHA | DD | − | − | − | − | |
| DA | −0.017 (−0.024, −0.01) | −0.019 (−0.025, −0.013) | −0.015 (−0.021, −0.009) | −0.004 (−0.011, 0.002) | ||
| AA | −0.036 (−0.044, −0.028) | −0.042 (−0.051, −0.032) | −0.023 (−0.035, −0.012) | −0.005 (−0.018, 0.008) | ||
| Total ω-3 | DD | − | − | − | − | |
| DA | −0.012 (−0.019, −0.005) | −0.005 (−0.011, 0.001) | −0.003 (−0.009, 0.003) | 0 (−0.006, 0.006) | ||
| AA | −0.021 (−0.03, −0.013) | −0.005 (−0.015, 0.005) | −0.007 (−0.018, 0.005) | 0.004 (−0.008, 0.016) | ||
| LA | DD | − | − | − | − | |
| DA | −0.018 (−0.024, −0.012) | −0.021 (−0.028, −0.015) | −0.017 (−0.023, −0.011) | −0.013 (−0.019, −0.006) | ||
| AA | −0.038 (−0.049, −0.027) | −0.047 (−0.058, −0.037) | −0.038 (−0.048, −0.029) | −0.028 (−0.037, −0.02) | ||
| Non-LA | DD | − | − | − | − | |
| DA | 0.008 (0.002, 0.015) | 0.005 (−0.001, 0.012) | −0.009 (−0.015, −0.003) | −0.019 (−0.026, −0.013) | ||
| AA | 0.015 (0.007, 0.023) | 0.001 (−0.008, 0.011) | −0.027 (−0.037, −0.016) | −0.04 (−0.052, −0.027) | ||
| logTG | DHA | DD | − | − | − | − |
| DA | −0.029 (−0.043, −0.014) | −0.041 (−0.053, −0.029) | −0.023 (−0.035, −0.012) | −0.009 (−0.02, 0.002) | ||
| AA | −0.063 (−0.082, −0.045) | −0.105 (−0.125, −0.085) | −0.078 (−0.099, −0.057) | −0.033 (−0.053, −0.012) | ||
| Non-DHA | DD | − | − | − | − | |
| DA | 0.1 (0.087, 0.113) | 0.115 (0.102, 0.127) | 0.088 (0.076, 0.101) | 0.014 (0, 0.028) | ||
| AA | 0.211 (0.195, 0.227) | 0.24 (0.22, 0.259) | 0.137 (0.113, 0.161) | −0.009 (−0.038, 0.019) | ||
| Total ω-3 | DD | − | − | − | − | |
| DA | 0.075 (0.06, 0.091) | 0.059 (0.046, 0.073) | 0.029 (0.016, 0.042) | −0.013 (−0.026, 0) | ||
| AA | 0.145 (0.126, 0.164) | 0.089 (0.066, 0.112) | 0.012 (−0.013, 0.037) | −0.052 (−0.078, −0.026) | ||
| LA | DD | − | − | − | − | |
| DA | 0.063 (0.049, 0.076) | 0.073 (0.061, 0.086) | 0.05 (0.038, 0.062) | 0.033 (0.022, 0.044) | ||
| AA | 0.131 (0.107, 0.154) | 0.148 (0.128, 0.169) | 0.119 (0.101, 0.138) | 0.062 (0.046, 0.077) | ||
| Non-LA | DD | − | − | − | − | |
| DA | −0.062 (−0.073, −0.05) | −0.076 (−0.086, −0.066) | −0.061 (−0.071, −0.052) | −0.024 (−0.033, −0.014) | ||
| AA | −0.135 (−0.151, −0.12) | −0.165 (−0.181, −0.15) | −0.127 (−0.144, −0.11) | −0.074 (−0.093, −0.054) |
| Outcome | FA | FADS-DD vs. -DA | ||||
|---|---|---|---|---|---|---|
| FADS-Only Model 1 Mean (CI) | p-Value 2 | Additive Model 3 Mean (CI) | p-Value 4 | % Mediation | ||
| TC | DHA | −1.3 (−1.9, −0.7) | 5.5 × 10−14 | −1.9 (−2.5, −1.4) | <2.0 × 10−16 | −51% |
| Non-DHA | 1.2 (0.6, 1.8) | 3.3 × 10−12 | 192% | |||
| ω-3 PUFA | 0.2 (−0.4, 0.7) | 0.35 | 112% | |||
| LA | −1.7 (−2.3, −1.2) | <2.0 × 10−16 | −34% | |||
| Non-LA | −6.3 (−6.8, −5.8) | <2.0 × 10−16 | −392% | |||
| ω-6 PUFA | −1.1 (−1.7, −0.6) | 8.2 × 10−12 | 11% | |||
| SFA | −1 (−1.6, −0.5) | 1.5 × 10−9 | 21% | |||
| LDL-c | DHA | −0.7 (−1.2, −0.3) | 1.2 × 10−8 | −1.4 (−1.8, −1) | <2.0 × 10−16 | −87% |
| Non-DHA | 0.8 (0.4, 1.3) | 4.0×10−10 | 210% | |||
| ω-3 PUFA | 0.1 (−0.4, 0.5) | 0.70 | 107% | |||
| LA | −1.2 (−1.6, −0.8) | <2.0 × 10−16 | −61% | |||
| Non-LA | −4.5 (−4.9, −4.2) | <2.0 × 10−16 | −507% | |||
| ω-6 PUFA | −0.6 (−1.1, −0.2) | 5.0 × 10−7 | 13% | |||
| SFA | −0.6 (−1.1, −0.2) | 1.9 × 10−6 | 17% | |||
| logHDL | DHA | −0.011 (−0.014, −0.008) | <2.0 × 10−16 | 0.003 (0, 0.006) | 1.2 × 10−3 | 127% |
| Non-DHA | −0.013 (−0.016, −0.01) | <2.0 × 10−16 | −18% | |||
| ω-3 PUFA | −0.004 (−0.008, −0.001) | 2.3 × 10−6 | 59% | |||
| LA | −0.017 (−0.02, −0.014) | <2.0 × 10−16 | −58% | |||
| Non-LA | −0.004 (−0.007, −0.001) | 4.9 × 10−6 | 61% | |||
| ω-6 PUFA | −0.012 (−0.015, −0.009) | <2.0 × 10−16 | −7% | |||
| SFA | −0.01 (−0.013, −0.007) | <2.0 × 10−16 | 10% | |||
| logTG | DHA | 0.024 (0.017, 0.031) | <2.0 × 10−16 | −0.025 (−0.031, −0.019) | <2.0 × 10−16 | 204% |
| Non-DHA | 0.075 (0.068, 0.081) | <2.0 × 10−16 | −212% | |||
| ω-3 PUFA | 0.033 (0.026, 0.04) | <2.0 × 10−16 | −37% | |||
| LA | 0.056 (0.05, 0.062) | <2.0 × 10−16 | −134% | |||
| Non-LA | −0.056 (−0.061, −0.051) | <2.0 × 10−16 | 334% | |||
| ω-6 PUFA | 0.03 (0.025, 0.034) | <2.0 × 10−16 | −23% | |||
| SFA | 0.028 (0.022, 0.035) | <2.0 × 10−16 | −19% | |||
| FADS-DD vs. -AA | ||||||
| TC | DHA | −4.2 (−5, −3.3) | <2.0 × 10−16 | −5.6 (−6.5, −4.7) | <2.0 × 10−16 | −35% |
| Non-DHA | 1.7 (0.8, 2.6) | 2.6 × 10−10 | 141% | |||
| ω-3 PUFA | −0.8 (−1.7, 0.2) | 6.0 × 10−3 | 82% | |||
| LA | −5.2 (−6, −4.3) | <2.0 × 10−16 | −24% | |||
| Non-LA | −15 (−15.8, −14.2) | <2.0 × 10−16 | −261% | |||
| ω-6 PUFA | −3.7 (−4.6, −2.8) | <2.0 × 10−16 | 11% | |||
| SFA | −3.6 (−4.5, −2.7) | <2.0 × 10−16 | 14% | |||
| LDL-c | DHA | −2.5 (−3.2, −1.9) | <2.0 × 10−16 | −4 (−4.7, −3.3) | <2.0 × 10−16 | −57% |
| Non-DHA | 1.3 (0.6, 2) | 1.0 × 10−9 | 150% | |||
| ω-3 PUFA | −0.6 (−1.3, 0.1) | 4.4 × 10−3 | 76% | |||
| LA | −3.6 (−4.3, −2.9) | <2.0 × 10−16 | −41% | |||
| Non-LA | −10.7 (−11.3, −10.1) | <2.0 × 10−16 | −323% | |||
| ω-6 PUFA | −2.2 (−2.9, −1.5) | <2.0 × 10−16 | 13% | |||
| SFA | −2.3 (−2.9, −1.6) | <2.0 × 10−16 | 11% | |||
| logHDL | DHA | −0.023 (−0.028, −0.018) | <2.0 × 10−16 | 0.008 (0.004, 0.013) | 3.9 × 10−9 | 136% |
| Non-DHA | −0.029 (−0.034, −0.024) | <2.0 × 10−16 | −24% | |||
| ω-3 PUFA | −0.009 (−0.014, −0.004) | 1.7 × 10−9 | 61% | |||
| LA | −0.037 (−0.042, −0.032) | <2.0 × 10−16 | −60% | |||
| Non-LA | −0.008 (−0.012, −0.003) | 2.0 × 10−7 | 67% | |||
| ω-6 PUFA | −0.025 (−0.03, −0.021) | <2.0 × 10−16 | −10% | |||
| SFA | −0.021 (−0.026, −0.016) | <2.0 × 10−16 | 10% | |||
| logTG | DHA | 0.042 (0.031, 0.052) | <2.0 × 10−16 | −0.068 (−0.078, −0.058) | <2.0 × 10−16 | 264% |
| Non-DHA | 0.163 (0.153, 0.173) | <2.0 × 10−16 | −293% | |||
| ω-3 PUFA | 0.066 (0.055, 0.077) | <2.0 × 10−16 | −59% | |||
| LA | 0.112 (0.102, 0.121) | <2.0 × 10−16 | −169% | |||
| Non-LA | −0.131 (−0.139, −0.123) | <2.0 × 10−16 | 416% | |||
| ω-6 PUFA | 0.059 (0.052, 0.066) | <2.0 × 10−16 | −43% | |||
| SFA | 0.051 (0.042, 0.061) | <2.0 × 10−16 | −24% | |||
| FADS Genotype | FA Quartile | % Difference in LDL-c from Quartile 1 | ||||||
|---|---|---|---|---|---|---|---|---|
| DHA | Non-DHA | ω-3 PUFA | LA | Non-LA | ω-6 PUFA | SFA | ||
| DD | 1 | − | − | − | − | − | − | − |
| DD | 2 | −1.1% | 8.1% | 4.9% | 2.0% | −9.8% | −3.3% | 3.2% |
| DD | 3 | −3.2% | 12.3% | 6.9% | 3.6% | −17.0% | −7.3% | 4.7% |
| DD | 4 | −4.9% | 14.4% | 7.9% | 6.3% | −27.3% | −13.4% | 8.0% |
| DA | 1 | − | − | − | − | − | − | − |
| DA | 2 | −2.6% | 7.3% | 3.8% | 2.3% | −11.2% | −3.0% | 2.6% |
| DA | 3 | −5.2% | 9.7% | 4.5% | 3.4% | −18.8% | −6.8% | 4.0% |
| DA | 4 | −6.5% | 10.3% | 4.3% | 7.1% | −28.1% | −12.3% | 6.9% |
| AA | 1 | − | − | − | − | − | − | − |
| AA | 2 | −4.0% | 5.6% | 2.2% | 1.1% | −12.1% | −3.4% | 1.9% |
| AA | 3 | −6.6% | 6.4% | 1.1% | 3.3% | −19.8% | −6.8% | 3.2% |
| AA | 4 | −7.7% | 5.7% | 1.1% | 6.2% | −28.0% | −11.6% | 6.3% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrington, C.E.; Westra, J.; Tintle, N.L.; Shearer, G.C. The Effect of Fatty Acid Desaturase on Cardiovascular Lipid Biomarkers Depends on Circulating ω-3 and ω-6 Polyunsaturated Fatty Acids in the UK Biobank. Nutrients 2025, 17, 1089. https://doi.org/10.3390/nu17061089
Arrington CE, Westra J, Tintle NL, Shearer GC. The Effect of Fatty Acid Desaturase on Cardiovascular Lipid Biomarkers Depends on Circulating ω-3 and ω-6 Polyunsaturated Fatty Acids in the UK Biobank. Nutrients. 2025; 17(6):1089. https://doi.org/10.3390/nu17061089
Chicago/Turabian StyleArrington, Carmen E., Jason Westra, Nathan L. Tintle, and Gregory C. Shearer. 2025. "The Effect of Fatty Acid Desaturase on Cardiovascular Lipid Biomarkers Depends on Circulating ω-3 and ω-6 Polyunsaturated Fatty Acids in the UK Biobank" Nutrients 17, no. 6: 1089. https://doi.org/10.3390/nu17061089
APA StyleArrington, C. E., Westra, J., Tintle, N. L., & Shearer, G. C. (2025). The Effect of Fatty Acid Desaturase on Cardiovascular Lipid Biomarkers Depends on Circulating ω-3 and ω-6 Polyunsaturated Fatty Acids in the UK Biobank. Nutrients, 17(6), 1089. https://doi.org/10.3390/nu17061089