
High-Fat Diet-Induced Diabetic Cardiomyopathy in Female Zebrafish: Cardiac Pathology and Functional Decline Mediated by Type 2 Diabetes
 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Subjects and Grouping
2.2. Intraperitoneal Glucose Tolerance Test (IPGTT) and Biochemical Analysis
2.3. Tissue Collection
2.4. Echocardiography
2.5. H&E Staining
2.6. Masson Staining
2.7. Oil Red O Staining
2.8. DHE Staining
2.9. Transmission Electron Microscopy (TEM)
2.10. Mitochondrial Respiration Assay
2.11. Western Blot
2.12. Real-Time Quantitative PCR
2.13. Statistical Analysis
3. Results
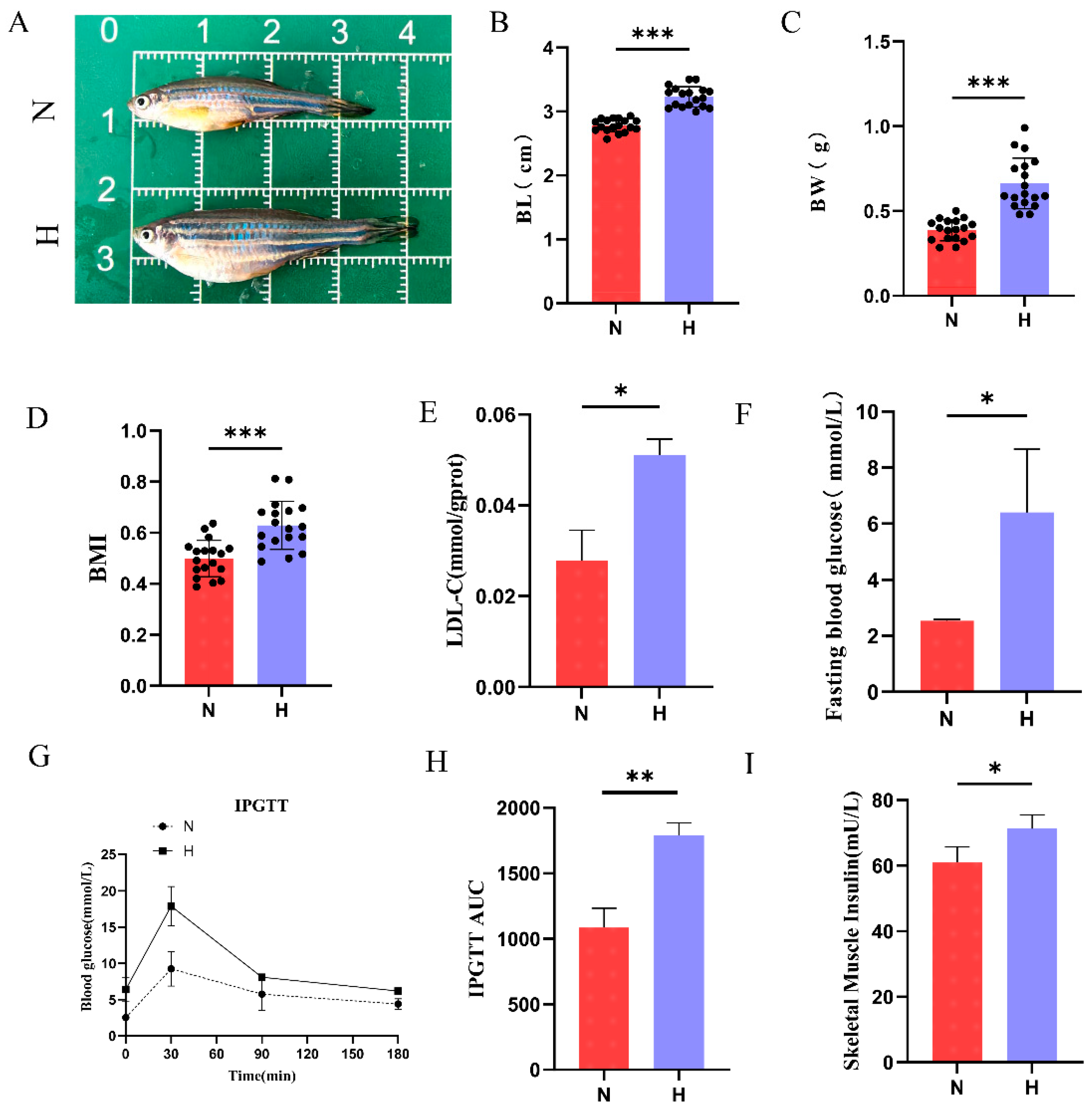
3.1. HFD Induces Obesity and Type 2 Diabetes in Zebrafish
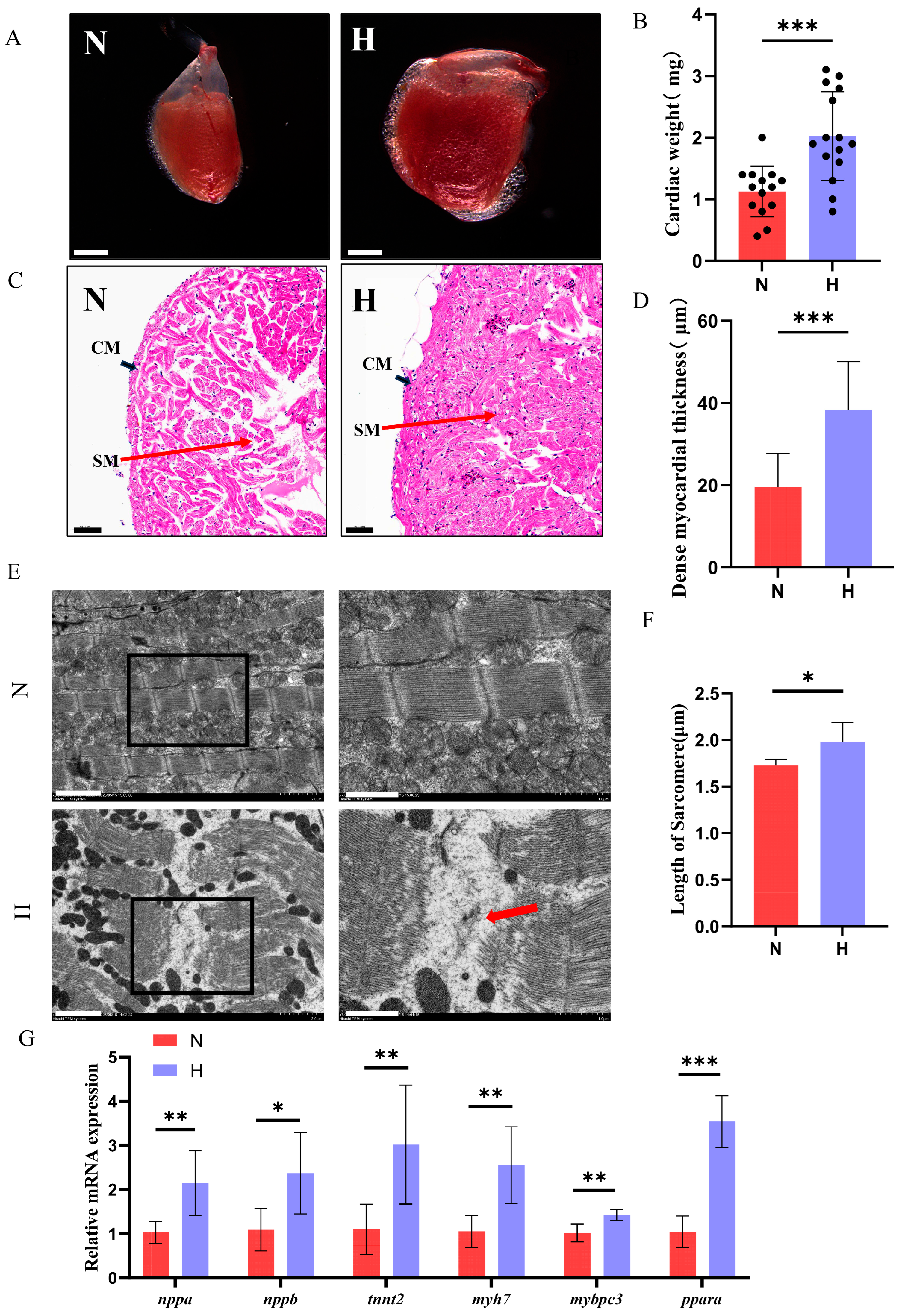
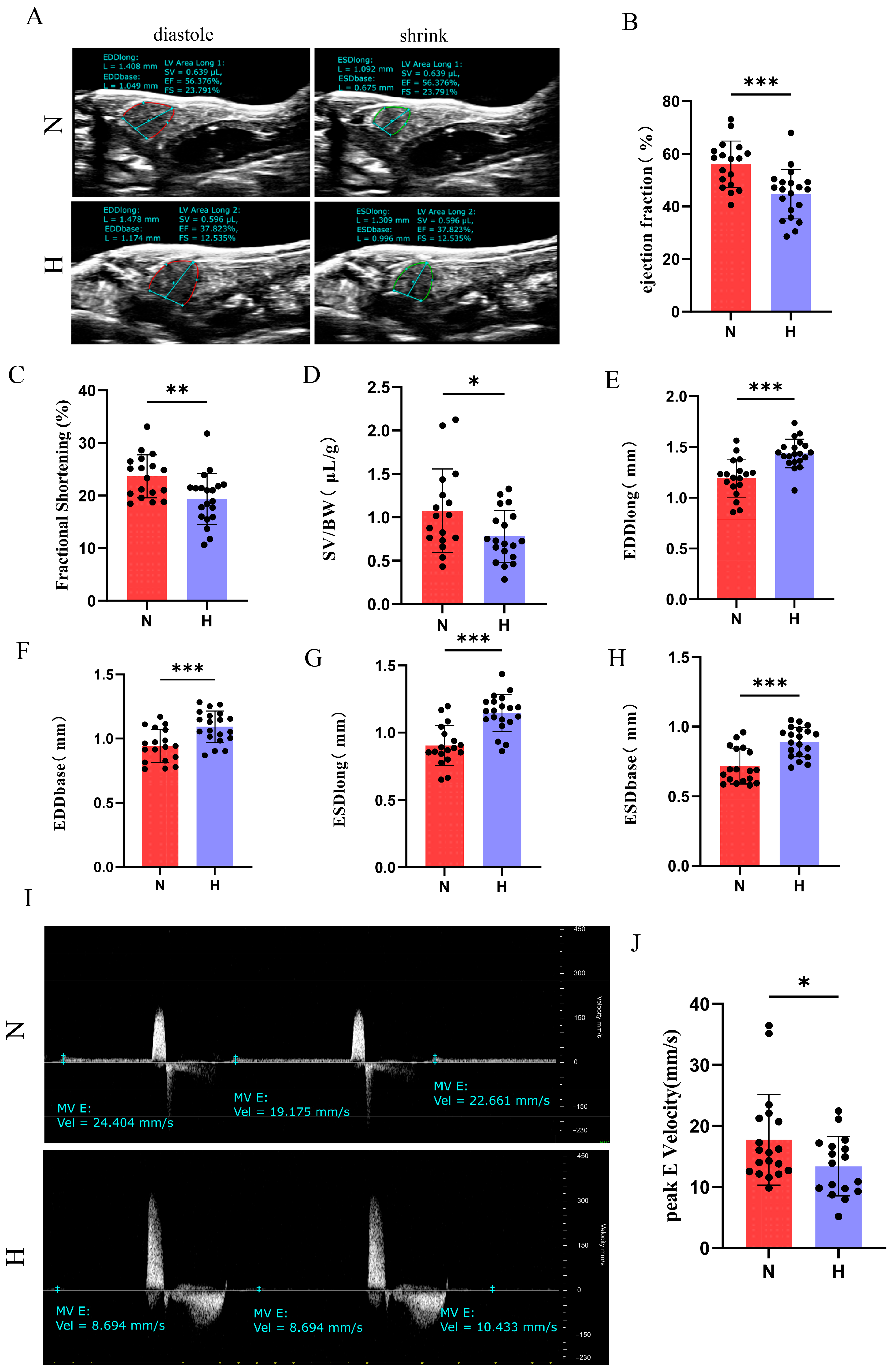
3.2. T2DM Induces Cardiac Hypertrophy and Functional Impairment in Zebrafish
3.3. T2DM Disrupts Cardiac Glucolipid Metabolism in Zebrafish
3.4. T2DM Triggers Cardiac Inflammation and Fibrosis in Zebrafish
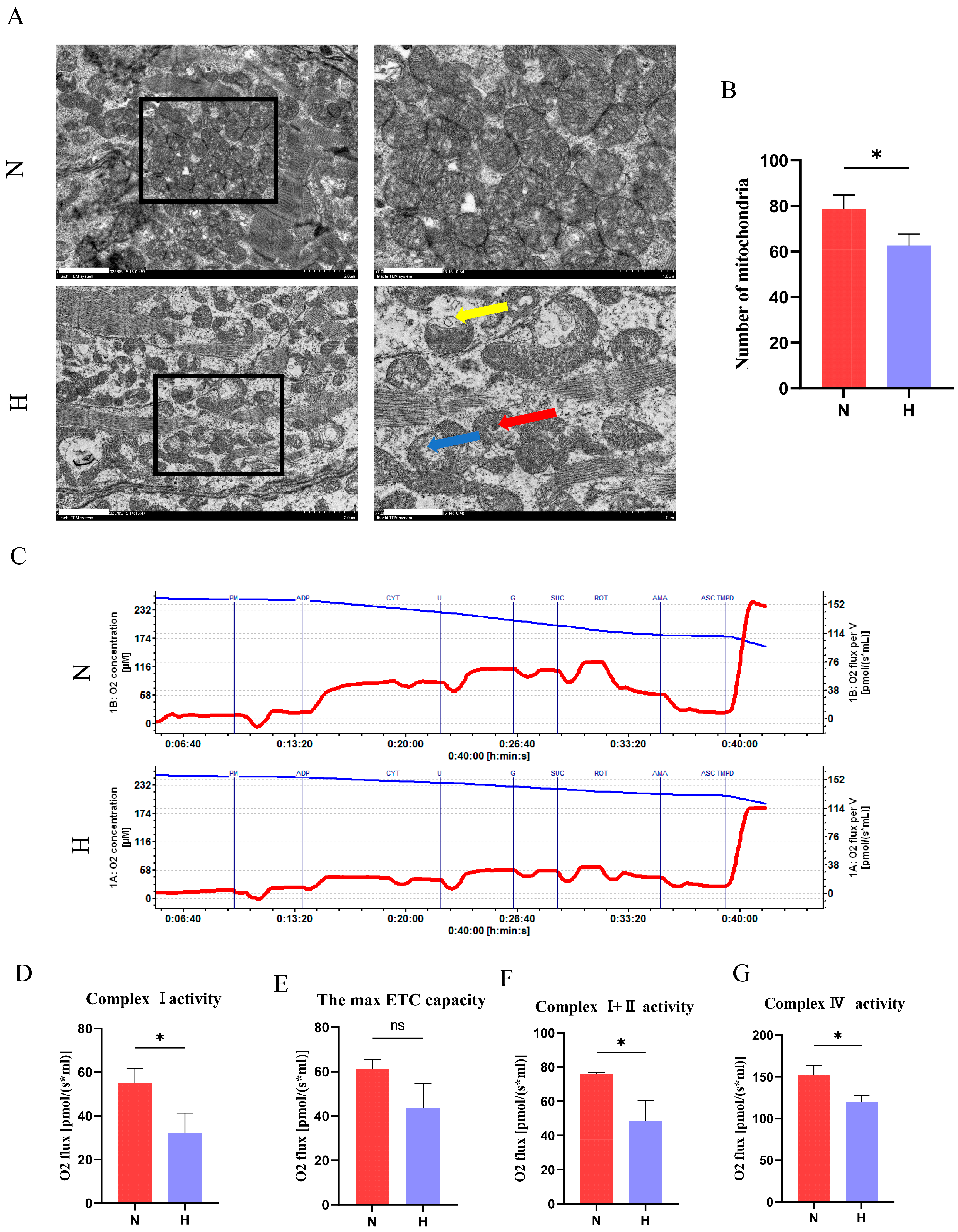
3.5. T2DM Impairs Mitochondrial Structure and Function in Zebrafish Hearts
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DCM | Diabetic Cardiomyopathy |
| HFD | High-Fat Diet |
| T2DM | Type 2 Diabetes Mellitus |
| FFA | Free Fatty Acid |
| STZ | Streptozotocin |
| BMI | Body Mass Index |
| RT-qPCR | Real-Time Quantitative Polymerase Chain Reaction |
| mtDNA | Mitochondrial DNA |
| ROS | Reactive Oxygen Species |
| ECM | Extracellular Matrix |
| EF | Ejection Fraction |
| FS | Fractional Shortening |
| EDD | End-Diastolic Diameter |
| ESD | End-Systolic Diameter |
| SV | Stroke Volume |
| IPGTT | Intraperitoneal Glucose Tolerance Test |
| AUC | Area Under the Curve |
| LDL-C | Low-Density Lipoprotein Cholesterol |
| GLUT4 | Glucose Transporter Type 4 |
| ETC | Electron Transport Chain |
| IL-1β | Interleukin-1 Beta |
| IL-18 | Interleukin-18 |
| α-SMA | Alpha-Smooth Muscle Actin |
| IRS-1 | Insulin Receptor Substrate 1 |
| NF-κB | Nuclear Factor Kappa-B |
| NOX4 | NADPH Oxidase 4 |
| NRF2 | Nuclear Factor Erythroid 2-Related Factor 2 |
| PGC1-α | Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-alpha |
| PPARα | Peroxisome Proliferator-Activated Receptor Alpha |
| CPT1a | Carnitine Palmitoyltransferase 1A |
| SIRT3 | Sirtuin 3 |
| SOD | Superoxide Dismutase |
| TGF-β1 | Transforming Growth Factor-Beta1 |
| TLR4 | Toll-Like Receptor 4 |
References
- Ke, J.; Pan, J.; Lin, H.; Gu, J. Diabetic cardiomyopathy: A brief summary on lipid toxicity. ESC Heart Fail. 2023, 10, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Whaley-Connell, A.; Sowers, J.R. Diabetic cardiomyopathy: A hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia 2018, 61, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Ouwens, D.M.; Boer, C.; Fodor, M.; de Galan, P.; Heine, R.J.; Maassen, J.A.; Diamant, M. Cardiac dysfunction induced by high-fat diet is associated with altered myocardial insulin signalling in rats. Diabetologia 2005, 48, 1229–1237. [Google Scholar] [CrossRef]
- Chavali, V.; Tyagi, S.C.; Mishra, P.K. Predictors and prevention of diabetic cardiomyopathy. Diabetes Metab. Syndr. Obes. Targets Ther. 2013, 6, 151–160. [Google Scholar] [CrossRef]
- Murarka, S.; Movahed, M.R. Diabetic cardiomyopathy. J. Card. Fail. 2010, 16, 971–979. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, G.; Virk, H.U.H.; Khalid, M.; Lavie, C.J.; Ventura, H.; Mukherjee, D.; Ramu, V.; Bhogal, S.; Kumar, G.; Shanmugasundaram, M.; et al. Diabetic cardiomyopathy—A comprehensive updated review. Prog. Cardiovasc. Dis. 2019, 62, 315–326. [Google Scholar] [CrossRef]
- Gollmer, J.; Zirlik, A.; Bugger, H. Mitochondrial Mechanisms in Diabetic Cardiomyopathy. Diabetes Metab. J. 2020, 44, 33–53. [Google Scholar] [CrossRef]
- El Hadi, H.; Vettor, R.; Rossato, M. Cardiomyocyte mitochondrial dysfunction in diabetes and its contribution in cardiac arrhythmogenesis. Mitochondrion 2019, 46, 6–14. [Google Scholar] [CrossRef]
- Xiang, K.; Qin, Z.; Zhang, H.; Liu, X. Energy Metabolism in Exercise-Induced Physiologic Cardiac Hypertrophy. Front. Pharmacol. 2020, 11, 1133. [Google Scholar] [CrossRef]
- Ye, W.; Han, K.; Xie, M.; Li, S.; Chen, G.; Wang, Y.; Li, T. Mitochondrial energy metabolism in diabetic cardiomyopathy: Physiological adaption, pathogenesis, and therapeutic targets. Chin. Med. J. 2024, 137, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Tan, J.; Cao, Z.; Cai, Y.; Huang, Z.; Chen, Z.; He, W.; Liu, X.; Jiang, Y.; Gao, Q.; et al. Sirt5 improves cardiomyocytes fatty acid metabolism and ameliorates cardiac lipotoxicity in diabetic cardiomyopathy via CPT2 de-succinylation. Redox Biol. 2024, 73, 103184. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, D.; Zhang, T.; Tong, Q.; Ye, R.D.; Lin, L. SIRT3 protects hepatocytes from oxidative injury by enhancing ROS scavenging and mitochondrial integrity. Cell Death Dis. 2017, 8, e3158. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, Y.D.; Kim, H.J.; Lee, Z.H.; Kim, H.H. SOD2 and Sirt3 Control Osteoclastogenesis by Regulating Mitochondrial ROS. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2017, 32, 397–406. [Google Scholar] [CrossRef]
- Yurre, A.R.; Martins, E.G.L.; Lopez-Alarcon, M.; Cabral, B.; Vera, N.; Lopes, J.A.; Galina, A.; Takiya, C.M.; Lindoso, R.S.; Vieyra, A.; et al. Type 2 diabetes mellitus alters cardiac mitochondrial content and function in a non-obese mice model. An. Acad. Bras. Cienc. 2020, 92, e20191340. [Google Scholar] [CrossRef]
- Lee, W.S.; Kim, J. Application of Animal Models in Diabetic Cardiomyopathy. Diabetes Metab. J. 2021, 45, 129–145. [Google Scholar] [CrossRef]
- Seth, A.; Stemple, D.L.; Barroso, I. The emerging use of zebrafish to model metabolic disease. Dis. Models Mech. 2013, 6, 1080–1088. [Google Scholar] [CrossRef]
- Carnovali, M.; Banfi, G.; Mariotti, M. Zebrafish Models of Human Skeletal Disorders: Embryo and Adult Swimming Together. BioMed Res. Int. 2019, 2019, 1253710. [Google Scholar] [CrossRef]
- Kannel, W.B.; McGee, D.L. Diabetes and cardiovascular disease. The Framingham study. Jama 1979, 241, 2035–2038. [Google Scholar] [CrossRef]
- Mira Hernandez, J.; Shen, E.Y.; Ko, C.Y.; Hourani, Z.; Spencer, E.R.; Smoliarchuk, D.; Bossuyt, J.; Granzier, H.; Bers, D.M.; Hegyi, B. Differential sex-dependent susceptibility to diastolic dysfunction and arrhythmia in cardiomyocytes from obese diabetic heart failure with preserved ejection fraction model. Cardiovasc. Res. 2025, 121, 254–266. [Google Scholar] [CrossRef]
- Visanji, M.; Venegas-Pino, D.E.; Werstuck, G.H. Understanding One Half of the Sex Difference Equation: The Modulatory Effects of Testosterone on Diabetic Cardiomyopathy. Am. J. Pathol. 2024, 194, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Toedebusch, R.; Belenchia, A.; Pulakat, L. Diabetic Cardiomyopathy: Impact of Biological Sex on Disease Development and Molecular Signatures. Front. Physiol. 2018, 9, 453. [Google Scholar] [CrossRef]
- Wang, L.W.; Huttner, I.G.; Santiago, C.F.; Kesteven, S.H.; Yu, Z.Y.; Feneley, M.P.; Fatkin, D. Standardized echocardiographic assessment of cardiac function in normal adult zebrafish and heart disease models. Dis. Models Mech. 2017, 10, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Song, S.; Attia, R.R.; Connaughton, S.; Niesen, M.I.; Ness, G.C.; Elam, M.B.; Hori, R.T.; Cook, G.A.; Park, E.A. Peroxisome proliferator activated receptor alpha (PPARalpha) and PPAR gamma coactivator (PGC-1alpha) induce carnitine palmitoyltransferase IA (CPT-1A) via independent gene elements. Mol. Cell. Endocrinol. 2010, 325, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Dimauro, I.; Paronetto, M.P.; Caporossi, D. Exercise, redox homeostasis and the epigenetic landscape. Redox Biol. 2020, 35, 101477. [Google Scholar] [CrossRef]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef]
- Chen, Z.; Zhou, Z.; Deng, Q.; Zou, Y.; Wang, B.; Huang, S.; Tian, J.; Zheng, L.; Peng, X.; Tang, C. Type 2 Diabetes Induces Mitochondrial Dysfunction in Zebrafish Skeletal Muscle Leading to Diabetic Myopathy via the miR-139-5p/NAMPT Pathway. Int. J. Mol. Sci. 2025, 26, 752. [Google Scholar] [CrossRef]
- Taylor, R. Type 2 diabetes: Etiology and reversibility. Diabetes Care 2013, 36, 1047–1055. [Google Scholar] [CrossRef]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef]
- McGee, S.L.; Hargreaves, M. Exercise performance and health: Role of GLUT4. Free. Radic. Biol. Med. 2024, 224, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Chaweewannakorn, C.; Nyasha, M.R.; Chen, W.; Sekiai, S.; Tsuchiya, M.; Hagiwara, Y.; Bouzakri, K.; Sasaki, K.; Kanzaki, M. Exercise-evoked intramuscular neutrophil-endothelial interactions support muscle performance and GLUT4 translocation: A mouse gnawing model study. J. Physiol. 2020, 598, 101–122. [Google Scholar] [CrossRef]
- Knudsen, J.R.; Steenberg, D.E.; Hingst, J.R.; Hodgson, L.R.; Henriquez-Olguin, C.; Li, Z.; Kiens, B.; Richter, E.A.; Wojtaszewski, J.F.P.; Verkade, P.; et al. Prior exercise in humans redistributes intramuscular GLUT4 and enhances insulin-stimulated sarcolemmal and endosomal GLUT4 translocation. Mol. Metab. 2020, 39, 100998. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Chen, B.; Behnke, B.J.; Ghosh, P.; Stabley, J.N.; Bramy, J.A.; Sepulveda, J.L.; Delp, M.D.; Muller-Delp, J.M. Exercise training reverses age-induced diastolic dysfunction and restores coronary microvascular function. J. Physiol. 2017, 595, 3703–3719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, J.; Yan, L.; He, Q.; Xie, H.; Chen, M. Resveratrol Ameliorates Cardiac Remodeling in a Murine Model of Heart Failure With Preserved Ejection Fraction. Front. Pharmacol. 2021, 12, 646240. [Google Scholar] [CrossRef]
- Chen, M.M.; Lam, A.; Abraham, J.A.; Schreiner, G.F.; Joly, A.H. CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: A potential role in heart fibrosis. J. Mol. Cell. Cardiol. 2000, 32, 1805–1819. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Deminice, R.; Ozdemir, M.; Yoshihara, T.; Bomkamp, M.P.; Hyatt, H. Exercise-induced oxidative stress: Friend or foe? J. Sport Health Sci. 2020, 9, 415–425. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| Glyceraldehyde-3-phosphate dehydrogenase (gapdh) | ATCATCTCTGCCCCAAGTGC | ACGGTCTTCTGTGTTGCTGT |
| Natriuretic peptide A (nppa) | ACAGAGACCGAGAGGAAG | AGGGTGCTGGAAGACCCTAT |
| Natriuretic peptide B (nppb) | CATGGGTGTTTTAAAGTTTCTCC | CTTCAATATTTGCCGCCTTTAC |
| Peroxisome proliferator-activated receptor alpha (ppara) | ATGTCCCACAATGCCATCCG | TCTGCTTGGCCAGGGTTTTC |
| Troponin T2, cardiac type (tnnt2) | GAAGGCCAGTGAAATGTGGC | CGACCTTTGGCACTCTGGT |
| Myosin heavy chain 7 (myh7) | CTTGGTGCACATCAGACAAGG | CTGGGGGTGAATGTCAGCTT |
| Myosin-binding protein C, cardiac-type (mybpc3) | CTCCACCAGCGAGCCTATTG | ACGTCTCTCATTTCTTGATGTCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.; Chen, Z.; Li, H.; Zou, Y.; Wang, B.; Zhao, W.; Zheng, L.; Zhou, Z.; Peng, X.; Tang, C. High-Fat Diet-Induced Diabetic Cardiomyopathy in Female Zebrafish: Cardiac Pathology and Functional Decline Mediated by Type 2 Diabetes. Nutrients 2025, 17, 2209. https://doi.org/10.3390/nu17132209
Huang S, Chen Z, Li H, Zou Y, Wang B, Zhao W, Zheng L, Zhou Z, Peng X, Tang C. High-Fat Diet-Induced Diabetic Cardiomyopathy in Female Zebrafish: Cardiac Pathology and Functional Decline Mediated by Type 2 Diabetes. Nutrients. 2025; 17(13):2209. https://doi.org/10.3390/nu17132209
Chicago/Turabian StyleHuang, Shuaiwang, Zhanglin Chen, Haoming Li, Yunyi Zou, Bihan Wang, Wenjun Zhao, Lan Zheng, Zuoqiong Zhou, Xiyang Peng, and Changfa Tang. 2025. "High-Fat Diet-Induced Diabetic Cardiomyopathy in Female Zebrafish: Cardiac Pathology and Functional Decline Mediated by Type 2 Diabetes" Nutrients 17, no. 13: 2209. https://doi.org/10.3390/nu17132209
APA StyleHuang, S., Chen, Z., Li, H., Zou, Y., Wang, B., Zhao, W., Zheng, L., Zhou, Z., Peng, X., & Tang, C. (2025). High-Fat Diet-Induced Diabetic Cardiomyopathy in Female Zebrafish: Cardiac Pathology and Functional Decline Mediated by Type 2 Diabetes. Nutrients, 17(13), 2209. https://doi.org/10.3390/nu17132209