
Ketogenic Diet Protects from Experimental Colitis in a Mouse Model Regardless of Dietary Fat Source

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
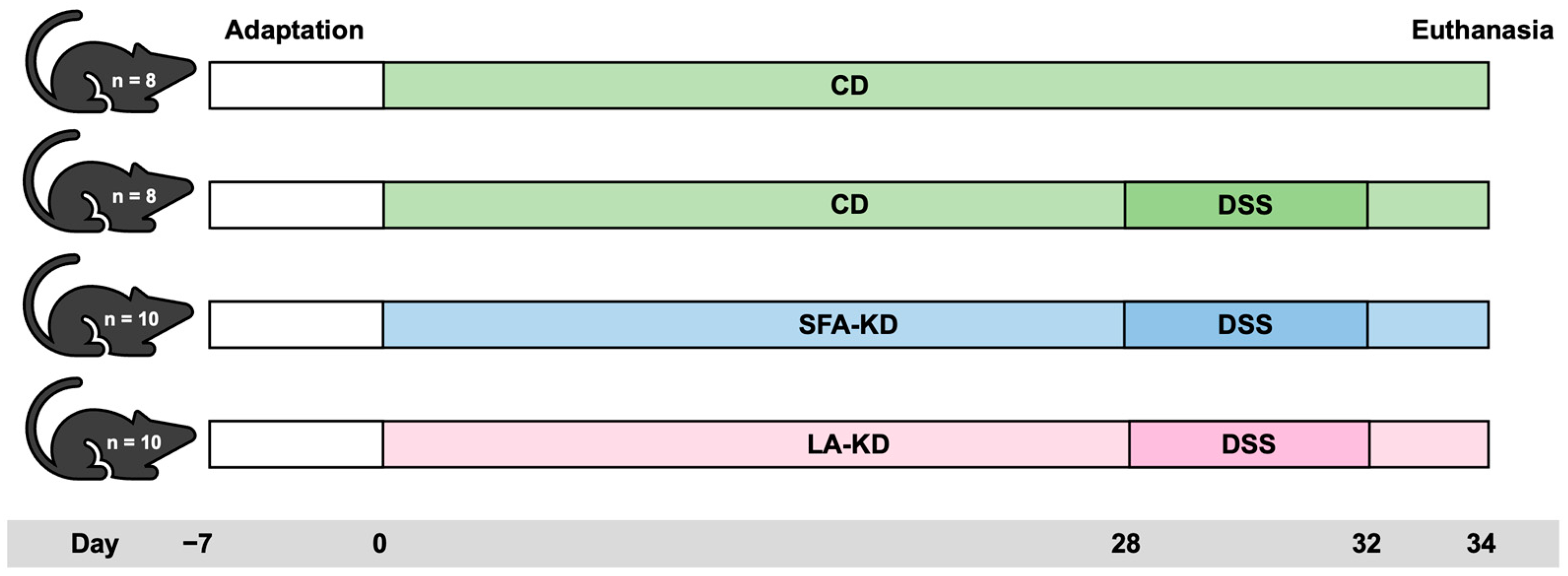
2.1. Animal Experiment
2.2. Collection of Samples
2.3. Measurement of Intestinal Permeability
2.4. Macroscopical Evaluation
2.5. Plasma β-Hydroxybutyrate
2.6. Histological Analyses
2.7. Reverse Transcription Quantitative Polymerase Chain Reaction
2.8. Western Blot
2.9. Statistical Analyses
3. Results
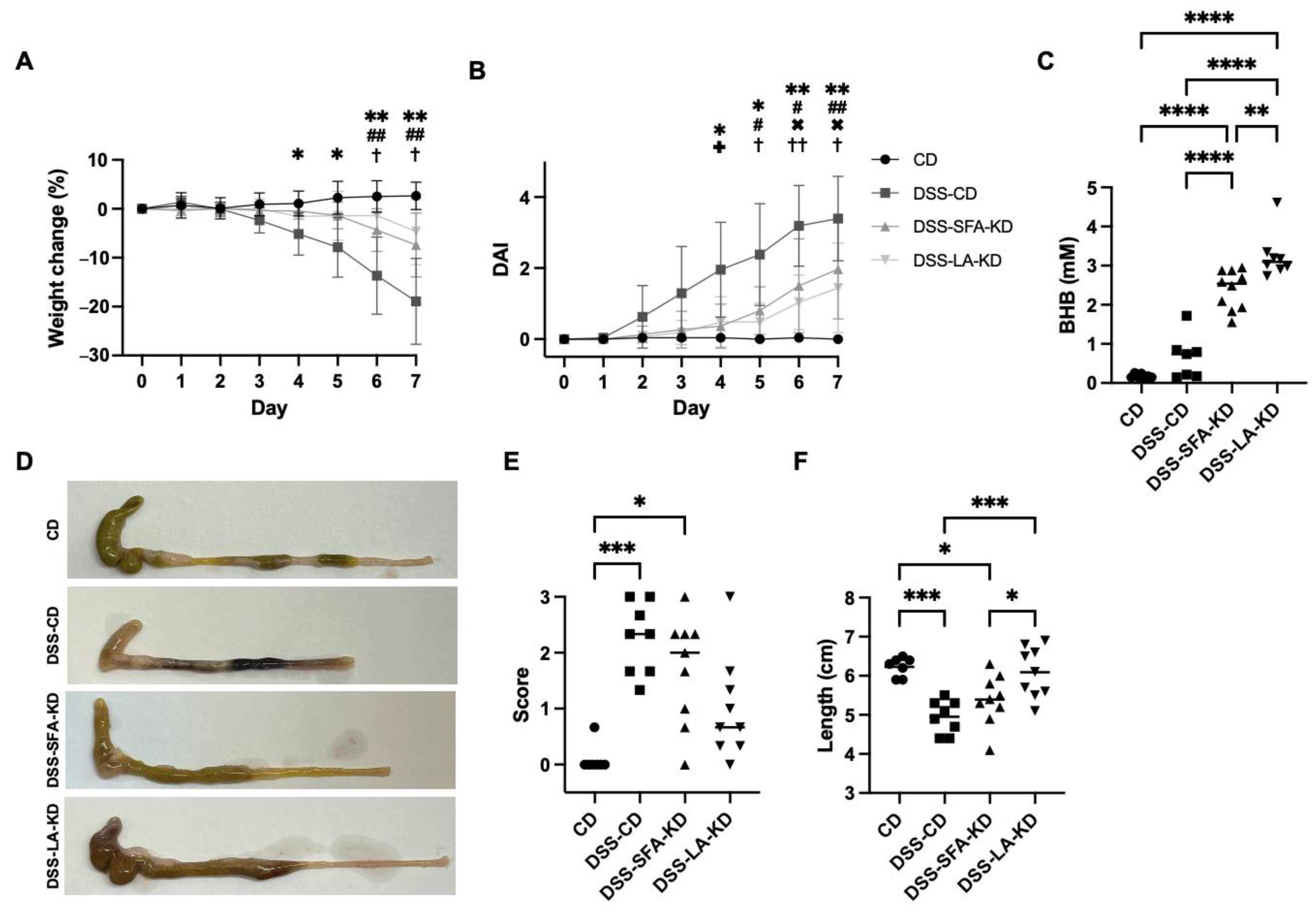
3.1. Ketogenic Diets Mitigate DSS-Induced Weight Loss and Macroscopic Signs of Inflammation
3.2. Ketogenic Diets Protect from DSS-Induced Mucosal Damage
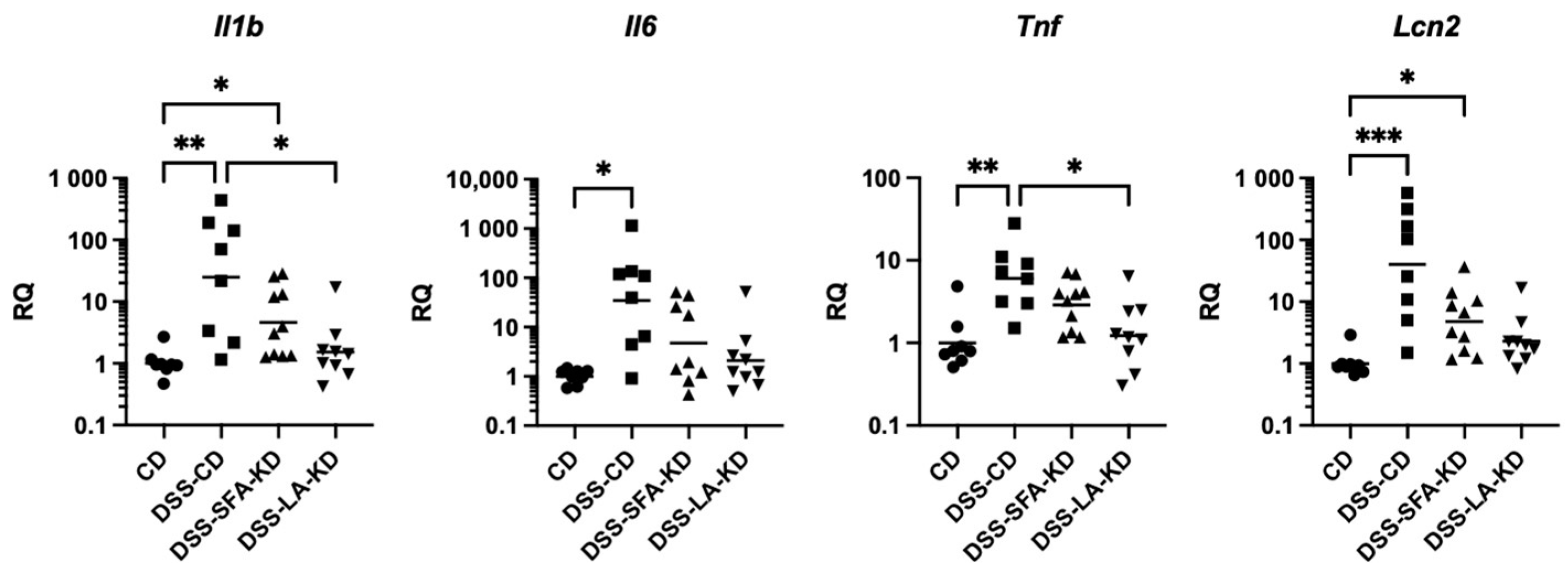
3.3. Ketogenic Diet High in Linoleic Acid Prevents Increases in Inflammatory Markers
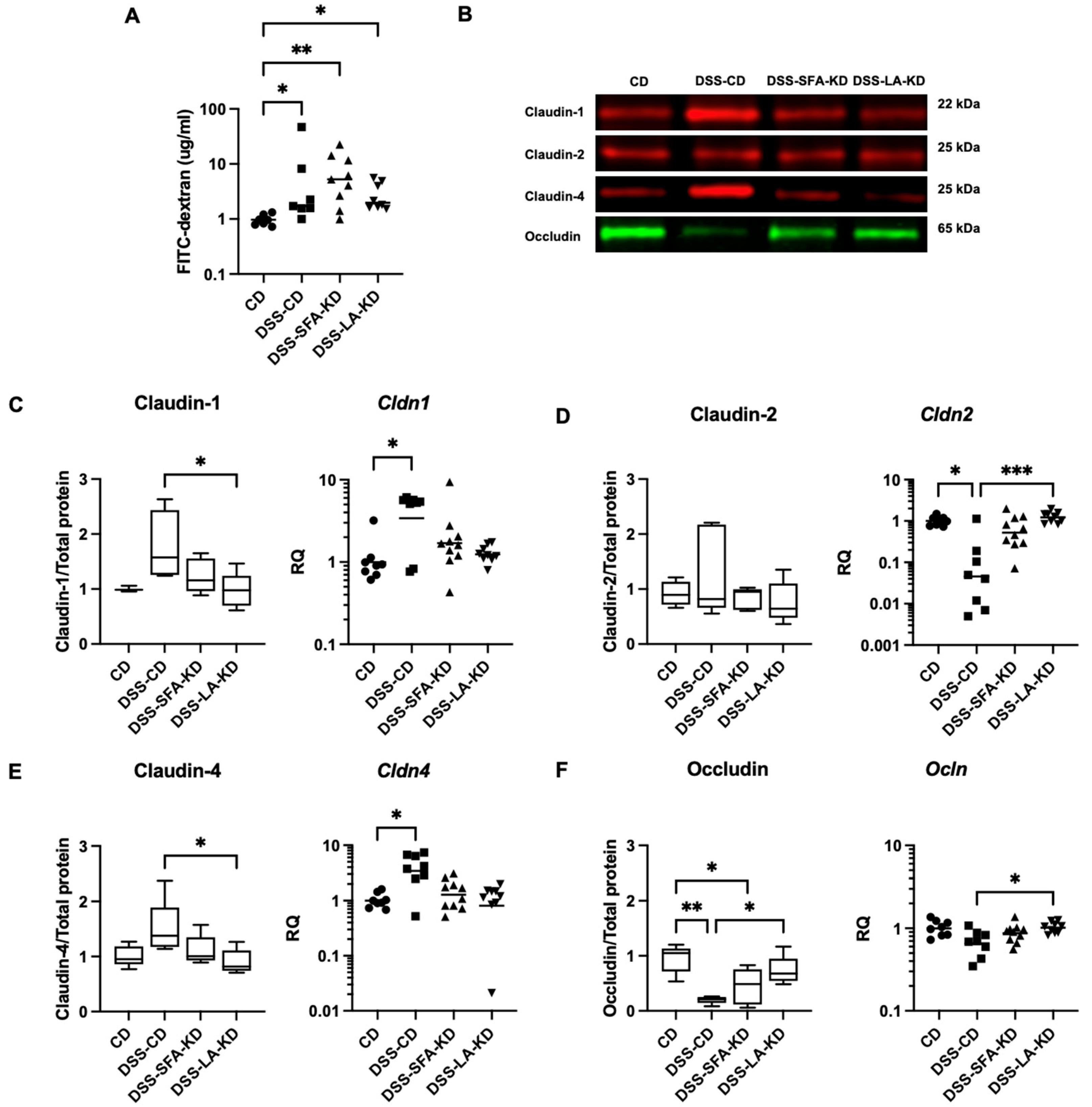
3.4. Ketogenic Diets Normalize DSS-Induced Abnormalities in Tight Junction Protein Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Wang, R.; Li, Z.; Liu, S.; Zhang, D. Global, regional and national burden of inflammatory bowel disease in 204 countries and territories from 1990 to 2019: A systematic analysis based on the Global Burden of Disease Study 2019. BMJ Open 2023, 13, e065186. [Google Scholar] [CrossRef] [PubMed]
- Kuenzig, M.E.; Manuel, D.G.; Donelle, J.; Benchimol, E.I. Life expectancy and health-adjusted life expectancy in people with inflammatory bowel disease. CMAJ 2020, 192, E1394–E1402. [Google Scholar] [CrossRef] [PubMed]
- Gubatan, J.; Kulkarni, C.V.; Talamantes, S.M.; Temby, M.; Fardeen, T.; Sinha, S.R. Dietary Exposures and Interventions in Inflammatory Bowel Disease: Current Evidence and Emerging Concepts. Nutrients 2023, 15, 579. [Google Scholar] [CrossRef] [PubMed]
- Windmueller, H.G.; Spaeth, A.E. Identification of ketone bodies and glutamine as the major respiratory fuels in vivo for postabsorptive rat small intestine. J. Biol. Chem. 1978, 253, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Shippy, D.C.; Wilhelm, C.; Viharkumar, P.A.; Raife, T.J.; Ulland, T.K. beta-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer’s disease pathology. J. Neuroinflamm. 2020, 17, 280. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Dmitrieva-Posocco, O.; Wong, A.C.; Lundgren, P.; Golos, A.M.; Descamps, H.C.; Dohnalova, L.; Cramer, Z.; Tian, Y.; Yueh, B.; Eskiocak, O.; et al. beta-Hydroxybutyrate suppresses colorectal cancer. Nature 2022, 605, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Gigante, I.; Tutino, V.; Russo, F.; De Nunzio, V.; Coletta, S.; Armentano, R.; Crovace, A.; Caruso, M.G.; Orlando, A.; Notarnicola, M. Cannabinoid Receptors Overexpression in a Rat Model of Irritable Bowel Syndrome (IBS) after Treatment with a Ketogenic Diet. Int. J. Mol. Sci. 2021, 22, 2880. [Google Scholar] [CrossRef]
- Abdelhady, R.; Saber, S.; Ahmed Abdel-Reheim, M.; Mohammad, S.A.M.; Alfaifi, J.; Adam, M.I.E.; Saleh, L.A.; Farag, A.I.; Elmorsy, E.A.; El-Wakeel, H.S.; et al. Unveiling the therapeutic potential of exogenous beta-hydroxybutyrate for chronic colitis in rats: Novel insights on autophagy, apoptosis, and pyroptosis. Front. Pharmacol. 2023, 14, 1239025. [Google Scholar] [CrossRef]
- Kong, C.; Yan, X.; Liu, Y.; Huang, L.; Zhu, Y.; He, J.; Gao, R.; Kalady, M.F.; Goel, A.; Qin, H.; et al. Ketogenic diet alleviates colitis by reduction of colonic group 3 innate lymphoid cells through altering gut microbiome. Signal Transduct. Target. Ther. 2021, 6, 154. [Google Scholar] [CrossRef]
- Li, S.; Zhuge, A.; Wang, K.; Lv, L.; Bian, X.; Yang, L.; Xia, J.; Jiang, X.; Wu, W.; Wang, S.; et al. Ketogenic diet aggravates colitis, impairs intestinal barrier and alters gut microbiota and metabolism in DSS-induced mice. Food Funct. 2021, 12, 10210–10225. [Google Scholar] [CrossRef]
- Kono, H.; Fujii, H.; Asakawa, M.; Yamamoto, M.; Matsuda, M.; Maki, A.; Matsumoto, Y. Protective Effects of Medium-Chain Triglycerides on the Liver and Gut in Rats Administered Endotoxin. Ann. Surg. 2003, 237, 246–255. [Google Scholar] [CrossRef]
- Kirpich, I.A.; Feng, W.; Wang, Y.; Liu, Y.; Barker, D.F.; Barve, S.S.; McClain, C.J. The type of dietary fat modulates intestinal tight junction integrity, gut permeability, and hepatic toll-like receptor expression in a mouse model of alcoholic liver disease. Alcohol. Clin. Exp. Res. 2012, 36, 835–846. [Google Scholar] [CrossRef]
- Toivio, L.; Launonen, H.; Linden, J.; Lehto, M.; Vapaatalo, H.; Salmenkari, H.; Korpela, R. Ketogenic Diet High in Saturated Fat Promotes Colonic Claudin Expression without Changes in Intestinal Permeability to Iohexol in Healthy Mice. Nutrients 2023, 16, 18. [Google Scholar] [CrossRef]
- Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 2017, 12, 1295–1309. [Google Scholar] [CrossRef] [PubMed]
- Melgar, S.; Karlsson, A.; Michaelsson, E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: Correlation between symptoms and inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1328–G1338. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Eissa, N.; Hussein, H.; Wang, H.; Rabbi, M.F.; Bernstein, C.N.; Ghia, J.E. Stability of Reference Genes for Messenger RNA Quantification by Real-Time PCR in Mouse Dextran Sodium Sulfate Experimental Colitis. PLoS ONE 2016, 11, e0156289. [Google Scholar] [CrossRef] [PubMed]
- Salmenkari, H.; Pasanen, L.; Linden, J.; Korpela, R.; Vapaatalo, H. Beneficial anti-inflammatory effect of angiotensin-converting enzyme inhibitor and angiotensin receptor blocker in the treatment of dextran sulfate sodium-induced colitis in mice. J. Physiol. Pharmacol. 2018, 69, 561–572. [Google Scholar] [CrossRef]
- Li, G.Z.; Wang, Z.H.; Cui, W.; Fu, J.L.; Wang, Y.R.; Liu, P. Tumor necrosis factor alpha increases intestinal permeability in mice with fulminant hepatic failure. World J. Gastroenterol. 2012, 18, 5042–5050. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Wu, S.; Lu, R.; Zhou, D.; Zhou, J.; Carmeliet, G.; Petrof, E.; Claud, E.C.; Sun, J. Tight junction CLDN2 gene is a direct target of the vitamin D receptor. Sci. Rep. 2015, 5, 10642. [Google Scholar] [CrossRef] [PubMed]
- Song, C.H.; Kim, N.; Sohn, S.H.; Lee, S.M.; Nam, R.H.; Na, H.Y.; Lee, D.H.; Surh, Y.J. Effects of 17beta-Estradiol on Colonic Permeability and Inflammation in an Azoxymethane/Dextran Sulfate Sodium-Induced Colitis Mouse Model. Gut Liver 2018, 12, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Salmenkari, H.; Laitinen, A.; Forsgard, R.A.; Holappa, M.; Linden, J.; Pasanen, L.; Korhonen, M.; Korpela, R.; Nystedt, J. The use of unlicensed bone marrow-derived platelet lysate-expanded mesenchymal stromal cells in colitis: A pre-clinical study. Cytotherapy 2019, 21, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Nakano, D.; Kitada, K.; Wan, N.; Zhang, Y.; Wiig, H.; Wararat, K.; Yanagita, M.; Lee, S.; Jia, L.; Titze, J.M.; et al. Lipopolysaccharide induces filtrate leakage from renal tubular lumina into the interstitial space via a proximal tubular Toll-like receptor 4-dependent pathway and limits sensitivity to fluid therapy in mice. Kidney Int. 2020, 97, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, S.; Zhang, Y.; Chen, J.; Wu, F.; Liu, G.; Chen, G.Q. Applications and Mechanism of 3-Hydroxybutyrate (3HB) for Prevention of Colonic Inflammation and Carcinogenesis as a Food Supplement. Mol. Nutr. Food Res. 2021, 65, e2100533. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, J.; Liu, H.; Huang, R.; Yan, X.; Song, M.; Tan, G.; Zhi, F. Ketone body beta-hydroxybutyrate ameliorates colitis by promoting M2 macrophage polarization through the STAT6-dependent signaling pathway. BMC Med. 2022, 20, 148. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Hata, M.; Furuse, K.; Yoshida, Y.; Haratake, A.; Sugitani, Y.; Noda, T.; Kubo, A.; Tsukita, S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: A lesson from claudin-1-deficient mice. J. Cell Biol. 2002, 156, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Michikawa, H.; Fujita-Yoshigaki, J.; Sugiya, H. Enhancement of barrier function by overexpression of claudin-4 in tight junctions of submandibular gland cells. Cell Tissue Res. 2008, 334, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.R.; Nalle, S.C.; Tretiakova, M.; Rubin, D.T.; Turner, J.R. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab. Investig. 2008, 88, 1110–1120. [Google Scholar] [CrossRef]
- Pope, J.L.; Bhat, A.A.; Sharma, A.; Ahmad, R.; Krishnan, M.; Washington, M.K.; Beauchamp, R.D.; Singh, A.B.; Dhawan, P. Claudin-1 regulates intestinal epithelial homeostasis through the modulation of Notch-signalling. Gut 2014, 63, 622–634. [Google Scholar] [CrossRef]
- Ang, Q.Y.; Alexander, M.; Newman, J.C.; Tian, Y.; Cai, J.; Upadhyay, V.; Turnbaugh, J.A.; Verdin, E.; Hall, K.D.; Leibel, R.L.; et al. Ketogenic Diets Alter the Gut Microbiome Resulting in Decreased Intestinal Th17 Cells. Cell 2020, 181, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, W.; Yu, T.; Yu, Y.; Cui, X.; Zhou, Z.; Yang, H.; Yu, Y.; Bilotta, A.J.; Yao, S.; et al. Th17 Cell-Derived Amphiregulin Promotes Colitis-Associated Intestinal Fibrosis Through Activation of mTOR and MEK in Intestinal Myofibroblasts. Gastroenterology 2023, 164, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Schoeler, M.; Ellero-Simatos, S.; Birkner, T.; Mayneris-Perxachs, J.; Olsson, L.; Brolin, H.; Loeber, U.; Kraft, J.D.; Polizzi, A.; Marti-Navas, M.; et al. The interplay between dietary fatty acids and gut microbiota influences host metabolism and hepatic steatosis. Nat. Commun. 2023, 14, 5329. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Jiang, Y.; Wu, L.; Fu, J.; Du, J.; Luo, Z.; Guo, L.; Xu, J.; Liu, Y. Porphyromonas gingivalis aggravates colitis via a gut microbiota-linoleic acid metabolism-Th17/Treg cell balance axis. Nat. Commun. 2024, 15, 1617. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD | SFA-KD | LA-KD | |
|---|---|---|---|
| Protein (E%) | 9.7 | 9.4 | 9.4 |
| Carbohydrate (E%) | 77.8 | 0.5 | 0.5 |
| Fat (E%) | 12.5 | 90.1 | 90.1 |
| Saturated fat (E%) | 2.6 | 56.6 | 22.8 |
| Monounsaturated fat (E%) | 3.0 | 26.0 | 17.9 |
| Polyunsaturated fat (E%) | 6.9 | 4.5 | 49.3 |
| Linoleic acid (E%) | 6.4 | 4.0 | 44.5 |
| Gene | Forward Primer | Reverse Primer | Ref. |
|---|---|---|---|
| 18S | AACGAACGAGACTCTGGCAT | ACGCCACTTGTCCCTCTAAG | |
| Eef2 | TGTCAGTCATCGCCCATGTG | CATCCTTGCGAGTGTCAGTGA | [18] |
| Rplp0 | TAACCCTGAAGTGCTCGACA | GGTACCCGATCTGCAGACA | [19] |
| Cldn1 | AGACCTGGATTTGCATCTTGGTG | TGCAACATAGGCAGGACAAGAGTTA | [20] |
| Cldn2 | GCAAACAGGCTCCGAAGATACT | GAGATGATGCCCAAGTACAGAG | [21] |
| Cldn4 | TGAGCGATGGCGTCTATGG | GATGTTGCTGCCGATGAAGG | |
| Ocln | CGGTACAGCAGCAATGGTAA | CTCCCCACCTGTCGTGTAGT | [22] |
| Il1b | CTCCAGCCAAGCTTCCTTGT | TCATCACTGTCAAAAGGTGGCA | [23] |
| Il6 | ATCGTGGAAATGAGAAAAGAGTTGT | CTGCAAGTGCATCATCGTTGT | |
| Tnf | TGGCACCACTAGTTGGTTGTCT | AGCCTGTAGCCCACGTCGTA | [24] |
| Lcn2 | CCACCACGGACTACAACCAG | AGCTCCTTGGTTCTTCCATACAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toivio, L.; Lindén, J.; Lehto, M.; Salmenkari, H.; Korpela, R. Ketogenic Diet Protects from Experimental Colitis in a Mouse Model Regardless of Dietary Fat Source. Nutrients 2024, 16, 1348. https://doi.org/10.3390/nu16091348
Toivio L, Lindén J, Lehto M, Salmenkari H, Korpela R. Ketogenic Diet Protects from Experimental Colitis in a Mouse Model Regardless of Dietary Fat Source. Nutrients. 2024; 16(9):1348. https://doi.org/10.3390/nu16091348
Chicago/Turabian StyleToivio, Lotta, Jere Lindén, Markku Lehto, Hanne Salmenkari, and Riitta Korpela. 2024. "Ketogenic Diet Protects from Experimental Colitis in a Mouse Model Regardless of Dietary Fat Source" Nutrients 16, no. 9: 1348. https://doi.org/10.3390/nu16091348
APA StyleToivio, L., Lindén, J., Lehto, M., Salmenkari, H., & Korpela, R. (2024). Ketogenic Diet Protects from Experimental Colitis in a Mouse Model Regardless of Dietary Fat Source. Nutrients, 16(9), 1348. https://doi.org/10.3390/nu16091348