
Alcohol Consumption and Liver Metabolism in the Era of MASLD: Integrating Nutritional and Pathophysiological Insights
 ,
,  , , , and
, , , and
Abstract
1. Introduction
Updated Nomenclature and Definitions of Steatotic Liver Disease
- MASLD is diagnosed in the presence of hepatic steatosis (≥5% liver fat as determined by imaging or histology) and at least one of the following five cardiometabolic risk factors:
- ○
- Overweight or obesity (BMI ≥ 25 kg/m2 or ethnicity-specific thresholds);
- ○
- Type 2 diabetes or prediabetes;
- ○
- Arterial hypertension (≥130/85 mmHg or current antihypertensive treatment);
- ○
- Hypertriglyceridemia (≥150 mg/dL) or treatment with lipid-lowering agents;
- ○
- Low HDL-cholesterol (≤40 mg/dL in men, ≤50 mg/dL in women) or lipid-lowering therapy [3].
- MetALD (Metabolic and Alcohol-related Liver Disease) refers to individuals with hepatic steatosis and metabolic dysfunction who consume alcohol in amounts above established threshold levels (typically > 20 g/day for women and >30 g/day for men). This category acknowledges the clinical overlap between metabolic and alcohol-induced liver injury [4].
- The term MASH (metabolic dysfunction-associated steatohepatitis) replaces “NASH” and designates steatohepatitis, with or without fibrosis, arising in the context of MASLD [3].
2. Hepatic Alcohol Metabolism and Metabolic Adaptations
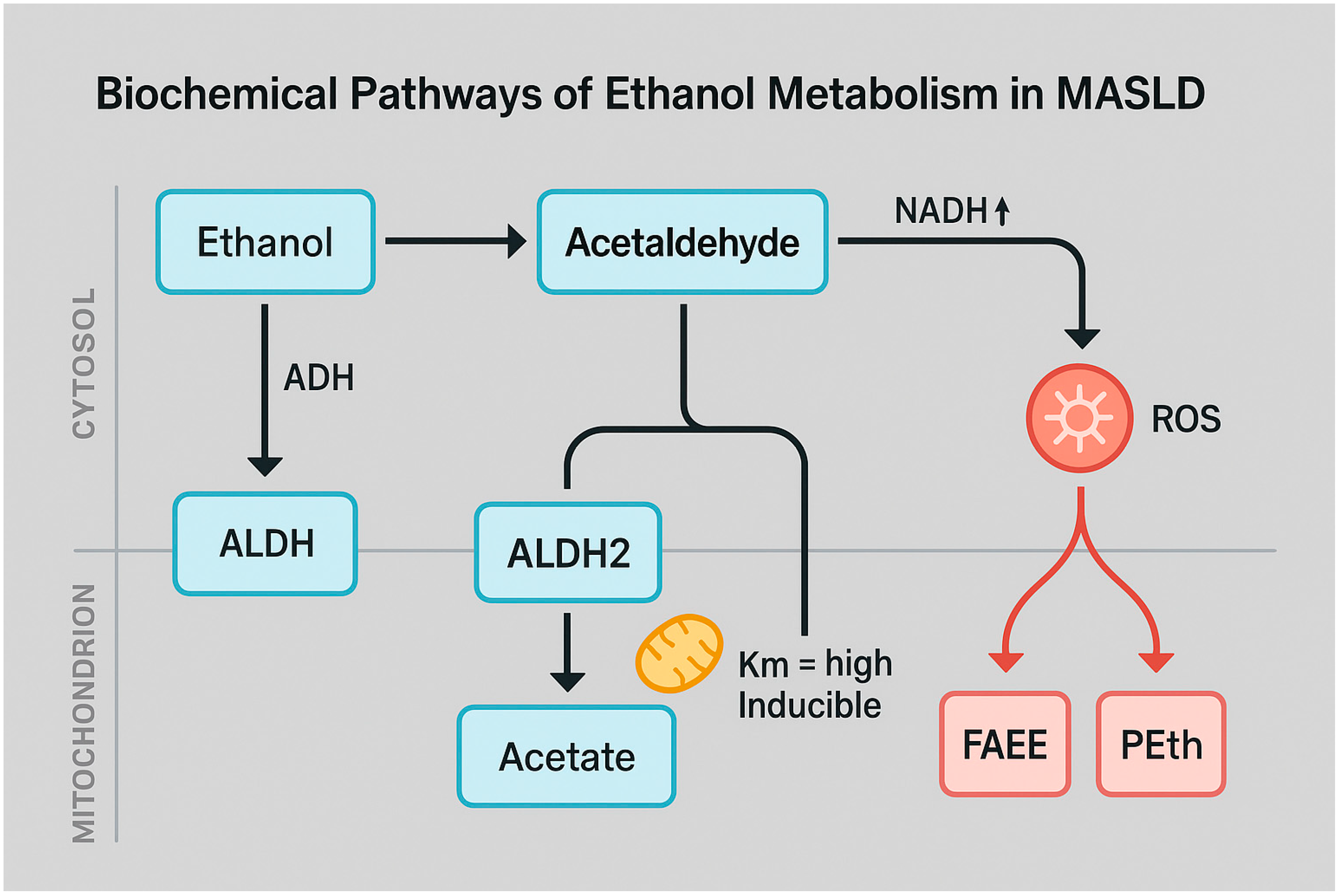
2.1. Biochemical Pathways of Ethanol Oxidation
2.2. Metabolic Alterations Induced by Chronic Alcohol Exposure
2.3. Distinct and Shared Mechanisms of Hepatic Injury in Alcoholic vs. Metabolic Dysfunction
3. Mitochondrial Dysfunction and Oxidative Stress: The Converging Axis of Alcohol and MASLD
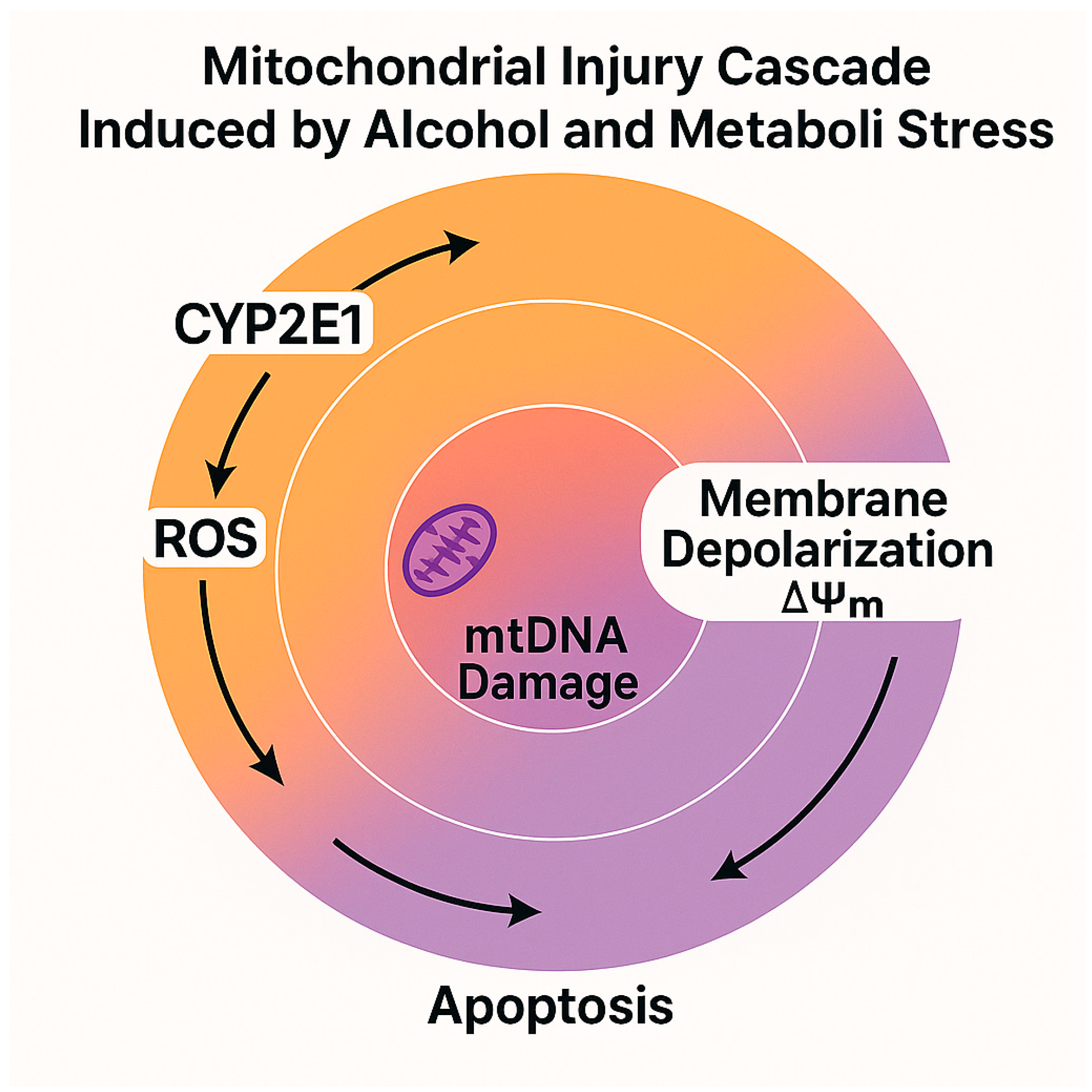
3.1. Impact on Mitochondrial Bioenergetics and ROS Generation
3.2. Lipid Homeostasis Disruption and Hepatocellular Injury
4. Gut–Liver Axis and Intestinal Permeability in MASLD and Alcohol-Related Liver Disease
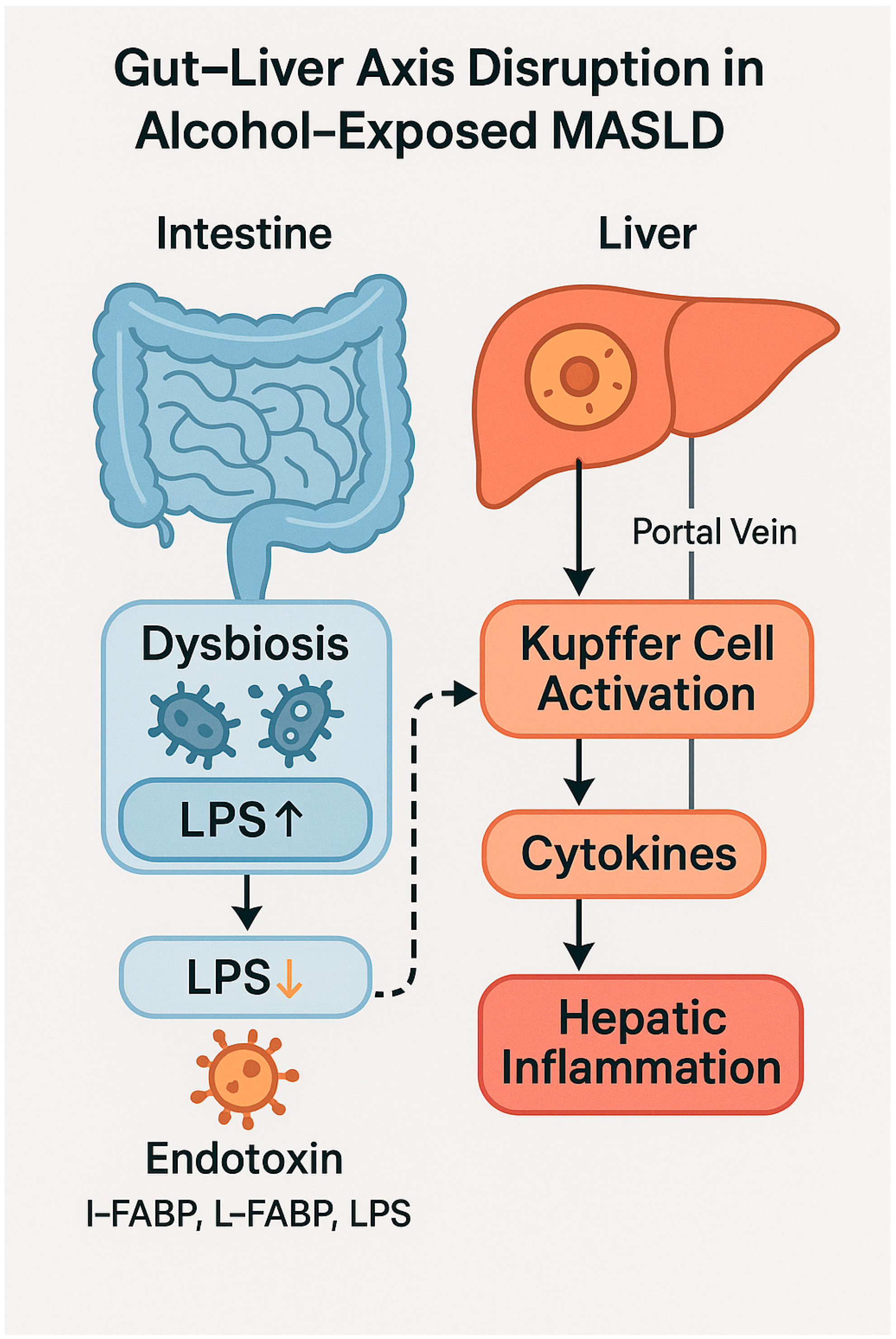
4.1. Pathophysiological Role of the Gut–Liver Axis
4.2. Intestinal Barrier Dysfunction and Bacterial Translocation
4.3. Clinical Implications and Therapeutic Opportunities
5. Pathophysiological Interaction Between Alcohol and MASLD
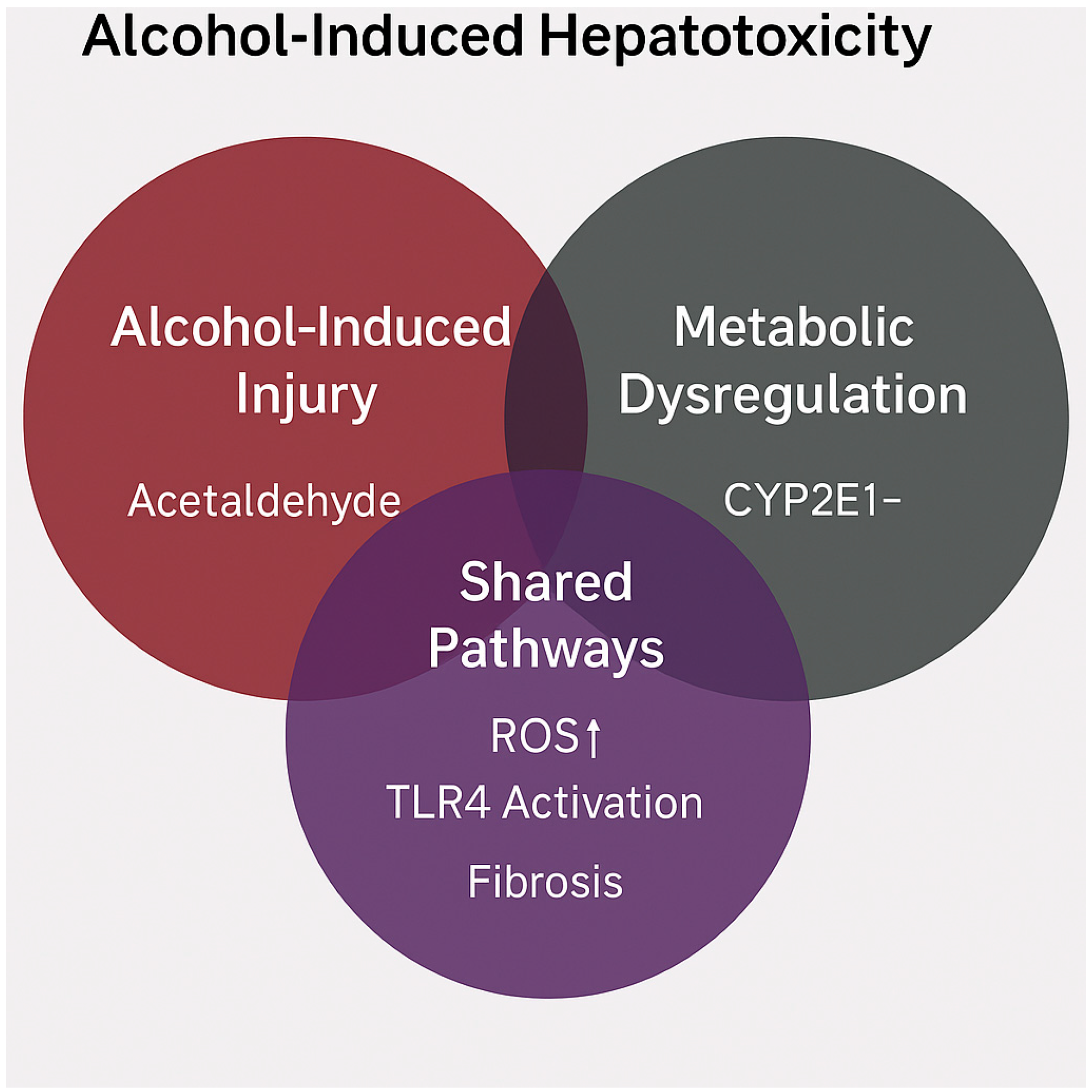
5.1. Shared Mechanisms and Synergistic Hepatotoxicity
5.2. From Steatosis to Inflammation and Fibrosis: A Convergent Model
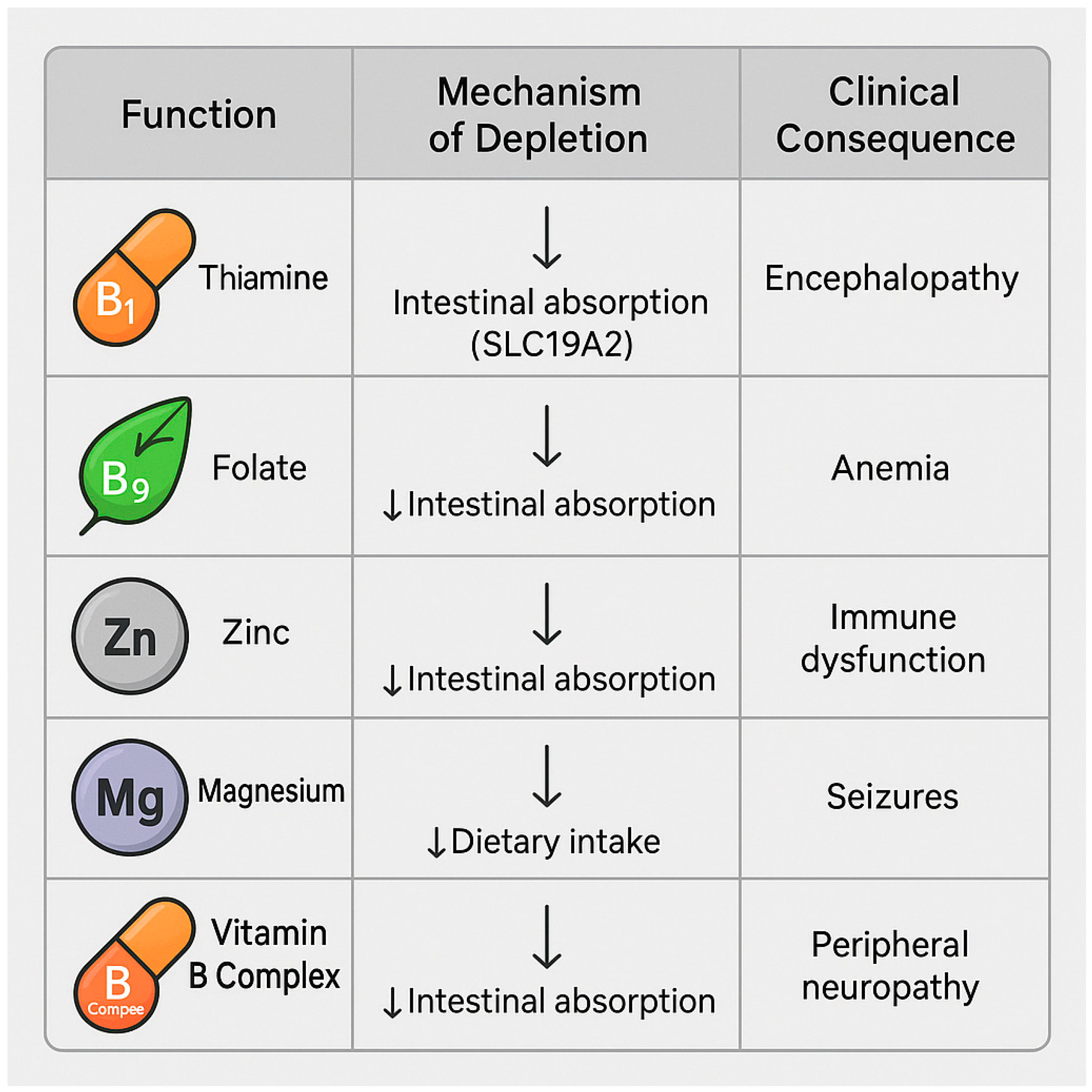
6. Micronutrient Depletion and Malabsorption Syndromes
6.1. Deficiencies in Folate, Thiamine, Zinc, Magnesium, and B Vitamins
- Thiamine (vitamin B1) is a cofactor for pyruvate oxidative decarboxylation and the pentose phosphate pathway. Thiamine deficiency impairs oxidative glucose metabolism, promoting lactate accumulation and contributing to hepatic and cerebral dysfunction [86]. In patients with MASLD, it may worsen insulin resistance and promote progression to steatohepatitis [87].
- Zinc is essential for the activity of numerous hepatic metalloenzymes, innate immune function (NK cells and neutrophils), redox homeostasis, and ammonia detoxification [91]. Zinc deficiency is associated with hepatic encephalopathy, insulin resistance, and impaired intestinal barrier integrity [92].
6.2. Nutritional Interactions of Alcohol and Clinical Implications
7. Alcohol, Insulin Resistance, and Sarcopenia
7.1. Liver–Muscle Axis in MASLD
7.2. Synergistic Effects of Alcohol on Muscle Mass
7.3. Emerging Biomarkers and Clinical Implications
8. Moderate Alcohol Consumption: Residual Risk in Metabolic Diseases
8.1. Threshold Effects and Recent Evidence
8.2. Mechanisms of Alcohol Vulnerability in MASLD
8.3. Critical Conclusions on the Clinical Use of the “Moderation” Concept
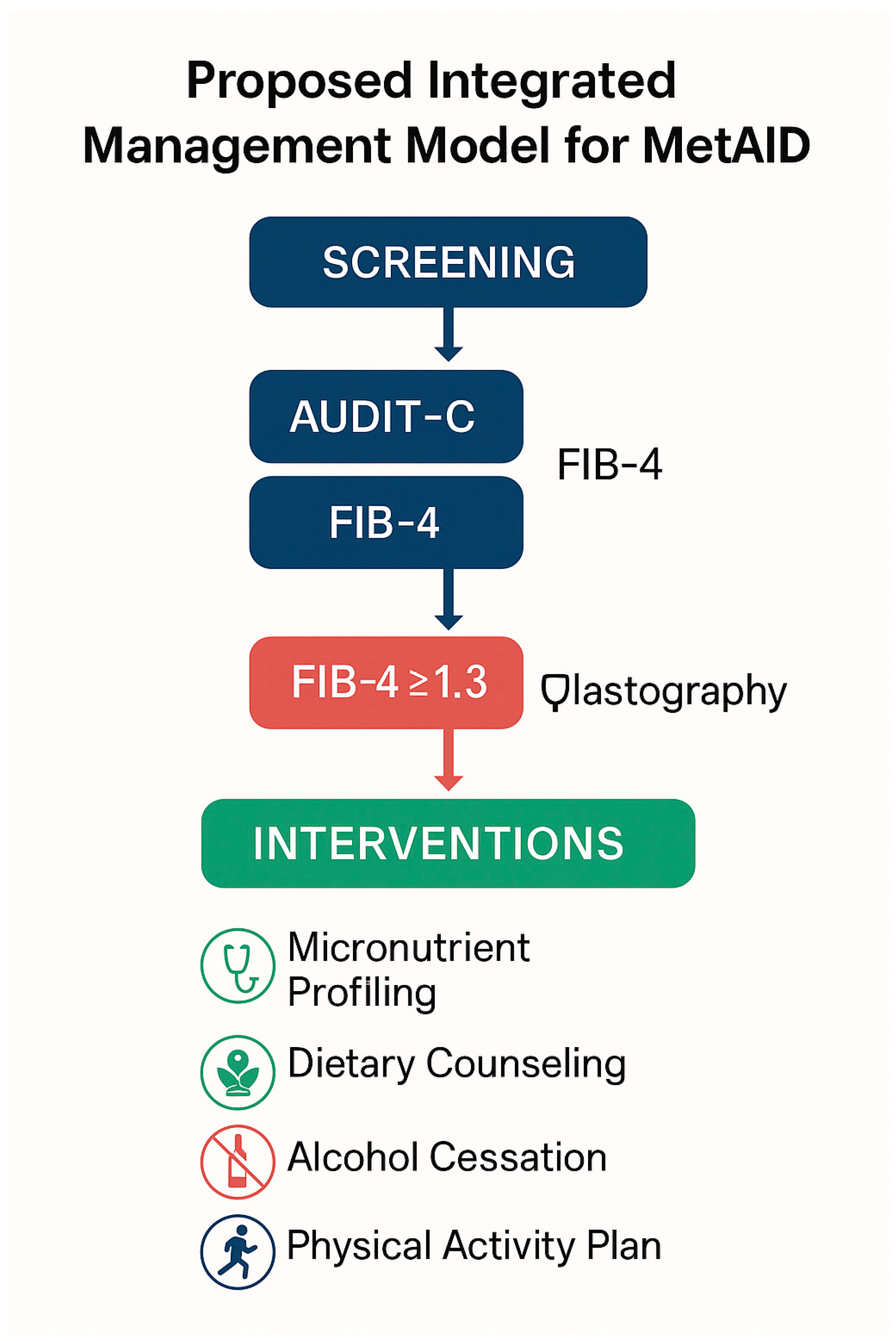
9. Nutritional and Clinical Implications
9.1. Counseling Strategies and Risk Communication
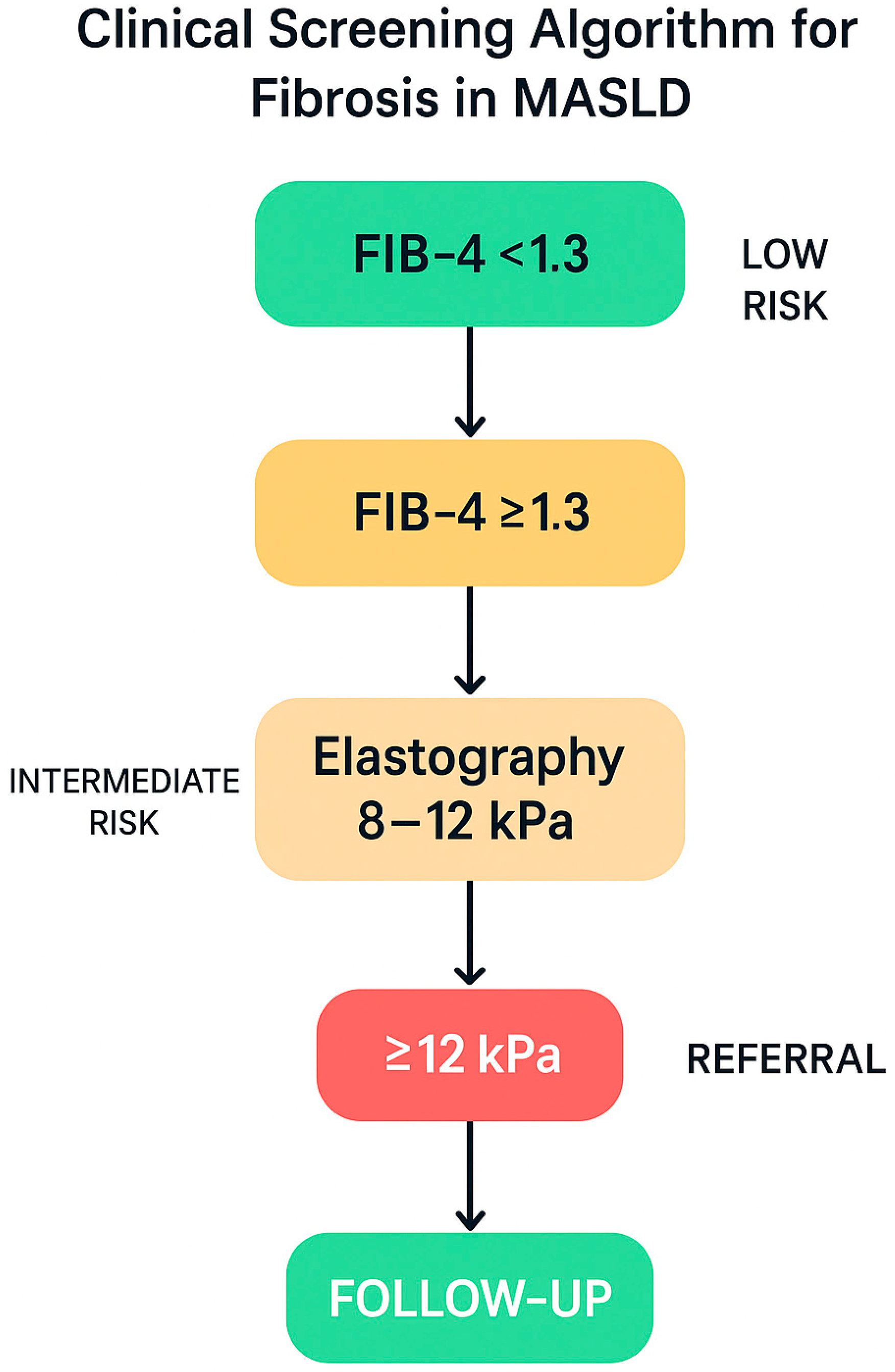
9.2. Clinical Screening for Fibrosis in Patients with MASLD
9.3. Dietary Interventions and Nutritional Strategies: Comparative Effectiveness of Dietary Models in MASLD Management
10. Conclusions and Future Perspectives
- The validation of specific biomarkers for alcohol-induced liver injury susceptibility in MASLD patients, leveraging multi-omics technologies, quantitative imaging, and immuno-nutritional profiling;
- The development of non-invasive diagnostic tools tailored to the MetALD phenotype, capable of capturing the specificity of ethanol–metabolism interactions;
- The evaluation of personalized nutritional and pharmacological interventions, potentially in combination with structured psychological counseling;
- The integration of liver health into cardiovascular and metabolic prevention programs, recognizing MASLD as a systemic, multi-organ condition.
Funding
Conflicts of Interest
References
- Targher, G.; Byrne, C.D.; Tilg, H. MASLD: A systemic metabolic disorder with cardiovascular and malignant complications. Gut 2024, 73, 691–702. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Adinolfi, L.E.; Marrone, A.; Rinaldi, L.; Nevola, R.; Izzi, A.; Sasso, F.C. Metabolic dysfunction-associated steatotic liver disease (MASLD): A systemic disease with a variable natural history and challenging management. Explor. Med. 2025, 6, 1001281. [Google Scholar] [CrossRef]
- Cao, L.; An, Y.; Liu, H.; Jiang, J.; Liu, W.; Zhou, Y.; Shi, M.; Dai, W.; Lv, Y.; Zhao, Y.; et al. Global epidemiology of type 2 diabetes in patients with NAFLD or MAFLD: A systematic review and meta-analysis. BMC Med. 2024, 22, 101. [Google Scholar] [CrossRef] [PubMed]
- Le, P.; Tatar, M.; Dasarathy, S.; Alkhouri, N.; Herman, W.H.; Taksler, G.B.; Deshpande, A.; Ye, W.; Adekunle, O.A.; McCullough, A.; et al. Estimated Burden of Metabolic Dysfunction-Associated Steatotic Liver Disease in US Adults, 2020 to 2050. JAMA Netw. Open 2025, 8, e2454707. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023, 78, 1966–1986. [Google Scholar] [CrossRef]
- Nguyen, V.H.; Le, M.H.; Cheung, R.C.; Nguyen, M.H. Differential Clinical Characteristics and Mortality Outcomes in Persons with NAFLD and/or MAFLD. Clin. Gastroenterol. Hepatol. 2021, 19, 2172–2181.e6. [Google Scholar] [CrossRef] [PubMed]
- Marti-Aguado, D.; Calleja, J.L.; Vilar-Gomez, E.; Iruzubieta, P.; Rodríguez-Duque, J.C.; Del Barrio, M.; Puchades, L.; Rivera-Esteban, J.; Perelló, C.; Puente, A.; et al. Low-to-moderate alcohol consumption is associated with increased fibrosis in individuals with metabolic dysfunction-associated steatotic liver disease. J. Hepatol. 2024, 81, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Babuta, M.; Morel, C.; Ribeiro, M.d.C.; Datta, A.A.; Calenda, C.; Copeland, C.; Nasser, I.; Szabo, G. A novel experimental model of MetALD in male mice recapitulates key features of severe alcohol-associated hepatitis. Hepatol. Commun. 2024, 8, e0450. [Google Scholar] [CrossRef]
- Åberg, F.; Puukka, P.; Salomaa, V.; Männistö, S.; Lundqvist, A.; Valsta, L.; Perola, M.; Färkkilä, M.; Jula, A. Risks of Light and Moderate Alcohol Use in Fatty Liver Disease: Follow-Up of Population Cohorts. Hepatology 2020, 71, 835–848. [Google Scholar] [CrossRef]
- Choi, J.H.; Sohn, W.; Cho, Y.K. The effect of moderate alcohol drinking in nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2020, 26, 662–669. [Google Scholar] [CrossRef]
- Magherman, L.; Van Parys, R.; Pauwels, N.S.; Verhelst, X.; Devisscher, L.; Van Vlierberghe, H.; Geerts, A.; Lefere, S. Meta-analysis: The impact of light-to-moderate alcohol consumption on progressive non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2023, 57, 820–836. [Google Scholar] [CrossRef]
- Tadokoro, T.; Morishita, A.; Himoto, T.; Masaki, T. Nutritional Support for Alcoholic Liver Disease. Nutrients 2023, 15, 1360. [Google Scholar] [CrossRef]
- Chrysavgis, L.; Cholongitas, E. From NAFLD to MASLD: What does it mean? Expert Rev. Gastroenterol. Hepatol. 2024, 18, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S.; Neuman, M.; Seitz, H.K. The role of cytochrome P4502E1 in ethanol mediated diseases: A narrative update. Alcohol Alcohol. Oxf. Oxfs. 2025, 60, agaf014. [Google Scholar] [CrossRef] [PubMed]
- Monari, P.; Venturuzzo, A.; Moro, R.; Calzavara-Pinton, P.; Gualdi, G. Nested graft for chronic ulcer in scar tissue after heroin extravasation in a drug addict. J. Tissue Viability 2021, 30, 121–123. [Google Scholar] [CrossRef]
- Arumugam, M.K.; Gopal, T.; Kalari Kandy, R.R.; Boopathy, L.K.; Perumal, S.K.; Ganesan, M.; Rasineni, K.; Donohue, T.M., Jr.; Osna, N.A.; Kharbanda, K.K. Mitochondrial Dysfunction-Associated Mechanisms in the Development of Chronic Liver Diseases. Biology 2023, 12, 1311. [Google Scholar] [CrossRef] [PubMed]
- Subramaniyan, V.; Lubau, N.S.A.; Mukerjee, N.; Kumarasamy, V. Alcohol-induced liver injury in signalling pathways and curcumin’s therapeutic potential. Toxicol. Rep. 2023, 11, 355–367. [Google Scholar] [CrossRef]
- Kumar, S.; Singla, B.; Singh, A.K.; Thomas-Gooch, S.M.; Zhi, K.; Singh, U.P. Hepatic, Extrahepatic and Extracellular Vesicle Cytochrome P450 2E1 in Alcohol and Acetaminophen-Mediated Adverse Interactions and Potential Treatment Options. Cells 2022, 11, 2620. [Google Scholar] [CrossRef]
- Yan, C.; Hu, W.; Tu, J.; Li, J.; Liang, Q.; Han, S. Pathogenic mechanisms and regulatory factors involved in alcoholic liver disease. J. Transl. Med. 2023, 21, 300. [Google Scholar] [CrossRef]
- Hyun, J.; Han, J.; Lee, C.; Yoon, M.; Jung, Y. Pathophysiological Aspects of Alcohol Metabolism in the Liver. Int. J. Mol. Sci. 2021, 22, 5717. [Google Scholar] [CrossRef]
- You, M.; Arteel, G.E. Effect of ethanol on lipid metabolism. J. Hepatol. 2019, 70, 237–248. [Google Scholar] [CrossRef]
- Sanders, F.W.B.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Vetrano, E.; Rinaldi, L.; Mormone, A.; Giorgione, C.; Galiero, R.; Caturano, A.; Nevola, R.; Marfella, R.; Sasso, F.C. Non-alcoholic Fatty Liver Disease (NAFLD), Type 2 Diabetes, and Non-viral Hepatocarcinoma: Pathophysiological Mechanisms and New Therapeutic Strategies. Biomedicines 2023, 11, 468. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Roden, M. Mitochondrial alterations in fatty liver diseases. J. Hepatol. 2023, 78, 415–429. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Kaplowitz, N.; Wong, N.; Arya, A.; Win, Z.T.; Win, S.H.; Phyu, E.H.; Kuemerle, C.; Suh, J.; et al. The central role of mitochondrial metabolism in hepatic steatosis. Explor. Dig. Dis. 2024, 3, 42–68. [Google Scholar] [CrossRef]
- Simon, L.; Molina, P.E. Cellular Bioenergetics: Experimental Evidence for Alcohol-induced Adaptations. Funct. Oxf. Engl. 2022, 3, zqac039. [Google Scholar] [CrossRef]
- LeFort, K.R.; Rungratanawanich, W.; Song, B.J. Contributing roles of mitochondrial dysfunction and hepatocyte apoptosis in liver diseases through oxidative stress, post-translational modifications, inflammation, and intestinal barrier dysfunction. Cell. Mol. Life. Sci. 2024, 81, 34. [Google Scholar] [CrossRef]
- Ma, X.; Chen, A.; Melo, L.; Clemente-Sanchez, A.; Chao, X.; Ahmadi, A.R.; Peiffer, B.; Sun, Z.; Sesaki, H.; Li, T.; et al. Loss of hepatic DRP1 exacerbates alcoholic hepatitis by inducing megamitochondria and mitochondrial maladaptation. Hepatology 2023, 77, 159–175. [Google Scholar] [CrossRef]
- Colell, A.; García-Ruiz, C.; Miranda, M.; Ardite, E.; Marí, M.; Morales, A.; Corrales, F.; Kaplowitz, N.; Fernández-Checa, J.C. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology 1998, 115, 1541–1551. [Google Scholar] [CrossRef]
- Bradford, B.U.; Rusyn, I. Swift increase in alcohol metabolism (SIAM): Understanding the phenomenon of hypermetabolism in liver. Alcohol 2005, 35, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Thoudam, T.; Gao, H.; Jiang, Y.; Huda, N.; Yang, Z.; Ma, J.; Liangpunsakul, S. Mitochondrial quality control in alcohol-associated liver disease. Hepatol. Commun. 2024, 8, e0534. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Rasineni, K.; Ganesan, M.; Donohue, T.M.; Kharbanda, K.K. Pathogenesis of Alcohol-Associated Liver Disease. J. Clin. Exp. Hepatol. 2022, 12, 1492–1513. [Google Scholar] [CrossRef] [PubMed]
- Acierno, C.; Caturano, A.; Pafundi, P.C.; Nevola, R.; Adinolfi, L.E.; Sasso, F.C. Nonalcoholic fatty liver disease and type 2 diabetes: Pathophysiological mechanisms shared between the two faces of the same coin. Explor. Med. 2020, 1, 287–306. [Google Scholar] [CrossRef]
- Bansal, S.K.; Bansal, M.B. Pathogenesis of MASLD and MASH–role of insulin resistance and lipotoxicity. Aliment. Pharmacol. Ther. 2024, 59, S10–S22. [Google Scholar] [CrossRef]
- Badmus, O.O.; Hillhouse, S.A.; Anderson, C.D.; Hinds, T.D.; Stec, D.E. Molecular mechanisms of metabolic associated fatty liver disease (MAFLD): Functional analysis of lipid metabolism pathways. Clin. Sci. 2022, 136, 1347–1366. [Google Scholar] [CrossRef]
- Rada, P.; González-Rodríguez, Á.; García-Monzón, C.; Valverde, Á.M. Understanding lipotoxicity in NAFLD pathogenesis: Is CD36 a key driver? Cell. Death Dis. 2020, 11, 802. [Google Scholar] [CrossRef]
- Arab, J.P.; Díaz, L.A.; Rehm, J.; Im, G.; Arrese, M.; Kamath, P.S.; Lucey, M.R.; Mellinger, J.; Thiele, M.; Thursz, M.; et al. Metabolic dysfunction and alcohol-related liver disease (MetALD): Position statement by an expert panel on alcohol-related liver disease. J. Hepatol. 2025, 82, 744–756. [Google Scholar] [CrossRef]
- Qu, W.; Ma, T.; Cai, J.; Zhang, X.; Zhang, P.; She, Z.; Wan, F.; Li, H. Liver Fibrosis and MAFLD: From Molecular Aspects to Novel Pharmacological Strategies. Front Med. 2021, 8, 761538. [Google Scholar] [CrossRef]
- Hoek, J.B.; Cahill, A.; Pastorino, J.G. Alcohol and Mitochondria: A Dysfunctional Relationship. Gastroenterology 2002, 122, 2049–2063. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, H.; Han, F.; Guo, P. Reactive Oxygen Species as Key Molecules in the Pathogenesis of Alcoholic Fatty Liver Disease and Nonalcoholic Fatty Liver Disease: Future Perspectives. Curr. Issues Mol. Biol. 2025, 47, 464. [Google Scholar] [CrossRef]
- Rice, J.; Lautrup, S.; Fang, E.F. NAD+ Boosting Strategies. Subcell. Biochem. 2024, 107, 63–90. [Google Scholar]
- Shin, S.; Kim, J.; Lee, J.Y.; Kim, J.; Oh, C.M. Mitochondrial Quality Control: Its Role in Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD). J. Obes. Metab. Syndr. 2023, 32, 289–302. [Google Scholar] [CrossRef]
- Zhao, P.; Kalhorn, T.F.; Slattery, J.T. Selective mitochondrial glutathione depletion by ethanol enhances acetaminophen toxicity in rat liver. Hepatology 2002, 36, 326–335. [Google Scholar] [CrossRef]
- Xiang, L.; Shao, Y.; Chen, Y. Mitochondrial dysfunction and mitochondrion-targeted therapeutics in liver diseases. J. Drug Target. 2021, 29, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Iturbe-Rey, S.; Maccali, C.; Arrese, M.; Aspichueta, P.; Oliveira, C.P.; Castro, R.E.; Lapitz, A.; Izquierdo-Sanchez, L.; Bujanda, L.; Perugorria, M.J.; et al. Lipotoxicity-driven metabolic dysfunction-associated steatotic liver disease (MASLD). Atherosclerosis. Gennaio 2025, 400, 119053. [Google Scholar] [CrossRef]
- Veluthakal, R.; Esparza, D.; Hoolachan, J.M.; Balakrishnan, R.; Ahn, M.; Oh, E.; Jayasena, C.S.; Thurmond, D.C. Mitochondrial Dysfunction, Oxidative Stress, and Inter-Organ Miscommunications in T2D Progression. Int. J. Mol. Sci. 2024, 25, 1504. [Google Scholar] [CrossRef] [PubMed]
- Ferdouse, A.; Clugston, R.D. Pathogenesis of Alcohol-Associated Fatty Liver: Lessons from Transgenic Mice. Front. Physiol. 2022, 13, 940974. [Google Scholar] [CrossRef] [PubMed]
- Sawada, K.; Chung, H.; Softic, S.; Moreno-Fernandez, M.E.; Divanovic, S. The bidirectional immune crosstalk in metabolic dysfunction-associated steatotic liver disease. Cell. Metab. 2023, 35, 1852–1871. [Google Scholar] [CrossRef]
- Peiseler, M.; Schwabe, R.; Hampe, J.; Kubes, P.; Heikenwälder, M.; Tacke, F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease–novel insights into cellular communication circuits. J. Hepatol. 2022, 77, 1136–1160. [Google Scholar] [CrossRef]
- Zdanowicz, K.; Kowalczuk-Kryston, M.; Olanski, W.; Werpachowska, I.; Mielech, W.; Lebensztejn, D.M. Increase in Serum MMP-9 and TIMP-1 Concentrations during Alcohol Intoxication in Adolescents—A Preliminary Study. Biomolecules 2022, 12, 710. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.Q.; Chen, Y.F.; Ma, C.; Cheng, X.; Guo, W.; Li, S. Advances in identifying risk factors of metabolic dysfunction-associated alcohol-related liver disease. Biomed. Pharmacother. 2025, 188, 118191. [Google Scholar] [CrossRef] [PubMed]
- Acharya, P.; Chouhan, K.; Weiskirchen, S.; Weiskirchen, R. Cellular Mechanisms of Liver Fibrosis. Front. Pharmacol. 2021, 12, 671640. [Google Scholar] [CrossRef] [PubMed]
- Acierno, C.; Nevola, R.; Barletta, F.; Rinaldi, L.; Sasso, F.C.; Adinolfi, L.E.; Caturano, A. Multidrug-Resistant Infections and Metabolic Syndrome: An Overlooked Bidirectional Relationship. Biomedicines 2025, 13, 1343. [Google Scholar] [CrossRef]
- Martín-Mateos, R.; Albillos, A. The Role of the Gut-Liver Axis in Metabolic Dysfunction-Associated Fatty Liver Disease. Front. Immunol. 2021, 12, 660179. [Google Scholar] [CrossRef]
- Malnick, S.D.H.; Alin, P.; Somin, M.; Neuman, M.G. Fatty Liver Disease-Alcoholic and Non-Alcoholic: Similar but Different. Int. J. Mol. Sci. 2022, 23, 16226. [Google Scholar] [CrossRef]
- Day, A.W.; Kumamoto, C.A. Gut Microbiome Dysbiosis in Alcoholism: Consequences for Health and Recovery. Front. Cell. Infect. Microbiol. 2022, 12, 840164. [Google Scholar] [CrossRef]
- Shu, J.Z.; Huang, Y.H.; He, X.H.; Liu, F.Y.; Liang, Q.Q.; Yong, X.T.; Xie, Y.-F. Gut microbiota differences, metabolite changes, and disease intervention during metabolic-dysfunction-related fatty liver progression. World J. Hepatol. 2025, 17, 103854. [Google Scholar] [CrossRef]
- Leal-Lassalle, H.; Estévez-Vázquez, O.; Cubero, F.J.; Nevzorova, Y.A. Metabolic and alcohol-associated liver disease (MetALD): A representation of duality. npj Gut Liver 2025, 2, 1. [Google Scholar] [CrossRef]
- Shukla, S.; Hsu, C.L. Alcohol Use Disorder and the Gut–Brain Axis: A Narrative Review of the Role of Gut Microbiota and Implications for Treatment. Microorganisms 2025, 13, 67. [Google Scholar] [CrossRef]
- Bettermann, K.; Hohensee, T.; Haybaeck, J. Steatosis and Steatohepatitis: Complex Disorders. Int. J. Mol. Sci. 2014, 15, 9924–9944. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhong, W. Targeting the gut barrier for the treatment of alcoholic liver disease. Liver Res. 2017, 1, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Torralba, M.; Tan, J.; Embree, M.; Zengler, K.; Stärkel, P.; van Pijkeren, J.-P.; DePew, J.; Loomba, R.; Ho, S.B.; et al. Supplementation of Saturated Long-chain Fatty Acids Maintains Intestinal Eubiosis and Reduces Ethanol-induced Liver Injury in Mice. Gastroenterology 2015, 148, 203–214.e16. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Gu, Y.; Li, L.; Liu, T.; Song, X.; Sun, Y.; Cao, X.; Wang, B.; Jiang, K.; Cao, H. Bile Acid–Gut Microbiota Axis in Inflammatory Bowel Disease: From Bench to Bedside. Nutrients 2021, 13, 3143. [Google Scholar] [CrossRef]
- Fleishman, J.S.; Kumar, S. Bile acid metabolism and signaling in health and disease: Molecular mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 2024, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Acierno, C.; Nevola, R.; Rinaldi, L.; Sasso, F.C.; Adinolfi, L.E.; Caturano, A. The Intestinal Thread of Fate: How the Microbiota Shapes the Story of Liver Disease. Livers 2025, 5, 17. [Google Scholar] [CrossRef]
- Wierzbicka-Rucińska, A.; Konopka, E.; Więckowski, S.; Jańczyk, W.; Świąder-Leśniak, A.; Świderska, J.; Trojanek, J.; Kułaga, Z.; Socha, P.; Bierła, J. Evaluation of Defensins as Markers of Gut Microbiota Disturbances in Children with Obesity and Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD). J. Clin. Med. 2025, 14, 3505. [Google Scholar] [CrossRef]
- Dumitru, A.; Tocia, C.; Bădescu, A.C.; Trandafir, A.; Alexandrescu, L.; Popescu, R.; Dumitru, E.; Chisoi, A.; Manea, M.; Matei, E.; et al. Linking gut permeability to liver steatosis: Noninvasive biomarker evaluation in MASLD patients—A prospective cross-sectional study. Medicine 2025, 104, e42476. [Google Scholar] [CrossRef]
- Williams, D.M.; Nagaraj, J.; Stephens, J.W.; Min, T. The Use of Non-invasive Biomarkers to Screen for Advanced Fibrosis Associated with Metabolic Dysfunction-associated Steatotic Liver Disease in People with Type 2 Diabetes: A Narrative Review. Touchreviews Endocrinol. 2025, 21, 24. [Google Scholar] [CrossRef]
- Yan, M.; Man, S.; Sun, B.; Ma, L.; Guo, L.; Huang, L.; Gao, W. Gut liver brain axis in diseases: The implications for therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 443. [Google Scholar] [CrossRef]
- Mishra, G.; Singh, P.; Molla, M.; Yimer, Y.S.; Dinda, S.C.; Chandra, P.; Singh, B.K.; Dagnew, S.B.; Assefa, A.N.; Ewunetie, A. Harnessing the potential of probiotics in the treatment of alcoholic liver disorders. Front. Pharmacol. 2023, 14, 1212742. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.Y.; Wu, G.S.; Li, C.; Ma, W.; Luo, H.R. Clinical efficacy of probiotics in the treatment of alcoholic liver disease: A systematic review and meta-analysis. Front. Cell. Infect. Microbiol. 2024, 14, 1358063. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.; Vergis, N. Mitochondrial dysfunction and liver disease: Role, relevance, and potential for therapeutic modulation. Ther. Adv. Gastroenterol. 2021, 14, 17562848211031394. [Google Scholar] [CrossRef]
- Gueguen, N.; Lenaers, G.; Reynier, P.; Weissig, V.; Edeas, M. Mitochondrial Dysfunction in Mitochondrial Medicine: Current Limitations, Pitfalls, and Tomorrow. Methods Mol. Biol. 2021, 2276, 1–29. [Google Scholar]
- Meyer, M.; Schwärzler, J.; Jukic, A.; Tilg, H. Innate Immunity and MASLD. Biomolecules 2024, 14, 476. [Google Scholar] [CrossRef]
- Thomes, P.G.; Rasineni, K.; Saraswathi, V.; Kharbanda, K.K.; Clemens, D.L.; Sweeney, S.A.; Kubik, J.L.; Donohue, T.M., Jr.; Casey, C.A. Natural Recovery by the Liver and Other Organs after Chronic Alcohol Use. Alcohol Res. Curr. Rev. 2021, 41, 05. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Valvano, C.M.; De Nardo, W.; Watt, M.J. Integrative Metabolism in MASLD and MASH: Pathophysiology and Emerging Mechanisms. J. Hepatol. 2025. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Wu, L.; Wei, J.; Wu, J.; Guo, C. The Gut Microbiome and Ferroptosis in MAFLD. J. Clin. Transl. Hepatol. 2022, 11, 174–187. [Google Scholar] [CrossRef]
- Syed-Abdul, M.M. Lipid Metabolism in Metabolic-Associated Steatotic Liver Disease (MASLD). Metabolites 2023, 14, 12. [Google Scholar] [CrossRef]
- Vesković, M.; Šutulović, N.; Hrnčić, D.; Stanojlović, O.; Macut, D.; Mladenović, D. The Interconnection between Hepatic Insulin Resistance and Metabolic Dysfunction-Associated Steatotic Liver Disease-The Transition from an Adipocentric to Liver-Centric Approach. Curr. Issues Mol. Biol. 2023, 45, 9084–9102. [Google Scholar] [CrossRef]
- Geng, Y.; Faber, K.N.; De Meijer, V.E.; Blokzijl, H.; Moshage, H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol. Int. 2021, 15, 21–35. [Google Scholar] [CrossRef]
- Prasun, P.; Ginevic, I.; Oishi, K. Mitochondrial dysfunction in nonalcoholic fatty liver disease and alcohol related liver disease. Hepatol. 2021, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhou, Y.; Wang, D.; Huang, Z.; Xiao, X.; Zheng, Q.; Li, S.; Long, D.; Feng, L. Mitochondrial Dysfunction in Metabolic Dysfunction Fatty Liver Disease (MAFLD). Int. J. Mol. Sci. 2023, 24, 17514. [Google Scholar] [CrossRef]
- Ciardullo, S.; Mantovani, A.; Morieri, M.L.; Muraca, E.; Invernizzi, P.; Perseghin, G. Impact of MASLD and MetALD on clinical outcomes: A meta-analysis of preliminary evidence. Liver Int. 2024, 44, 1762–1767. [Google Scholar] [CrossRef] [PubMed]
- Ayares, G.; Diaz, L.A.; Idalsoaga, F.; Alkhouri, N.; Noureddin, M.; Bataller, R.; Loomba, R.; Arab, J.P.; Arrese, M. MetALD: New Perspectives on an Old Overlooked Disease. Liver Int. 2025, 45, e70017. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J.; Johnson, C.R.; Koshy, R.; Hess, S.Y.; Qureshi, U.A.; Mynak, M.L.; Fischer, P.R. Thiamine deficiency disorders: A clinical perspective. Ann. N. Y. Acad. Sci. 2021, 1498, 9–28. [Google Scholar] [CrossRef]
- Kalyesubula, M.; Mopuri, R.; Asiku, J.; Rosov, A.; Yosefi, S.; Edery, N.; Bocobza, S.; Moallem, U.; Dvir, H. High-dose vitamin B1 therapy prevents the development of experimental fatty liver driven by overnutrition. Dis. Model. Mech. 2021, 14, dmm048355. [Google Scholar] [CrossRef]
- Duthie, S.J.; Narayanan, S.; Brand, G.M.; Pirie, L.; Grant, G. Impact of Folate Deficiency on DNA Stability. J. Nutr. 2002, 132, 2444S–2449S. [Google Scholar] [CrossRef]
- Sodum, N.; Kumar, G.; Bojja, S.L.; Kumar, N.; Rao, C.M. Epigenetics in NAFLD/NASH: Targets and therapy. Pharmacol. Res. Maggio 2021, 167, 105484. [Google Scholar] [CrossRef]
- Duthie, S.J.; Grant, G.; Pirie, L.P.; Watson, A.J.; Margison, G.P. Folate deficiency alters hepatic and colon MGMT and OGG-1 DNA repair protein expression in rats but has no impact on genome-wide DNA methylation. Cancer Prev. Res. 2010, 3, 92–100. [Google Scholar] [CrossRef]
- Riggio, O.; Merli, M.; Capocaccia, L.; Caschera, M.; Zullo, A.; Pinto, G.; Gaudio, E.; Franchitto, A.; Spagnoli, R.; D’AQuilino, E.; et al. Zinc supplementation reduces blood ammonia and increases liver ornithine transcarbamylase activity in experimental cirrhosis. Hepatology 1992, 16, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Baj, J.; Flieger, W.; Teresiński, G.; Buszewicz, G.; Sitarz, R.; Forma, A.; Karakuła, K.; Maciejewski, R. Magnesium, Calcium, Potassium, Sodium, Phosphorus, Selenium, Zinc, and Chromium Levels in Alcohol Use Disorder: A Review. J. Clin. Med. 2020, 9, 1901. [Google Scholar] [CrossRef] [PubMed]
- Vanoni, F.O.; Milani, G.P.; Agostoni, C.; Treglia, G.; Faré, P.B.; Camozzi, P.; Lava, S.A.G.; Bianchetti, M.G.; Janett, S. Magnesium Metabolism in Chronic Alcohol-Use Disorder: Meta-Analysis and Systematic Review. Nutrients 2021, 13, 1959. [Google Scholar] [CrossRef]
- Yang, Z.; Li, J.; Zhang, J.; Sun, C. Magnesium Deficiency: The Insidious Executor of the Liver Disease. J. Am. Nutr. Assoc. 2024, 44, 439–453. [Google Scholar] [CrossRef]
- Butts, M.; Sundaram, V.L.; Murughiyan, U.; Borthakur, A.; Singh, S. The Influence of Alcohol Consumption on Intestinal Nutrient Absorption: A Comprehensive Review. Nutrients 2023, 15, 1571. [Google Scholar] [CrossRef]
- Muhamad, R.; Akrivaki, A.; Papagiannopoulou, G.; Zavridis, P.; Zis, P. The Role of Vitamin B6 in Peripheral Neuropathy: A Systematic Review. Nutrients 2023, 15, 2823. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.C.; Steinkamp, M.P.; Kawatsuji, R.; Tartaglini, E.; Pinkus, J.L.; Pinkus, G.S.; Fleming, M.D.; Neufeld, E.J. Characterization of a murine high-affinity thiamine transporter, Slc19a2. Mol. Genet. Metab. 2001, 74, 273–280. [Google Scholar] [CrossRef]
- Jophlin, L.; Liu, T.Y.; McClain, C.J. Nutritional deficiencies in alcohol use disorder/alcohol-associated liver disease. Curr. Opin. Gastroenterol. 2024, 40, 112–117. [Google Scholar] [CrossRef]
- Pohl, K.; Moodley, P.; Dhanda, A.D. Alcohol’s Impact on the Gut and Liver. Nutrients 2021, 13, 3170. [Google Scholar] [CrossRef]
- Butts, M.; Singh, S.; Haynes, J.; Arthur, S.; Sundaram, U. Moderate Alcohol Consumption Uniquely Regulates Sodium-Dependent Glucose Co-Transport in Rat Intestinal Epithelial Cells In Vitro and In Vivo. J. Nutr. 2020, 150, 747–755. [Google Scholar] [CrossRef]
- Tomaszewska, E.; Muszyński, S.; Arczewska-Włosek, A.; Domaradzki, P.; Pyz-Łukasik, R.; Donaldson, J.; Świątkiewicz, S. Cholesterol Content, Fatty Acid Profile and Health Lipid Indices in the Egg Yolk of Eggs from Hens at the End of the Laying Cycle, Following Alpha-Ketoglutarate Supplementation. Foods 2021, 10, 596. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.K.; Wong, R.J.; Dasarathy, S.; Abdelmalek, M.F.; Neuschwander-Tetri, B.A.; Limketkai, B.N.; Petrey, J.; McClain, C.J. ACG Clinical Guideline: Malnutrition and Nutritional Recommendations in Liver Disease. Am. J. Gastroenterol. 2025, 120, 950–972. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Alcohol Metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef]
- Genchi, V.A.; Cignarelli, A.; Sansone, A.; Yannas, D.; Dalla Valentina, L.; Renda Livraghi, D.; Spaggiari, G.; Santi, D. Understanding the Role of Alcohol in Metabolic Dysfunction and Male Infertility. Metabolites 2024, 14, 626. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Del Cerro, E.; Félix, J.; Martínez-Poyato, M.C.; De la Fuente, M. Supplementation with Bioactive Compounds Improves Health and Rejuvenates Biological Age in Postmenopausal Women. Biomolecules 2025, 15, 739. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; Maggini, S. Vitamin C and Immune Function. Nutrients 2017, 9, 1211. [Google Scholar] [CrossRef]
- Chen, C.; Xie, L.; Zhang, M.; Shama Cheng, K.K.Y.; Jia, W. The interplay between the muscle and liver in the regulation of glucolipid metabolism. J. Mol. Cell Biol. 2023, 15, mjad073. [Google Scholar] [CrossRef]
- Li, X.; He, J.; Sun, Q. The prevalence and effects of sarcopenia in patients with metabolic dysfunction-associated steatotic liver disease (MASLD): A systematic review and meta-analysis. Clin. Nutr. 2024, 43, 2005–2016. [Google Scholar] [CrossRef]
- Viswanath, A.; Fouda, S.; Fernandez, C.J.; Pappachan, J.M. Metabolic-associated fatty liver disease and sarcopenia: A double whammy. World J. Hepatol. 2024, 16, 152–163. [Google Scholar] [CrossRef]
- Isakov, V. Metabolic dysfunction-associated steatotic liver disease: A story of muscle and mass. World J. Gastroenterol. 2025, 31, 105346. [Google Scholar] [CrossRef]
- DiLeo, M.R.; Hall, R.E.; Vellers, H.L.; Daniels, C.L.; Levitt, D.E. Alcohol Alters Skeletal Muscle Bioenergetic Function: A Scoping Review. Int. J. Mol. Sci. 2024, 25, 12280. [Google Scholar] [CrossRef] [PubMed]
- Alcohol impairs skeletal muscle protein synthesis and mTOR signaling in a time-dependent manner following electrically stimulated muscle contraction. J. Appl. Physiol. 2014, 117, 1170–1179. [CrossRef]
- Levitt, D.E.; Chalapati, N.; Prendergast, M.J.; Simon, L.; Molina, P.E. Ethanol-Impaired Myogenic Differentiation is Associated with Decreased Myoblast Glycolytic Function. Alcohol. Clin. Exp. Res. 2020, 44, 2166–2176. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Vitamin D and Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD): An Update. Nutrients 2020, 12, 3302. [Google Scholar] [CrossRef]
- Agoncillo, M.; Yu, J.; Gunton, J.E. The Role of Vitamin D in Skeletal Muscle Repair and Regeneration in Animal Models and Humans: A Systematic Review. Nutrients 2023, 15, 437. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef]
- Ladang, A.; Beaudart, C.; Reginster, J.Y.; Al-Daghri, N.; Bruyère, O.; Burlet, N.; Cesari, M.; Cherubini, A.; da Silva, M.C.; Cooper, C.; et al. Biochemical Markers of Musculoskeletal Health and Aging to be Assessed in Clinical Trials of Drugs Aiming at the Treatment of Sarcopenia: Consensus Paper from an Expert Group Meeting Organized by the European Society for Clinical and Economic Aspects of Osteoporosis, Osteoarthritis and Musculoskeletal Diseases (ESCEO) and the Centre Académique de Recherche et d’Expérimentation en Santé (CARES SPRL), Under the Auspices of the World Health Organization Collaborating Center for the Epidemiology of Musculoskeletal Conditions and Aging. Calcif. Tissue Int. 2023, 112, 197–217. [Google Scholar]
- Zhao, M.; Zhou, X.; Yuan, C.; Li, R.; Ma, Y.; Tang, X. Association between serum irisin concentrations and sarcopenia in patients with liver cirrhosis: A cross-sectional study. Sci. Rep. 2020, 10, 16093. [Google Scholar] [CrossRef]
- Shen, C.; Wu, K.; Ke, Y.; Zhang, Q.; Chen, S.; Li, Q.; Ruan, Y.; Yang, X.; Liu, S.; Hu, J. Circulating irisin levels in patients with MAFLD: An updated systematic review and meta-analysis. Front. Endocrinol. 2024, 15, 1464951. [Google Scholar] [CrossRef]
- Xiong, X.Q.; Chen, D.; Sun, H.J.; Ding, L.; Wang, J.J.; Chen, Q.; Li, Y.-H.; Zhou, Y.-B.; Han, Y.; Zhang, F.; et al. FNDC5 overexpression and irisin ameliorate glucose/lipid metabolic derangements and enhance lipolysis in obesity. Biochim. Biophys Acta BBA-Mol Basis Dis. 2015, 1852, 1867–1875. [Google Scholar] [CrossRef]
- Robinson, S.; Granic, A.; Sayer, A.A. Micronutrients and sarcopenia: Current perspectives. Proc. Nutr. Soc. 2021, 80, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Liu, X.; Zhao, J.; Zhang, Y.; Qiu, L.; Zhang, J. Association of sarcopenia and physical activity on the severity of metabolic dysfunction-associated steatotic liver disease among United States adults: NHANES 2017-2018. Front. Aging 2025, 6, 1573170. [Google Scholar] [CrossRef]
- Drummond, C.; Hillyard, M.; Leonhardt, M.; Wurst, F.; Dom, G.; Mann, K.; Bramness, J.G. Comparison of European Clinical Guidelines on the Management of Alcohol Use Disorders. Eur. Addict. Res. 2021, 27, 227–236. [Google Scholar] [CrossRef]
- Serio, F.; Imbriani, G.; Acito, M.; Moretti, M.; Fanizzi, F.P.; De Donno, A.; Valacchi, G. Moderate red wine intake and cardiovascular health protection: A literature review. Food Funct. 2023, 14, 6346–6362. [Google Scholar] [CrossRef] [PubMed]
- Hoek, A.G.; van Oort, S.; Mukamal, K.J.; Beulens, J.W.J. Alcohol Consumption and Cardiovascular Disease Risk: Placing New Data in Context. Curr. Atheroscler. Rep. 2022, 24, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Llamosas-Falcón, L.; Rehm, J.; Bright, S.; Buckley, C.; Carr, T.; Kilian, C.; Lasserre, A.M.; Lemp, J.M.; Zhu, Y.; Probst, C. The Relationship Between Alcohol Consumption, BMI, and Type 2 Diabetes: A Systematic Review and Dose-Response Meta-analysis. Diabetes Care 2023, 46, 2076–2083. [Google Scholar] [CrossRef]
- Ajmera, V.H.; Terrault, N.A.; Harrison, S.A. Is moderate alcohol use in nonalcoholic fatty liver disease good or bad? A critical review. Hepatology 2017, 65, 2090–2099. [Google Scholar] [CrossRef]
- Wongtrakul, W.; Niltwat, S.; Charatcharoenwitthaya, P. The Effects of Modest Alcohol Consumption on Non-alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Front. Med. 2021, 8, 744713. [Google Scholar] [CrossRef]
- Cao, G.; Yi, T.; Liu, Q.; Wang, M.; Tang, S. Alcohol consumption and risk of fatty liver disease: A meta-analysis. PeerJ 2016, 4, e2633. [Google Scholar] [CrossRef]
- Abdelmegeed, M.A.; Ha, S.K.; Choi, Y.; Akbar, M.; Song, B.J. Role of CYP2E1 in mitochondrial dysfunction and hepatic tissue injury in alcoholic and non-alcoholic diseases. Curr. Mol. Pharmacol. 2017, 10, 207–225. [Google Scholar] [CrossRef]
- Abdelhameed, F.; Mustafa, A.; Kite, C.; Lagojda, L.; Dallaway, A.; Than, N.N.; Kassi, E.; Kyrou, I.; Randeva, H.S. Gut Microbiota and Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD): Emerging Pathogenic Mechanisms and Therapeutic Implications. Livers 2025, 5, 11. [Google Scholar] [CrossRef]
- Luo, L.; Ye, J.; Zhou, T.; Dong, Z.; Feng, S.; Wang, W.; Zhuo, S.; Zhong, B. Weight-Loss Plateau during Lifestyle Intervention Predicts Treatment Response in Patients with Metabolic Dysfunction-Associated Steatotic Liver Disease and Obesity. Obes. Facts. 2025, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Stockwell, T.; Naimi, T.; Churchill, S.; Clay, J.; Sherk, A. Association Between Daily Alcohol Intake and Risk of All-Cause Mortality: A Systematic Review and Meta-analyses. JAMA Netw. Open 2023, 6, e236185. [Google Scholar] [CrossRef]
- Boccatonda, A.; Andreetto, L.; D’Ardes, D.; Cocco, G.; Rossi, I.; Vicari, S.; Schiavone, O.; Cipollone, F.; Guagnano, M.T. From NAFLD to MAFLD: Definition, Pathophysiological Basis and Cardiovascular Implications. Biomedicines 2023, 11, 883. [Google Scholar] [CrossRef] [PubMed]
- Donroe, J.H.; Edelman, E.J. Alcohol Use. Ann. Intern. Med. 2022, 175, ITC145–ITC160. [Google Scholar] [CrossRef]
- Caballeria, E.; Pons-Cabrera, M.T.; Balcells-Oliveró, M.; Braddick, F.; Gordon, R.; Gual, A.; Matrai, S.; López-Pelayo, H. «Doctor, Can I Drink an Alcohol-Free Beer?» Low-Alcohol and Alcohol-Free Drinks in People with Heavy Drinking or Alcohol Use Disorders: Systematic Review of the Literature. Nutrients 2022, 14, 3925. [Google Scholar] [CrossRef]
- Botwright, S.; Sutawong, J.; Kingkaew, P.; Anothaisintawee, T.; Dabak, S.V.; Suwanpanich, C.; Promchit, N.; Kampang, R.; Isaranuwatchai, W. Which interventions for alcohol use should be included in a universal healthcare benefit package? An umbrella review of targeted interventions to address harmful drinking and dependence. BMC Public Health 2023, 23, 382. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). J. Hepatol. 2024, 81, 492–542. [Google Scholar] [CrossRef]
- Abdelhameed, F.; Kite, C.; Lagojda, L.; Dallaway, A.; Chatha, K.K.; Chaggar, S.S.; Dalamaga, M.; Kassi, E.; Kyrou, I.; Randeva, H.S. Non-invasive Scores and Serum Biomarkers for Fatty Liver in the Era of Metabolic Dysfunction-associated Steatotic Liver Disease (MASLD): A Comprehensive Review from NAFLD to MAFLD and MASLD. Curr. Obes. Rep. 2024, 13, 510–531. [Google Scholar] [CrossRef]
- Lombardi, R.; Petta, S.; Pisano, G.; Dongiovanni, P.; Rinaldi, L.; Adinolfi, L.E.; Acierno, C.; Valenti, L.; Boemi, R.; Spatola, F.; et al. FibroScan Identifies Patients with Nonalcoholic Fatty Liver Disease and Cardiovascular Damage. Clin. Gastroenterol. Hepatol. 2020, 18, 517–519. [Google Scholar] [CrossRef]
- Peng, H.; Pan, L.; Ran, S.; Wang, M.; Huang, S.; Zhao, M.; Cao, Z.; Yao, Z.; Xu, L.; Yang, Q.; et al. Prediction of MAFLD and NAFLD using different screening indexes: A cross-sectional study in U.S. adults. Front Endocrinol. 2023, 14, 1083032. [Google Scholar] [CrossRef] [PubMed]
- Sarkar Das, T.; Meng, X.; Abdallah, M.; Bilal, M.; Sarwar, R.; Shaukat, A. An Assessment of the Feasibility, Patient Acceptance, and Performance of Point-of-Care Transient Elastography for Metabolic-Dysfunction-Associated Steatotic Liver Disease (MASLD): A Systematic Review and Meta-Analysis. Diagnostics 2024, 14, 2478. [Google Scholar] [CrossRef]
- Mambrini, S.P.; Grillo, A.; Colosimo, S.; Zarpellon, F.; Pozzi, G.; Furlan, D.; Amodeo, G.; Bertoli, S. Diet and physical exercise as key players to tackle MASLD through improvement of insulin resistance and metabolic flexibility. Front. Nutr. 2024, 11, 1426551. [Google Scholar] [CrossRef] [PubMed]
- Jamil, A.; Chivese, T.; Elshaikh, U.; Sendall, M. Efficacy of the Mediterranean diet in treating metabolic dysfunction-associated steatotic liver disease (MASLD) in children and adolescents: A systematic review and meta-analysis. BMC Public Health. 2024, 24, 2701. [Google Scholar] [CrossRef]
- Guasch-Ferré, M.; Willett, W.C. The Mediterranean diet and health: A comprehensive overview. J. Intern. Med. 2021, 290, 549–566. [Google Scholar] [CrossRef]
- Brouns, F. Overweight and diabetes prevention: Is a low-carbohydrate-high-fat diet recommendable? Eur. J. Nutr. 2018, 57, 1301–1312. [Google Scholar] [CrossRef]
- Akbari, M.; Vali, M.; Rezaei, S.; Bazmi, S.; Tabrizi, R.; Lankarani, K.B. Comparison of weight loss effects among overweight/obese adults: A network meta-analysis of mediterranean, low carbohydrate, and low-fat diets. Clin. Nutr. ESPEN 2024, 64, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Simonson, M.; Boirie, Y.; Guillet, C. Protein, amino acids and obesity treatment. Rev. Endocr. Metab. Disord. 2020, 21, 341–353. [Google Scholar] [CrossRef]
- García-Conesa, M.T.; Philippou, E.; Pafilas, C.; Massaro, M.; Quarta, S.; Andrade, V.; Jorge, R.; Chervenkov, M.; Ivanova, T.; Dimitrova, D.; et al. Exploring the Validity of the 14-Item Mediterranean Diet Adherence Screener (MEDAS): A Cross-National Study in Seven European Countries around the Mediterranean Region. Nutrients 2020, 12, 2960. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, S.; Lee, Y.; Kwon, Y.J.; Lee, J.W. Higher Adherence to the Mediterranean Diet Is Associated with a Lower Risk of Steatotic, Alcohol-Related, and Metabolic Dysfunction-Associated Steatotic Liver Disease: A Retrospective Analysis. Nutrients 2024, 16, 3551. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | MASLD | MetALD | MASH | ALD | References |
|---|---|---|---|---|---|
| Main Etiology | Metabolic dysfunction (e.g., obesity, T2DM) | Metabolic dysfunction + moderate alcohol | MASLD with inflammation and hepatocellular injury | Chronic heavy alcohol intake | [2,3,6] |
| Alcohol Intake | Low to none | Moderate (men < 50 g/day, women < 30 g/day) | Same as MASLD | High (men > 50 g/day, women > 30 g/day) | [6,8,10] |
| Steatosis | Yes | Yes | Yes | Yes | [2,6] |
| Inflammation and Ballooning | No (or minimal) | No (or minimal) | Yes | Yes | [3,7,8] |
| Fibrosis Risk | Variable, increases with comorbidities | Higher than MASLD alone | High (progressive) | High, esp. with sustained alcohol use | [8,9,10] |
| Progression Speed | Usually slow | Intermediate | Faster than MASLD | Variable; can be rapid | [5,8,9] |
| Clinical Risk Factors | Obesity, IR, T2DM, dyslipidemia | Same as MASLD + alcohol use | Same as MASLD | Alcohol use disorder, binge drinking | [3,6,9] |
| Diagnosis Criteria | Imaging or biopsy: steatosis + metabolic criteria | Steatosis + metabolic criteria + moderate alcohol | Histologic: steatohepatitis (inflammation + ballooning) | History of alcohol use + liver injury | [2,6,8] |
| Treatment Focus | Lifestyle: weight loss, control of metabolic comorbidities | Same as MASLD + reduce/stop alcohol | Intensive lifestyle + monitor inflammation/fibrosis | Abstinence, nutritional support | [8,9,13] |
| Histology Needed? | Not always | Not always | Yes (for definite diagnosis) | Often used to assess severity | [3,8,9] |
| Mechanism | Description | Impact on Liver | References |
|---|---|---|---|
| Excess NADH production | Ethanol oxidation via ADH/ALDH2 generates high NADH levels | Inhibits β-oxidation and gluconeogenesis | [15,17,22] |
| CYP2E1 overexpression | Induced by alcohol and insulin resistance | Promotes ROS generation, lipid peroxidation, and mitochondrial injury | [18,19,28] |
| Redox imbalance | Altered NAD+/NADH ratio affects metabolic homeostasis | Leads to oxidative stress and ATP production inefficiency | [15,26,31] |
| Megamitochondria formation | Observed in alcohol-exposed hepatocytes | Reflects impaired mitochondrial dynamics and fusion/fission | [29] |
| mtGSH depletion | Due to increased ROS and impaired transport | Reduces antioxidant capacity | [30,44] |
| Mitochondrial respiratory chain damage | Impairs complexes I and III of ETC | Triggers apoptosis via cytochrome c release | [28,43] |
| Mechanism | Description | Hepatic Consequences | References |
|---|---|---|---|
| Dysbiosis | Reduction in SCFA-producing bacteria, increase in pathogens | Promotes endotoxemia and inflammation | [57,58] |
| Barrier protein loss | Reduced ZO-1 and occludin expression | Increases intestinal permeability | [60] |
| LPS translocation | Entry of bacterial endotoxin into portal circulation | Activates TLR4 on Kupffer cells, induces cytokines | [60,61] |
| PAMPs and metabolites | Ethanol-derived and microbial toxins (e.g., DCA, PEth) | Promote hepatic inflammation and FXR/PXR modulation | [64,65] |
| Pathway | MASLD | ALD | MetALD Synergy | References |
|---|---|---|---|---|
| CYP2E1 induction | Moderate, driven by insulin resistance | High, ethanol induced | Synergistic ROS production and mitochondrial injury | [19,73] |
| Lipotoxicity | Excess nutrients activate SREBP-1c, ChREBP | Acetaldehyde and NADH promote steatosis | Enhanced inflammatory cascades | [24,80,82] |
| Intestinal permeability | Metabolic inflammation and dysbiosis | Alcohol disrupts tight junctions | Facilitates endotoxemia and fibrosis | [60,66] |
| Fibrosis progression | Driven by TGF-β, PPAR modulation | Mediated via HSC activation and apoptosis | Accelerated transition to MASH | [28,83,84] |
| Micronutrient | Function | Consequence of Deficiency | References |
|---|---|---|---|
| Thiamine (B1) | Cofactor in energy metabolism | Lactic acidosis, worsened insulin resistance | [86,87] |
| Folate | DNA synthesis and methylation | Genomic instability, carcinogenesis risk | [88,90] |
| Zinc | Enzymatic activity, ammonia detoxification | Encephalopathy, immune dysfunction | [91,92] |
| Magnesium | Mitochondrial stability, glucose metabolism | Insulin resistance, cramps | [93,94] |
| B-complex vitamins | Metabolic cofactors | Neuropathy, cognitive deficits | [95,96] |
| Biomarker/Score | Function | Clinical Use | Limitations | References |
|---|---|---|---|---|
| ALT/AST | Hepatocellular enzymes | Basic liver injury marker | Poor specificity; normal in advanced disease | [5,6,39,139] |
| Cytokeratin-18 (CK-18) | Apoptosis marker | Marker of steatohepatitis | Variable sensitivity | [39] |
| FIB-4 | Composite index (age, AST, ALT, platelets) | Fibrosis staging | Overlaps with other liver diseases | [39,139,140,141] |
| ELF score | Fibrosis biomarker panel | Non-invasive fibrosis staging | Limited availability | [39,139,140,141] |
| Pro-C3 | Collagen turnover marker | Advanced fibrosis indicator | Needs standardization | [39,139,140,141] |
| Hepamet, ADAPT, Agile 3+ | Risk scores integrating metabolic and fibrosis parameters | Predict progression, fibrosis | Require validation in MetALD | [39,139,140,141] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acierno, C.; Barletta, F.; Caturano, A.; Nevola, R.; Sasso, F.C.; Adinolfi, L.E.; Rinaldi, L. Alcohol Consumption and Liver Metabolism in the Era of MASLD: Integrating Nutritional and Pathophysiological Insights. Nutrients 2025, 17, 2229. https://doi.org/10.3390/nu17132229
Acierno C, Barletta F, Caturano A, Nevola R, Sasso FC, Adinolfi LE, Rinaldi L. Alcohol Consumption and Liver Metabolism in the Era of MASLD: Integrating Nutritional and Pathophysiological Insights. Nutrients. 2025; 17(13):2229. https://doi.org/10.3390/nu17132229
Chicago/Turabian StyleAcierno, Carlo, Fannia Barletta, Alfredo Caturano, Riccardo Nevola, Ferdinando Carlo Sasso, Luigi Elio Adinolfi, and Luca Rinaldi. 2025. "Alcohol Consumption and Liver Metabolism in the Era of MASLD: Integrating Nutritional and Pathophysiological Insights" Nutrients 17, no. 13: 2229. https://doi.org/10.3390/nu17132229
APA StyleAcierno, C., Barletta, F., Caturano, A., Nevola, R., Sasso, F. C., Adinolfi, L. E., & Rinaldi, L. (2025). Alcohol Consumption and Liver Metabolism in the Era of MASLD: Integrating Nutritional and Pathophysiological Insights. Nutrients, 17(13), 2229. https://doi.org/10.3390/nu17132229